经取代的多环性吡啶酮衍生物及其前药
文献发布时间:2023-06-19 11:24:21
本申请为2016年4月27日提交的申请号为PCT/JP2016/063139、发明名称为“经取代的多环性吡啶酮衍生物及其前药”的国际申请的分案申请,该国际申请于2017年12月27日进入中国国家阶段,申请号为201680037827.7。
技术领域
本发明涉及一种表现出帽依赖性核酸内切酶(Cap-dependent endonuclease)抑制活性的经取代的多环性吡啶酮衍生物、其前药、及含有这些的药物组合物。
背景技术
流行性感冒是由流行性感冒病毒的感染引起的急性呼吸道传染病。在日本每年冬季有数百万人的流行性感冒患者的报道,且流行性感冒伴随着较高的发病率与死亡率。对于婴幼儿、老年人等高危人群而言,流行性感冒是尤其重要的疾病,且在老年人中肺炎的并发率较高,因流行性感冒而导致死亡的大部分人为老年人。
作为抗流行性感冒药,公知有抑制病毒的脱核过程的盐酸金刚烷胺(Symmetrel;商品名:金刚烷胺(Amantadine))或盐酸金刚乙胺(Flumadine;商品名:金刚乙胺(Rimantadine))、抑制病毒自细胞出芽、释出之神经胺酸酶抑制剂即奥司他韦(Oseltamivir;商品名:达菲(Tamiflu))或扎那米韦(Zanamivir;商品名:瑞乐砂(Relenza))。然而,有耐药性菌株的出现、副作用的问题,又有病原性或致死性较高的新型流行性感冒病毒世界性大流行等的顾虑,因此期望开发出新型机理的抗流行性感冒药。
关于作为源自流行性感冒病毒的酶的帽依赖性核酸内切酶,由于其对于病毒增殖而言是必须的,且具有宿主所不具有的病毒特异性酶活性,故而认为该帽依赖性核酸内切酶适于抗流行性感冒药的目标物。流行性感冒病毒的帽依赖性核酸内切酶是生成以宿主mRNA前体为底物且包含帽结构的9~13个碱基(帽结构的碱基并不包含于上述碱基数量中)的片段的核酸内切酶活性。该片段作为病毒RNA聚合酶的引子发挥功能,用于编码病毒蛋白质的mRNA的合成。即,认为抑制帽依赖性核酸内切酶的物质通过抑制病毒mRNA的合成而抑制病毒蛋白质的合成,结果抑制病毒增殖。
作为抑制帽依赖性核酸内切酶的化合物,报道了Flutimide(专利文献1以及非专利文献1及2)、4-取代2,4-二氧代丁酸(专利文献2以及非专利文献3及4)及近年来专利文献3~12所记载的化合物等,但尚未在临床上用作抗流行性感冒药。专利文献9及12记载有具有与本发明化合物类似的结构的化合物,但并未记载本申请化合物。另外,专利文献13~15中记载有具有与本发明化合物类似的结构的化合物作为具有HIV整合酶抑制活性的化合物,但并无关于帽依赖性核酸内切酶的记载。需要说明的是,在专利文献16及17中记载有已由申请人提出申请的具有与具有帽依赖性核酸内切酶抑制活性的本发明化合物类似的结构的化合物及其前药,但并未记载本发明化合物。
现有技术文献
专利文献
专利文献1:GB第2280435号说明书
专利文献2:US第5475109号说明书
专利文献3:US第20130090300号说明书
专利文献4:国际公开第2013/057251号说明书
专利文献5:国际公开第2013/174930号说明书
专利文献6:国际公开第2014/023691号说明书
专利文献7:国际公开第2014/043252号说明书
专利文献8:国际公开第2014/074926号说明书
专利文献9:国际公开第2014/108406号说明书
专利文献10:国际公开第2014/108407号说明书
专利文献11:国际公开第2014/108408号说明书
专利文献12:国际公开第2015/038655号说明书
专利文献13:国际公开第2005/016927号说明书
专利文献14:国际公开第2006/066414号说明书
专利文献15:国际公开第2007/049675号说明书
专利文献16:国际公开第2010/147068号说明书
专利文献17:国际公开第2012/039414号说明书
非专利文献
非专利文献1:Tetrahedron Lett 1995,36(12),2005
非专利文献2:Tetrahedron Lett 1995,36(12),2009
非专利文献3:Antimicrobial Agents And Chemotherapy,Dec.1994,p.2827-2837
非专利文献4:Antimicrobial Agents And Chemotherapy,May 1996,p.1304–1307
发明内容
发明要解决的问题
本发明的目的在于提供一种具有抗病毒作用、尤其是流行性感冒病毒的增殖抑制活性的化合物。本发明的另一目的在于提供一种通过将用于向活体给药(例如,口服给药)的化合物进行前药化,从而在给药后被有效地吸收至体内、表现出高药理效果的化合物。
解决问题的方法
本发明提供以下所示的发明。
(1)以下的式(I)所示的化合物或其药学上可接受的盐,
[化学式1]
(式中,P为氢或形成前药的P
A
A
A
A
这里,由A
R
R
R
R
R
X为CH
R
m为0~2的整数
n为1~2的整数。)
并且,在式(I)所示的化合物中,以下的化合物除外,
[化学式2]
(式中,各符号与上述含义相同)。
(2)如上述(1)所述的化合物或其药学上可接受的盐,其中,
[化学式3]
(式中,各符号与上述(1)含义相同)
所示的基团为
[化学式4]
(式中,R
(3)如上述(1)所述的化合物或其药学上可接受的盐,其中,
[化学式5]
(式中,各符号与上述(1)含义相同)
所示的基团为
[化学式6]
(4)如上述(1)~(3)中任一项所述的化合物或其药学上可接受的盐,其中,
[化学式7]
(式中,各符号与上述(1)含义相同)
所示的基团为
[化学式8]
(式中,各符号与上述(1)含义相同)。
(5)如上述(1)所述的化合物或其药学上可接受的盐,其由以下的任意式表示,
[化学式9]
[化学式10]
(式中,各符号与上述(1)含义相同)。
(6)如上述(1)所述的化合物或其药学上可接受的盐,其由下式表示,
[化学式11]
(式中,各符号与上述(1)含义相同)。
(7)如上述(1)所述的化合物或其药学上可接受的盐,其由下式表示,
[化学式12]
(式中,各符号与上述(1)含义相同)。
(8)如上述(1)所述的化合物或其药学上可接受的盐,其由下式表示,
[化学式13]
(式中,各符号与上述(1)含义相同)。
(9)如上述(1)所述的化合物或其药学上可接受的盐,其由下式表示,
[化学式14]
(式中,各符号与上述(1)含义相同)。
(10)如上述(1)所述的化合物或其药学上可接受的盐,其由下式表示,
[化学式15]
(式中,各符号与上述(1)含义相同)。
(11)一种化合物或其药学上可接受的盐,其由下式表示,
[化学式16]
(式中,P为氢或形成前药的P
(12)如上述(1)~(11)中任一项所述的化合物或其药学上可接受的盐,其中,P
a)-C(=O)-P
b)-C(=O)-P
c)-C(=O)-L-P
d)-C(=O)-L-O-P
e)-C(=O)-L-O-L-O-P
f)-C(=O)-L-O-C(=O)-P
g)-C(=O)-O-P
h)-C(=O)-N(-K)(P
i)-C(=O)-O-L-O-P
j)-C(P
k)-C(P
l)-C(P
m)-C(P
n)-C(P
o)-C(P
p)-C(P
q)-C(P
r)-C(P
s)-C(P
t)-C(P
u)-C(P
v)-C(P
w)-C(=N
x)-C(P
y)-C(P
z)-P(=O)(-P
aa)-S(=O)
ab)-P
ac)-C(P
(式中,L为直链或支链状的亚烷基、或者直链或支链状的亚烯基,
K为氢、或任选被取代基组A取代的烷基,
P
P
P
P
P
P
P
P
P
P
并且,
P
P
P
取代基组A:氧代基、烷基、羟基烷基、氨基、烷基氨基、碳环基、杂环基、碳环烷基、烷基羰基、卤素、羟基、羧基、烷基羰基氨基、烷基羰基氨基烷基、烷基羰氧基、烷氧基羰基、烷氧基羰基烷基、烷氧基羰氧基、烷基氨基羰氧基、烷基氨基烷基、烷氧基、氰基、硝基、叠氮基、烷基磺酰基、三烷基甲硅烷基、及磷酰基)。
(13)如上述(12)所述的化合物或其药学上可接受的盐,其中,P
a)-C(=O)-P
b)-C(=O)-P
g)-C(=O)-O-P
h)-C(=O)-N(-K)(P
i)-C(=O)-O-L-O-P
l)-C(P
m)-C(P
o)-C(P
v)-C(P
x)-C(P
y)-C(P
z)-P(=O)(-P
(式中,L为直链或支链状的亚烷基;
K为氢、或任选被取代基组A取代的烷基,
P
P
P
P
P
P
P
P
并且,
P
取代基组A:氧代基、烷基、烷基氨基、碳环基、杂环基、烷基羰基、卤素、羟基、烷基羰基氨基、烷基羰氧基、烷氧基羰基、烷氧基羰基烷基、烷基氨基羰氧基、烷氧基、硝基、叠氮基、烷基磺酰基、及三烷基甲硅烷基)。
(14)以下述中的任意式表示的化合物或其药学上可接受的盐,
[化学式17]
(15)以下述中的任意式表示的化合物或其药学上可接受的盐,
[化学式18]
(16)一种药物组合物,其含有上述(1)~(15)中任一项所述的化合物、其药学上可接受的盐。
(17)如上述(16)所述的药物组合物,其具有抗病毒作用。
(18)如上述(16)所述的药物组合物,其具有帽依赖性核酸内切酶抑制作用。
(19)由具有帽依赖性核酸内切酶的病毒引起的疾病的治疗或预防方法,其特征在于:给药上述(1)~(15)中任一项所述的化合物、或其药学上可接受的盐。
(20)如上述(1)~(15)中任一项所述的化合物或其药学上可接受的盐,其用于治疗或预防由具有帽依赖性核酸内切酶的病毒引起的疾病。
(21)上述(1)~(15)中任一项所述的化合物或其药学上可接受的盐的用于制造由具有帽依赖性核酸内切酶的病毒引起的疾病的治疗或预防剂的用途。
(22)一种用于口服给药的药物组合物,其含有上述(1)~(15)中任一项所述的化合物、或其药学上可接受的盐。
(23)如(22)所述的药物组合物,其为片剂、粉剂、颗粒剂、胶囊剂、丸剂、膜剂、悬浮剂、乳剂、酏剂、糖浆剂、柠檬剂、醑剂、芳香水剂、提取剂、煎剂或酊剂。
(24)如(16)所述的药物组合物,其为糖衣片、膜衣片、肠溶性包衣片、缓释片、含片、舌下片、颊含片、咀嚼片、口腔崩解片、干糖浆剂、软胶囊剂、微胶囊剂或缓释性胶囊剂。
(25)一种用于非口服给药的药物组合物,其含有上述(1)~(15)中任一项所述的化合物、或其药学上可接受的盐。
(26)如(25)所述的药物组合物,其用于经皮、皮下、静脉内、动脉内、肌内、腹膜内、经黏膜、吸入、经鼻、滴眼、滴耳或阴道内给药。
(27)如(25)或(26)所述的药物组合物,其为注射剂、点滴剂、滴眼剂、滴鼻剂、滴耳剂、气雾剂、吸入剂、洗剂、注入剂、涂布剂、含漱剂、灌肠剂、软膏剂、硬膏剂、凝胶剂、霜剂、贴剂、糊剂、外用粉剂或栓剂。
(28)一种儿童用或老年人用的药物组合物,其含有上述(1)~(15)中任一项所述的化合物、或其药学上可接受的盐。
(29)一种药物组合物,其包含上述(1)~(15)中任一项所述的化合物或其药学上可接受的盐、与神经胺酸酶抑制剂、RNA依赖性RNA聚合酶抑制剂、M2蛋白质抑制剂、PB2 Cap结合抑制剂、抗HA抗体、或免疫作用药的组合。
(30)用于与神经胺酸酶抑制剂、RNA依赖性RNA聚合酶抑制剂、M2蛋白质抑制剂、PB2 Cap结合抑制剂、抗HA抗体、或免疫作用药的并用疗法的药物组合物,其含有上述(1)~(15)中任一项所述的化合物、或其药学上可接受的盐。
本发明进一步提供一种使用前药化合物的流行性感冒传染病的治疗法或预防法、以及具有抗流行性感冒作用的上述化合物。本发明进一步提供一种前药化合物的母化合物。该母化合物系作为抗流行性感冒剂、或该前药化合物的中间体有用。
发明的效果
本发明的化合物具有对帽依赖性核酸内切酶的抑制活性。更优选的化合物是前药,由于其在给药后在体内成为具有对帽依赖性核酸内切酶的抑制活性的母化合物,因此作为流行性感冒传染病的治疗剂和/或预防剂而有用。
附图说明
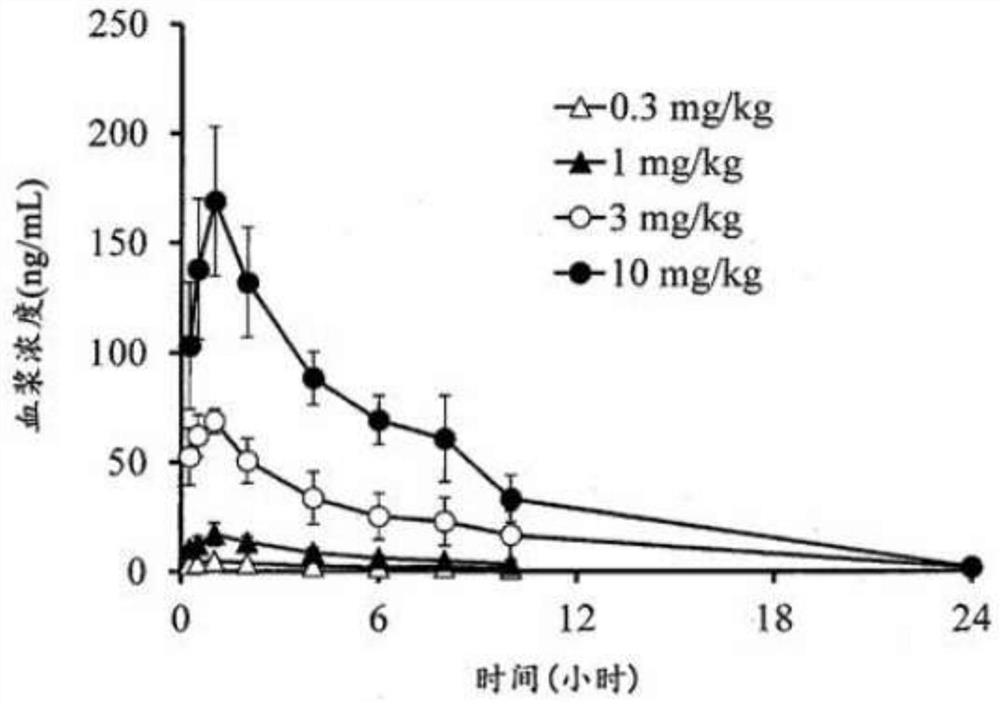
图1是针对将作为母化合物的化合物III-2进行前药化而成的化合物II-6,测定在非断食下向大鼠口服给药后化合物III-2的血浆中浓度变化而获得的结果。
图2是针对将作为母化合物的化合物III-2进行前药化而成的化合物II-6,在测定非断食下向大鼠口服给药后化合物II-6的血浆中浓度变化而获得的结果。
具体实施方式
以下对本说明书中所使用的各用语的含义进行说明。各用语只要没有特别事先说明,则无论是在单独使用的情况下,还是与其他用语组合使用的情况下,均以相同的含义使用。
“由…构成”的用语意指仅具有构成要素。
“包含”这一用语意指不限定于构成要素,不排除未记载的要素。
所谓“任选被取代基组A取代”,意指可在任意位置经选自取代基组A中的1个或2个以上的相同或不同的取代基取代。
本说明书中的所谓“前药”,是指以下的反应式中的式(II)所示的化合物或其药学上可接受的盐,表示的是在体内的生理条件下通过由药物代谢酶、水解酶、胃酸、肠内细菌等所引起的分解反应而转化为式(III)所示的化合物,从而表现出帽依赖性核酸内切酶(CEN)抑制活性、和/或CPE抑制效果的化合物。
[化学式19]
(式中,各符号与上述含义相同)
该前药更优选表示的是:体内给药时的生物利用率和/或AUC(血中浓度曲线下面积)较式(III)所示的化合物得到提高的化合物。
因此,该前药在给药至活体(例如,口服给药)后在胃和/或肠等中被高效地吸收至体内,其后被转化为式(III)所示的化合物,因而可优选地表现出较式(III)所示的化合物更高的流行性感冒治疗/或预防效果。
所谓“
[化学式20]
(式中,各符号与上述(1)含义相同)
所示的基团”的一个实施方式,可列举下式所示的基团:
[化学式21]
(式中,R
作为另一实施方式,可列举下式所示的基团:
[化学式22]
优选为下式所示的基团:
[化学式23]
特别优选为下式所示的基团:
[化学式24]
本说明书中的所谓“形成前药的P
[化学式25]
(式中,各符号系上述含义相同)
该“形成前药的P
作为形成前药的P
式(I)或式(II)的“P
a)-C(=O)-P
b)-C(=O)-P
c)-C(=O)-L-P
d)-C(=O)-L-O-P
e)-C(=O)-L-O-L-O-P
f)-C(=O)-L-O-C(=O)-P
g)-C(=O)-O-P
h)-C(=O)-N(-K)(P
i)-C(=O)-O-L-O-P
j)-C(P
k)-C(P
l)-C(P
m)-C(P
n)-C(P
o)-C(P
p)-C(P
q)-C(P
r)-C(P
s)-C(P
t)-C(P
u)-C(P
v)-C(P
w)-C(=N
x)-C(P
y)-C(P
z)-P(=O)(-P
aa)-S(=O)
ab)-P
ac)-C(P
(式中,L为直链或支链状的亚烷基、或者直链或支链状的亚烯基,
K为氢、或任选被取代基组A取代的烷基,
P
P
P
P
P
P
P
P
P
P
P
P
P
取代基组A:氧代基、烷基、羟基烷基、氨基、烷基氨基、碳环基、杂环基、碳环烷基、烷基羰基、卤素、羟基、羧基、烷基羰基氨基、烷基羰基氨基烷基、烷基羰氧基、烷氧基羰基、烷氧基羰基烷基、烷氧基羰氧基、烷基氨基羰氧基、烷基氨基烷基、烷氧基、氰基、硝基、叠氮基、烷基磺酰基、三烷基甲硅烷基、及磷酰基)。
形成前药的P
a)-C(=O)-P
b)-C(=O)-P
g)-C(=O)-O-P
h)-C(=O)-N(-K)(P
i)-C(=O)-O-L-O-P
l)-C(P
m)-C(P
o)-C(P
v)-C(P
x)-C(P
y)-C(P
z)-P(=O)(-P
(式中,L为直链或支链状的亚烷基,
K为氢、或任选被取代基组A取代的烷基,
P
P
P
P
P
P
P
P
P
取代基组A:氧代基、烷基、烷基氨基、碳环基、杂环基、烷基羰基、卤素、羟基、烷基羰基氨基、烷基羰氧基、烷氧基羰基、烷氧基羰基烷基、烷基氨基羰氧基、烷氧基、硝基、叠氮基、烷基磺酰基、及三烷基甲硅烷基)。
本说明书中的所谓“前药化”,意指如以下反应式所示那样,将式(III)或其药学上可接受的盐的羟基转化为-OP
[化学式26]
(式中,各符号与上述含义相同)
本说明书中的所谓“母化合物”,意指合成上述“前药”之前的成为原料的化合物、和/或在体内的生理条件下通过由酶、胃酸等引起的反应而由上述“前药”放出的化合物,具体而言,意指上述式(III)所示的化合物、或其药学上可接受的盐或者它们的溶剂合物。
所谓“卤素”,包含氟原子、氯原子、溴原子、及碘原子。特别优选为氟原子、及氯原子。
所谓“烷基”,包含碳原子数1~15、优选为碳原子数1~10、更优选为碳原子数1~6、进一步优选为碳原子数1~4的直链或支链状的烃基。例如可列举:甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、正戊基、异戊基、新戊基、正己基、异己基、正庚基、异庚基、正辛基、异辛基、正壬基、正癸基等。
作为“烷基”的优选实施方式,可列举:甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、正戊基。作为进一步优选的实施方式,可列举:甲基、乙基、正丙基、异丙基、叔丁基。
所谓“烯基”,包含在任意的位置具有1个以上双键的碳原子数2~15、优选为碳原子数2~10、更优选为碳原子数2~6、进一步优选为碳原子数2~4的直链或支链状的烃基。例如可列举:乙烯基、烯丙基、丙烯基、异丙烯基、丁烯基、异丁烯基、戊烯基、丁二烯基、戊烯基、异戊烯基、戊二烯基、己烯基、异己烯基、己二烯基、庚烯基、辛烯基、壬烯基、癸烯基、十一烯基、十二烯基、十三烯基、十四烯基、十五烯基等。
作为“烯基”的优选实施方式,可列举:乙烯基、烯丙基、丙烯基、异丙烯基、丁烯基。
所谓“亚烷基”,包含碳原子数1~15、优选为碳原子数1~10、更优选为碳原子数1~6、进一步优选为碳原子数1~4的直链或支链状的2价烃基。例如可列举:亚甲基、亚乙基、三亚甲基、亚丙基、四亚甲基、五亚甲基、六亚甲基等。
所谓“亚烯基”,包含在任意的位置具有1个以上双键的碳原子数2~15、优选为碳原子数2~10、更优选为碳原子数2~6、进一步优选为碳原子数2~4的直链或支链状的2价烃基。例如可列举:亚乙烯基、亚丙烯基、亚丁烯基、亚戊烯基等。
所谓“羟基烷基”,意指1个以上的羟基置换上述“烷基”的与碳原子键合的氢原子而成的基团。例如可列举:羟基甲基、1-羟基乙基、2-羟基乙基、1-羟基丙基、2-羟基丙基、1,2-羟基乙基等。
作为“羟基烷基”的优选实施方式,可列举:羟基甲基。
所谓“烷氧基”,意指上述“烷基”键合于氧原子而成的基团。例如可列举:甲氧基、乙氧基、正丙氧基、异丙氧基、正丁氧基、叔丁氧基、异丁氧基、仲丁氧基、戊氧基、异戊氧基、己氧基等。
作为“烷氧基”的优选实施方式,可列举:甲氧基、乙氧基、正丙氧基、异丙氧基、叔丁氧基。
所谓“卤代烷基”包括:1个以上的上述“卤素”置换上述“烷基”的与碳原子键合的氢原子而成的基团。例如可列举:单氟甲基、单氟乙基、单氟丙基、2,2,3,3,3-五氟丙基、单氯甲基、三氟甲基、三氯甲基、2,2,2-三氟乙基、2,2,2-三氯乙基、1,2-二溴乙基、1,1,1-三氟丙烷-2-基等。
作为“卤代烷基”的一个实施方式,可列举:三氟甲基、三氯甲基。
所谓“烷基羰基”,意指上述“烷基”键合于羰基而成的基团。例如可列举:甲基羰基、乙基羰基、丙基羰基、异丙基羰基、叔丁基羰基、异丁基羰基、仲丁基羰基、戊基羰基、异戊基羰基、己基羰基等。
作为“烷基羰基”的优选实施方式,可列举:甲基羰基、乙基羰基、正丙基羰基。
所谓“烷基氨基”,意指上述“烷基”置换氨基的与氮原子键合的1个或2个氢原子而成的基团。2个烷基可相同也可不同。例如可列举:甲基氨基、乙基氨基、异丙基氨基、二甲基氨基、二乙基氨基、N,N-二异丙基氨基、N-甲基-N-乙基氨基、N-异丙基-N-乙基氨基等。
作为“烷基氨基”的优选实施方式,可列举:甲基氨基、乙基氨基、二甲基氨基、二乙基氨基。
所谓“烷基氨基烷基”,意指上述“烷基氨基”键合于上述“烷基”而成的基团。
所谓“烷基氨基羰基”,意指上述“烷基氨基”键合于羰基而成的基团。
所谓“烷基氨基羰氧基”,意指上述“烷基氨基羰基”键合于氧原子而成的基团。
所谓“烷基羰基氨基”,意指上述“烷基羰基”置换氨基的与氮原子键合的1个氢原子而成的基团。例如可列举:甲基羰基氨基、乙基羰基氨基、丙基羰基氨基、异丙基羰基氨基、叔丁基羰基氨基、异丁基羰基氨基、仲丁基羰基氨基等。
作为“烷基羰基氨基”的优选实施方式,可列举:甲基羰基氨基、乙基羰基氨基。
所谓“烷基羰氧基”,意指上述“烷基羰基”键合于氧原子而成的基团。例如可列举:甲基羰氧基、乙基羰氧基、丙基羰氧基、异丙基羰氧基、叔丁基羰氧基、异丁基羰氧基、仲丁基羰氧基等。
作为“烷基羰氧基”的优选实施方式,可列举:甲基羰氧基、乙基羰氧基。
所谓“烷基羰基氨基烷基”,意指上述“烷基羰基氨基”键合于上述“烷基”而成的基团。
所谓“烷氧基羰基”,意指上述“烷氧基”键合于羰基而成的基团。例如可列举:甲氧基羰基、乙氧基羰基、丙氧基羰基、异丙氧基羰基、叔丁氧基羰基、异丁氧基羰基、仲丁氧基羰基、戊氧基羰基、异戊氧基羰基、己氧基羰基等。
作为“烷氧基羰基”的优选实施方式,可列举:甲氧基羰基、乙氧基羰基、丙氧基羰基。
所谓“烷氧基羰基烷基”,意指上述“烷氧基羰基”键合于上述“烷基”而成的基团。
所谓“烷氧基羰氧基”,意指上述“烷氧基羰基”键合于氧原子而成的基团。
所谓“烷基硫基”,意指上述“烷基”置换烷硫基的与硫原子键合的氢原子而成的基团。例如可列举:甲基硫基、乙基硫基、正丙基硫基、异丙基硫基等。
所谓“烷基磺酰基”,包含上述“烷基”键合于磺酰基而成的基团。例如可列举:甲基磺酰基、乙基磺酰基、丙基磺酰基、异丙基磺酰基、叔丁基磺酰基、异丁基磺酰基、仲丁基磺酰基等。
作为“烷基磺酰基”的一个实施方式,可列举:甲基磺酰基、乙基磺酰基。
所谓“三烷基甲硅烷基”,意指上述“烷基”中的3个键合于硅原子而成的基团。3个烷基可相同也可不同。例如可列举:三甲基硅烷基、三乙基硅烷基、叔丁基二甲基硅烷基等。
所谓“碳环基”,意指碳原子数3~20、优选为碳原子数3~16、更优选为碳原子数4~12的碳环基,包含芳香族碳环基及非芳香族碳环基。
所谓“芳香族碳环基”,包含单环或2环以上的环状芳香族烃基。例如可列举:苯基、萘基、蒽基、菲基等。
作为“芳香族碳环基”的一个实施方式,可列举:苯基、1-萘基、2-萘基。作为另一实施方式,可列举:苯基。
所谓“非芳香族碳环基”,包含单环或2环以上的环状饱和烃基或环状非芳香族不饱和烃基。2环以上的“非芳香族碳环基”也包括在单环或2环以上的非芳香族碳环基上稠合上述“芳香族碳环基”的环而成的基团。
进一步,“非芳香族碳环基”也包括如下述这样桥连而成的环式基团、或形成螺环的环式基团。
[化学式27]
作为单环的非芳香族碳环基,优选为碳原子数3~16,更优选为碳原子数3~12,进一步优选为碳原子数3~8。例如可列举:环丙基、环丁基、环戊基、环己基、环庚基、环辛基、环壬基、环癸基、环丙烯基、环丁烯基、环戊烯基、环己烯基、环庚烯基、环己二烯基等。
作为2环以上的非芳香族碳环基,例如可列举:二氢茚基、茚基、苊烯基、四氢萘基、芴基等。
作为“碳环”,意指碳原子数3~20、优选为碳原子数3~16、更优选为碳原子数4~12的碳环,包含芳香族碳环及非芳香族碳环。
所谓“芳香族碳环”,包含单环或2环以上的环状芳香族烃。例如可列举:苯环、萘环、蒽环、菲环等。
作为“芳香族碳环”的一个实施方式,可列举:苯环、萘环。作为另一实施方式,可列举:苯环。
所谓“非芳香族碳环”,包含单环或2环以上的环状饱和烃或环状非芳香族不饱和烃。2环以上的“非芳香族碳环”也包括在单环或2环以上的非芳香族碳环上稠合上述“芳香族碳环”中的环而成的那些。
进一步,“非芳香族碳环”也包括如下述这样的交联的环、或形成螺环的环。
[化学式28]
作为单环的非芳香族碳环,优选为碳原子数3~16,更优选为碳原子数3~12,进一步优选为碳原子数3~8。例如可列举:环丙烷、环丁烷、环戊烷、环己烷、环庚烷、环辛烷、环壬烷、环癸烷、环丙烯、环丁烯、环戊烯、环己烯、环庚烯、环己二烯等。
作为2环以上的非芳香族碳环,例如可列举:茚满、茚、乙烯合萘、四氢化萘、芴等。
作为“杂环基”,包含于环内具有1个以上的任意地选自O、S及N中的相同或不同的杂原子的芳香族杂环基及非芳香族杂环基。
所谓“芳香族杂环基”,包含于环内具有1个以上的任意地选自O、S及N中的相同或不同的杂原子的单环或2环以上的芳香族环式基团。
2环以上的“芳香族杂环基”也包括在单环或2环以上的芳香族杂环基上稠合上述“芳香族碳环基”中的环而成的基团。
作为单环的芳香族杂环基,优选为5~8元,更优选为5元或6元。例如可列举:吡咯基、咪唑基、吡唑基、吡啶基、哒嗪基、嘧啶基、吡嗪基、三唑基、三嗪基、四唑基、呋喃基、噻吩基、异
作为2环的芳香族杂环基,例如可列举:吲哚基、异吲哚基、吲唑基、吲哚嗪基、喹啉基、异喹啉基、噌啉基、酞嗪基、喹唑啉基、萘啶基、喹喏啉基、嘌呤基、喋啶基、苯并咪唑基、苯并异
作为3环以上的芳香族杂环基,例如可列举:咔唑基、吖啶基、呫吨基、吩噻嗪基、吩
所谓“非芳香族杂环基”,包含在环内具有1个以上的任意地选自O、S及N中的相同或不同的杂原子的单环或2环以上的环状非芳香族环式基团。
2环以上的“非芳香族杂环基”也包括在单环或2环以上的非芳香族杂环基上稠合上述“芳香族碳环基”、“非芳香族碳环基”、和/或“芳香族杂环基”中的各环而成的基团。
进一步,“非芳香族杂环基”也包括如下述这样桥连而成的基团、或形成螺环的基团。
[化学式29]
作为单环的非芳香族杂环基,优选为3~8元,更优选为5元或6元。例如可列举:二氧杂环己基、硫杂环丙基、环氧乙烷基、氧杂环丁基、氧硫杂环戊基、氮杂环丁基、噻烷基、噻唑烷基、吡咯烷基、吡咯啉基、咪唑烷基、咪唑啉基、吡唑烷基、吡唑啉基、哌啶基、哌嗪基、吗啉基、N-吗啉基、硫代吗啉基、硫代N-吗啉基、二氢吡啶基、四氢吡啶基、四氢呋喃基、四氢吡喃基、二氢噻唑基、四氢噻唑基、四氢异噻唑基、二氢
作为2环以上的非芳香族杂环基,例如可列举:吲哚啉基、异吲哚啉基、苯并二氢吡喃基、异苯并二氢吡喃基等。
作为“杂环”,包含在环内具有1个以上的任意地选自O、S及N中的相同或不同的杂原子的芳香族杂环及非芳香族杂环。
所谓“芳香族杂环”,包含在环内具有1个以上的任意地选自O、S及N中的相同或不同的杂原子的单环或2环以上的芳香族环。
2环以上的“芳香族杂环”也包括在单环或2环以上的芳香族杂环上稠合上述“芳香族碳环”中的环而成的那些。
作为单环的芳香族杂环,优选为5~8元,更优选为5元或6元。例如可列举:吡咯、咪唑、吡唑、吡啶、哒嗪、嘧啶、吡嗪、三唑、三嗪、四唑、呋喃、噻吩、异
作为2环的芳香族杂环,例如可列举:吲哚啉、异吲哚啉、吲唑、吲哚嗪、喹啉、异喹啉、噌啉、酞嗪、喹唑啉、萘啶、喹
作为3环以上的芳香族杂环,例如可列举:咔唑、吖啶、呫吨、酚噻嗪、吩噻
所谓“非芳香族杂环”,包含在环内具有1个以上的任意地选自氧原子、硫原子及氮原子中的相同或不同的杂原子的单环或2环以上的环状非芳香族环。
2环以上的“非芳香族杂环”也包括在单环或2环以上的非芳香族杂环上稠合上述“芳香族碳环”、“非芳香族碳环”、和/或“芳香族杂环”中的各环而成的那些。
进一步,“非芳香族杂环”也包括如下述这样桥连而成的环、或形成螺环的环。
[化学式30]
作为单环的非芳香族杂环,优选为3~8元,更优选为5元或6元。例如可列举:二
作为2环以上的非芳香族杂环,例如可列举:吲哚啉、异吲哚啉、苯并二氢吡喃、异苯并二氢吡喃等。
“碳环烷基”、“碳环氧基”、及“碳环氨基”的碳环部分也与上述“碳环”相同
“杂环烷基”、“杂环氧基”、及“杂环氨基”的杂环部分也与上述“杂环”相同。
本发明的化合物的特征在于如下方面:通过将键合有2个3环的稠环的化合物进行手性拆分,帽依赖性核酸内切酶抑制活性得到提高。
本发明的化合物的其他特征在于如下方面:通过导入形成前药的P
本发明的化合物的一个以上的氢、碳和/或其他原子可分别被氢、碳和/或其他原子的同位素取代。作为这样的同位素的例子,分别如
本发明的化合物的放射性标记物可利用该技术领域中周知的方法进行制备。例如,本发明的化合物的氚标记化合物可通过利用使用氚的催化性脱卤反应,将氚导入至本发明的特定化合物中而进行制备。该方法包括如下工序:在适当的催化剂、例如Pd/C的存在下、碱的存在下或非存在下,使本发明的化合物适当经卤素取代而成的前体与氚气进行反应。用以制备氚标记化合物的其他适当的方法可参照“Isotopes in the Physical andBiomedical Sciences,Vol.1,Labeled Compounds(Part A),Chapter 6(1987年)”。
作为本发明的化合物的药学上可接受的盐,例如可列举:本发明的化合物与碱金属(例如,锂、钠、钾等)、碱土金属(例如,钙、钡等)、镁、过渡金属(例如,锌、铁等)、氨、有机碱(例如,三甲胺、三乙胺、二环己基胺、乙醇胺、二乙醇胺、三乙醇胺、葡甲胺、乙二胺、吡啶、甲基吡啶、喹啉等)及氨基酸的盐;或者与无机酸(例如,盐酸、硫酸、硝酸、碳酸、氢溴酸、磷酸、氢碘酸等)、及本发明的化合物与有机酸(例如,甲酸、乙酸、丙酸、三氟乙酸、柠檬酸、乳酸、酒石酸、草酸、马来酸、富马酸、扁桃酸、戊二酸、苹果酸、苯甲酸、邻苯二甲酸、抗坏血酸、苯磺酸、对甲苯磺酸、甲磺酸、乙磺酸等)的盐。尤其是可列举本发明的化合物与盐酸、硫酸、磷酸、酒石酸、甲磺酸的盐等。这些盐可通过通常进行的方法而形成。
本发明的化合物或其药学上可接受的盐存在形成溶剂合物(例如,水合物等)和/或多晶型的情况,本发明也包括这样的各种溶剂合物及多晶型。“溶剂合物”中相对于本发明的化合物,配位任意数量的溶剂分子(例如,水分子等)。通过将本发明的化合物或其药学上可接受的盐放置于大气中,存在吸收水分而附着吸附水的情况、或者形成水合物的情况。另外,通过将本发明的化合物或其药学上可接受的盐进行重结晶,存在形成多晶型的情况。
P
作为P
a)-C(=O)-P
b)-C(=O)-P
c)-C(=O)-L-P
d)-C(=O)-L-O-P
e)-C(=O)-L-O-L-O-P
f)-C(=O)-L-O-C(=O)-P
g)-C(=O)-O-P
h)-C(=O)-N(-K)(P
i)-C(=O)-O-L-O-P
j)-C(P
k)-C(P
l)-C(P
m)-C(P
n)-C(P
o)-C(P
p)-C(P
q)-C(P
r)-C(P
s)-C(P
t)-C(P
u)-C(P
v)-C(P
w)-C(=N
x)-C(P
y)-C(P
z)-P(=O)(-P
aa)-S(=O)
ab)-P
ac)-C(P
(式中,L为直链或支链状的亚烷基、或者直链或支链状的亚烯基,
K为氢、或任选被取代基组A取代的烷基,
P
P
P
P
P
P
P
P
P
P
P
P
P
取代基组A:氧代基、烷基、羟基烷基、氨基、烷基氨基、碳环基、杂环基、碳环烷基、烷基羰基、卤素、羟基、羧基、烷基羰基氨基、烷基羰基氨基烷基、烷基羰氧基、烷氧基羰基、烷氧基羰基烷基、烷氧基羰氧基、烷基氨基羰氧基、烷基氨基烷基、烷氧基、氰基、硝基、叠氮基、烷基磺酰基、三烷基甲硅烷基、及磷酰基)。
作为P
a)-C(=O)-P
b)-C(=O)-P
g)-C(=O)-O-P
h)-C(=O)-N(-K)(P
i)-C(=O)-O-L-O-P
l)-C(P
m)-C(P
o)-C(P
v)-C(P
x)-C(P
y)-C(P
z)-P(=O)(-P
(式中,L为直链或支链状的亚烷基,
K为氢、或任选被取代基组A取代的烷基,
P
P
P
P
P
P
P
P
P
取代基组A:氧代基、烷基、烷基氨基、碳环基、杂环基、烷基羰基、卤素、羟基、烷基羰基氨基、烷基羰氧基、烷氧基羰基、烷氧基羰基烷基、烷基氨基羰氧基、烷氧基、硝基、叠氮基、烷基磺酰基、及三烷基甲硅烷基)。
作为P
[化学式31]
(本发明的化合物的制造法)
将本发明的化合物的通常制造法例示于以下。另外,萃取、纯化等只要进行在通常的有机化学实验中进行的处理即可。
本发明的化合物的合成可参考该领域中公知的方法实施。
原料化合物可应用市售的化合物、本说明书中所记载的那些、本说明书中所引用的文献中记载的那些、及其他的公知化合物。
在欲取得本发明的化合物的盐时,在以盐的形式获得本发明的化合物的情况下,直接进行纯化即可,另外,在以游离的形式获得本发明的化合物的情况下,只要溶解或悬浮于适当的有机溶剂中,添加酸或碱并通过通常的方法形成盐即可。
另外,本发明的化合物及其药学上可接受的盐也有时以与水或各种溶剂的加成物(水合物或溶剂合物)的形式存在,但这些加成物也包括在本发明中。
在通常的合成法以及参考例、实施例、及中间体合成例中,各缩写的含义如下所述。
Boc:叔丁氧基羰基
DBU:二氮杂双环十一烯
DMA:N,N-二甲基乙酰胺
DMF:N,N-二甲基甲酰胺
HATU:O-(7-氮杂苯并三唑-1-基)-N,N,N',N'-四甲基脲鎓六氟磷酸盐
NMP:N-甲基吡咯烷酮
OBn:苄氧基
THF:四氢呋喃
T3P:丙基膦酸酐
WSC·HCl:N-乙基-N'-(3-二甲基氨基丙基)碳二亚胺盐酸盐
需要说明的是,“楔形”及“虚线”表示绝对构型(absolute configuration)。
(制法1)
[化学式32]
(式中,P
第1工序
于DMF、THF、二氯甲烷、乙腈等溶剂中或这些的混合溶剂中,且于二环己基碳二酰亚胺、羰基二咪唑、二环己基碳二酰亚胺-N-羟基苯并三唑、4-(4,6-二甲氧基-1,3,5,-三嗪-2-基)-4-甲基吗啉盐酸盐、2-(7-氮杂-1H-苯并三唑-1-基)-1,1,3,3-四甲基脲鎓六氟磷酸盐、WSC·HCl、HATU等脱水缩合剂存在下向化合物A1添加化合物A2,于-20℃~60℃、优选为-10℃~40℃反应0.1小时~24小时、优选为1小时~12小时,由此可获得化合物A3。
或者,于THF、二
第2工序
于DMF、DMA、NMP、THF等溶剂的存在下,向化合物A3添加碳酸钾、碳酸钠、O-(2,4-二硝基苯基)羟基胺,于10℃~60℃、优选为20℃~40℃反应0.1小时~48小时、优选为1小时~24小时,由此可获得化合物A4。
第3工序
化合物A4的缩醛保护基的脱保护反应可利用Protective Groups in OrganicSynthesis,Theodora W Green(John Wiley&Sons)等所记载的通常方法进行。其后,使所生成的醛基进行分子内反应,由此可获得化合物A5。
例如,于DMF、甲苯、THF等溶剂的存在下,向化合物A4添加乙酸和/或对甲苯磺酸、甲磺酸等,于10℃~80℃、优选为30℃~60℃反应0.5小时~12小时、优选为1小时~6小时,由此可获得化合物A5的外消旋体。利用SFC(Supercritical Fluid Chromatography,超临界流体色谱法)或HPLC(High performance liquid chromatography,高效液相色谱法)系统(手性色谱柱)将化合物A5的外消旋体进行手性拆分,由此可获得化合物A5。
第4工序
于DMF、DMA、NMP、THF等溶剂的存在下或这些的混合溶剂中,向化合物A5添加化合物A6及碳酸钠、碳酸钾、碳酸铯等碱,以0℃~60℃、优选为10℃~40℃反应0.1小时~48小时、优选为1小时~24小时反应,由此可获得化合物A7。
另外,于DMF、乙酸乙酯、乙酸丁酯、1,4-二
第5工序
化合物A7的羟基的保护基的脱保护反应可利用Protective Groups in OrganicSynthesis,Theodora W Green(John Wiley&Sons)等所记载的通常方法进行。
第6工序
可通过将化合物(II)的羟基转化为酯基或醚基的通常方法而获得化合物(III)。
例如,可应用Protective Groups in Organic Synthesis,Theodora W Green(John Wiley&Sons)、Prog.Med.5:2157-2161(1985)、及Supplied by The BritishLibrary-“The world's Knowledge”等所记载的方法。
(制法2)
[化学式33]
(式中,P
第1工序
于DMF、THF、二氯甲烷、乙腈等溶剂中或这些的混合溶剂中,向化合物B1添加碘甲烷等卤代烷,并于二氮杂双环十一烯等碱存在下添加化合物A2,以-20℃~60℃、优选为-10℃~40℃反应0.1小时~24小时、优选为1小时~12小时,由此可获得化合物B2。
或者,于THF、二
第2工序
于THF、二
第3工序
化合物B3的氨基的保护基的脱保护反应可利用Protective Groups in OrganicSynthesis,Theodora W Green(John Wiley&Sons)等所记载的通常方法进行。
第4工序
于THF、二
第5工序
于THF、二
第6工序
使化合物B7溶解于乙醇中,使之于0℃~100℃下与对甲苯磺酸或甲磺酸反应0.1小时~48小时、优选为1小时~24小时,由此可获得化合物B8。
第7工序
于THF、二
第8工序
使化合物B10溶解于乙醇中,并将碳电极(阳极)与铂电极(阴极)浸渍于碳酸钾等碱与四乙基高氯酸铵中,一边搅拌0.1小时~48小时、优选为1小时~24小时一边使0.1~1.0A的恒定电流流过,由此可获得化合物B8。
第9~10工序
使用化合物B4与化合物B8而实施制法1的第3~6工序,由此可获得化合物(I)。
本发明化合物具有帽依赖性核酸内切酶抑制作用。因此,本发明化合物作为流行性感冒的治疗剂和/或预防剂有用。
本发明化合物不仅具有帽依赖性核酸内切酶抑制作用,还具备作为药物的有用性,且具有下述中的任一或全部的优异特征。
a)对CYP酶(例如,CYP1A2、CYP2C9、CYP2C19、CYP2D6、CYP3A4等)的抑制作用弱。
b)表现出高生物利用率、适度的清除率等良好的药代动力学。
c)代谢稳定性高。
d)于本说明书中所记载的测定条件的浓度范围内,对CYP酶(例如,CYP3A4)未表现出不可逆的抑制作用。
e)不具有诱突变性。
f)心血管系统的危险性低。
g)表现出高溶解性。
h)不具有光毒性。
以治疗上述疾病为目的而将本发明化合物向人类给药的情况下,可以以粉剂、颗粒剂、片剂、胶囊剂、丸剂、液剂等的形式进行口服给药,或者以注射剂、栓剂、经皮吸收剂、吸入剂等的形式进行非口服给药。另外,可视需要向本化合物的有效量混合适合其剂型的赋形剂、结合剂、湿润剂、崩解剂、润滑剂等药物用添加剂,从而制成医药制剂。对于注射剂的情况而言,与适当的载体一起进行灭菌处理而制成制剂。
在给药本发明的药物组合物的情况下,可利用口服、非口服中的任意方法进行给药。在口服给药时,只要依据常规方法,制备为片剂、颗粒剂、粉剂、胶囊剂等通常所使用的剂型而给药即可。在非口服给药时,可以以注射剂等通常所使用的任意剂型而适宜地进行给药。本发明的化合物的口服吸收性高,因此可适宜地用作口服剂。
可视需要向本发明化合物的有效量中混合适合其剂型的赋形剂、结合剂、崩解剂、润滑剂等各种药物用添加剂而制成药物组合物。
给药量根据疾病的状态、给药路径、患者的年龄、或体重而不同,但在向成人口服给药的情况下,通常为0.1~100mg/kg/天,优选为1~20mg/kg/天。
本发明的药物组合物的给药量较理想为考虑患者的年龄、体重、疾病的种类或程度、给药途径等的情况下进行设定,在向成人口服给药的情况下,通常为0.05~100mg/kg/天,优选为0.1~10mg/kg/天的范围内。在非口服给药的情况下,本发明的药物组合物的给药量根据给药途径而大不相同,但通常为0.005~10mg/kg/天,优选为0.01~1mg/kg/天的范围内。将其以1天1次~分数次的方式进行给药即可。
本发明化合物可出于增强该化合物的作用或减少该化合物的给药量等目的而与其他药剂等(以下简称为并用药剂)组合使用。例如在流行性感冒的疾病中,可与神经胺酸酶抑制剂(例如,奥司他韦、扎那米韦、帕拉米韦及Inavir等)、RNA依赖性RNA聚合酶抑制剂(例如,法匹拉韦(Favipiravir))、M2蛋白质抑制剂(例如金刚胺(Amantadine))、PB2 Cap结合抑制剂(例如,VX-787)、抗HA抗体(例如,MHAA4549A)、或免疫作用药(例如,硝唑尼特(Nitazoxanide))组合使用。此时,本发明化合物与并用药剂的给药时期并无限定,可将这些同时给药至给药对象,也可隔开时间差进行给药。进一步,本发明化合物与并用药剂可以以包含各活性成分中的2种以上的制剂的形式进行给药,也可以以包含全部活性成分的单一制剂的形式进行给药。
并用药剂的给药量可以以临床上所使用的用量为基准而适当地进行选择。另外,本发明化合物与并用药剂的配比可根据给药对象、给药路径、对象疾病、症状、组合等而适当地进行选择。例如,在给药对象为人类的情况下,相对于本发明化合物1重量份,使用并用药剂0.01~100重量份即可。
以下列举本发明的实施例、参考例及中间体合成例、以及试验例而对本发明进一步详细地进行说明,但本发明并不受这些限定。
参考例及实施例中所获得的NMR分析是在300MHz下进行,且使用DMSO-d
RT表示LC/MS:液相色谱法/质谱分析中的保留时间,在以下的条件下进行测定。
(测定条件)
(1)色谱柱:ACQUITY UPLC(注册商标)BEH C18(1.7μm i.d.2.1x50mm)(Waters)
流速:0.8mL/分;UV检测波长:254nm;
流动相:[A]含0.1%甲酸的水溶液、[B]含0.1%甲酸的乙腈溶液
以3.5分钟进行5%-100%溶剂[B]的线性梯度后,保持100%溶剂[B]0.5分钟。
(2)色谱柱:Shim-pack XR-ODS(2.2μm,i.d.50x3.0 mm)(Shimadzu)
流速:1.6mL/分;UV检测波长:254nm;
流动相:[A]含0.1%甲酸的水溶液、[B]含0.1%甲酸的乙腈溶液
梯度:以3分钟进行10%-100%溶剂[B]的线性梯度,保持100%溶剂[B]0.5分钟。
参考例1
[化学式34]
第1工序
在氮气氛围中且于利用干冰-丙酮冷却至-78℃下,向化合物1(5.0g,49.5mmol)的THF(100mL)溶液滴加1.62mol/L正丁基锂-己烷溶液(30.5mL,49.5mmol),于-78℃下搅拌2小时。向反应液滴加氯甲酸烯丙酯(5.96g,49.5mmol)的THF(20mL)溶液,于-78℃下搅拌2小时。将反应液于饱和氯化铵水溶液中进行骤冷,升温至室温后,利用乙酸乙酯进行萃取。利用饱和食盐水洗涤有机层,利用无水硫酸镁进行干燥后,减压蒸馏除去溶剂,而获得化合物2(5.66g,产率62%)。
1H-NMR(CDCl3)δ:3.83(t,J=8.0Hz,2H),3.92(t,J=8.0Hz,2H),4.26(s,2H),4.78(d,J=8.0Hz,2H),5.30(d,J=12.0Hz,1H),5.44(d,J=16.0Hz,1H),5.93-6.03(m,1H),
第2工序
在氮气氛围中且于利用干冰-丙酮冷却至-78℃下,向化合物2(6.6g,35.6mmol)的THF(66mL)溶液滴加1.03mol/L DIBAL-H己烷溶液(45.0mL,46.3mmol),于-78℃下搅拌1小时。将反应液于丙酮中骤冷后,添加罗谢耳盐水溶液并进行搅拌,利用乙酸乙酯进行萃取。利用饱和食盐水洗涤有机层,利用无水硫酸镁进行干燥后,减压蒸馏除去溶剂,而获得化合物3(6.21g,产率93%)。
1H-NMR(CDCl3)δ:3.44(br,1H),3.50-3.64(m,2H),3.71(br,1H),3.95(d,J=8.0Hz,2H),4.64(d,J=8.0Hz,2H),5.24(d,J=12.0Hz,1H),5.40(d,J=16.0Hz,1H),5.47(d,J=4Hz,1H),5.87-6.00(m,1H)
第3工序
向化合物3(6.2g,33.1mmol)的甲醇(65mL)溶液添加对甲苯磺酸一水合物(0.63g,3.31mmol)并于室温下搅拌整夜。将反应液于碳酸氢钠水溶液中进行骤冷后,进行浓缩,利用乙酸乙酯进行萃取。利用饱和食盐水洗涤有机层,利用无水硫酸镁进行干燥后,减压蒸馏除去溶剂,而获得化合物4(5.77g,产率87%)。
1H-NMR(CDCl3)δ:3.34(s,3H),3.55(br,2H),3.73-3.99(m,3H),4.64(d,J=8.0Hz,2H),5.10-5.20(m,1H),5.25(d,J=8.0Hz,1H),5.33(d,J=16Hz,1H),5.88-6.05(m,1H)
第4工序
向化合物5(20.0g,81mmol)的DMF(100mL)溶液添加碘乙烷(22.8g,146mmol)与二氮杂双环十一烯(18.4mL,122mmol)并于室温下搅拌整夜。将反应液注入10%氯化铵水溶液中,利用乙酸乙酯进行萃取。利用饱和食盐水洗涤有机层,利用无水硫酸镁进行干燥后,减压蒸馏除去溶剂,而获得化合物6(22.3g,产率100%)。
1H-NMR(CDCl3)δ:1.23(t,J=8.0Hz,3H),4.28(q,J=8.0Hz,2H),5.16(s,2H),6.57(d,J=4.0Hz,1H),7.28-7.48(m,5H),8.21(d,J=4.0Hz,1H).
第5工序
向化合物6(500mg,1.82mmol)的DMA(5.0mL)溶液添加对甲苯磺酸吡啶鎓(1.37g,5.47mmol)与Boc肼(361mg,2.74mmol)并于60℃下搅拌14小时。将反应液添加于水中,利用乙酸乙酯进行萃取。利用饱和氯化铵水溶液与饱和食盐水洗涤有机层,利用无水硫酸镁进行干燥后,减压蒸馏除去溶剂。通过硅胶柱色谱法(氯仿-甲醇)对所获得的残渣进行纯化而获得化合物7(519mg,产率73%)。
1H-NMR(CDCl3)δ:1.24(t,J=8.0Hz,3H),1.46(s,9H),4.26(q,J=8.0Hz,2H),5.28(s,2H),6.40(d,J=8.0Hz,1H),7.27-7.38(m,4H),7.40-7.45(m,2H).
第6工序
使化合物7(500mg,1.29mmol)溶解于4mol/L盐酸乙酸乙酯溶液(5mL)中,于室温下搅拌1小时。将反应液的溶剂减压蒸馏除去,向所获得的残渣添加饱和碳酸氢钠水溶液,利用二氯甲烷进行萃取。利用饱和食盐水洗涤有机层,利用无水硫酸镁进行干燥后,减压蒸馏除去溶剂,而获得化合物8(369mg,产率99%)。
1H-NMR(CDCl3)δ:1.26(t,J=8.0Hz,3H),4.31(q,J=8.0Hz,2H),5.24(s,2H),6.47(d,J=8.0,1H),7.28-7.44(m,5H),7.64(d,J=8.0,1H).
第7工序
在氮气氛围中且于利用干冰-四氯化碳冷却至-25℃下,向化合物7(365mg,1.27mmol)与化合物4(306mg,1.52mmol)的乙腈(8mL)溶液滴加四氯化锡(0.223mL,1.90mmol),并于-25℃下搅拌45分钟。将反应液于饱和碳酸氢钠水溶液中骤冷后,添加二氯甲烷,于室温下进行搅拌,进行硅藻土过滤,利用二氯甲烷萃取滤液。利用饱和食盐水将所获得的有机层洗涤,利用硫酸镁进行干燥,减压蒸馏除去溶剂,而获得化合物9的粗产物。使所获得的化合物9溶解于THF(8mL)中,添加吗啉(1.10mL,12.7mmol)、四(三苯基膦)钯(146mg,0.127mmol),于室温下搅拌2小时。向反应液添加乙醚(16mL),滤取所析出的固体,使所获得的固体干燥,而获得化合物10(418mg,产率100%)。
1H-NMR(CDCl3)δ:2.90-2.99(m,1H),3.13(t,J=12.0Hz,1H),3.40-3.46(m,1H),4.00-4.08(m,1H),4.14(d,J=12.0Hz,1H),5.07(s,2H),6.22(d,J=8.0Hz,1H),7.29-7.40(m,3H),7.56(d,J=8.0Hz,2H),7.71(d,J=8.0Hz,1H)
第8工序
于室温下依次向(R)-四氢呋喃-2-羧酸(855mg,7.36mmol)、化合物10(2.00g,6.11mmol)的乙酸乙酯(9ml)悬浮液添加吡啶(4.00ml,49.6mmol)及T3P(50%乙酸乙酯溶液,11.0ml,18.5mmol)并搅拌整夜。滤取所析出的固体后,利用乙酸乙酯(4ml)、乙醇(4ml)依次进行洗涤。使所获得的固体悬浮于乙醇(6ml)中,于室温下搅拌6.5小时。过滤悬浮液,利用乙醇(2ml)将所获得的固体洗涤2次,而获得化合物11(1.18g,产率45.4%)。
第9工序
于室温下向化合物11(500mg,1.18mmol)的乙醇(3.5ml)悬浮液添加DBU(0.0035ml,0.023mmol)并搅拌30分钟。向所获得的悬浮液添加二异丙基醚(6.5ml),于室温下搅拌30分钟。滤取所析出的固体后,利用乙酸乙酯(1.5ml)洗涤2次,而获得化合物i1(346mg,产率89.9%)。
参考例2
[化学式35]
第1工序
于冰浴下向化合物13(8.0g,50.8mmol)的二氯甲烷(120mL)悬浮液添加三乙胺(17.6mL,127mmol),并滴加氯甲酸烯丙酯(6.44mL,60.9mmol),于0℃下搅拌1小时。向反应液添加水,利用二氯甲烷进行萃取。利用5%柠檬酸水溶液与饱和碳酸氢钠水溶液洗涤有机层,利用无水硫酸镁进行干燥后,减压蒸馏除去溶剂,而获得化合物14(10.1g,产率97%)。
1H-NMR(CDCl3)δ:1.96(br,4H),3.62(s,4H),4.60(s,2H),5.22(d,J=12.0Hz,1H),5.30(d,J=16.0Hz,1H),5.86-5.99(m,1H)
第2工序
将碳电极(阳极)与铂电极(阴极)浸渍于化合物14(0.9g,4.39mmol)、碳酸钾(60mg,0.44mmol)、四乙基高氯酸铵(50mg,0.22mmol)的甲醇(30mL)溶液中,一边于室温下搅拌6小时一边使0.1A的恒定电流流过。向反应液添加乙酸乙酯与水,利用乙酸乙酯进行萃取。利用无水硫酸镁将有机层干燥后,减压蒸馏除去溶剂,而获得化合物15(992mg,产率96%)。
1H-NMR(CDCl3)δ:1.81-2.15(m,3H),2.39(t,J=12.0Hz,1H),3.27(s,3H),3.61(s,1H),4.11(br,1H),4.61(br,2H),5.20-5.36(m,2H),5.57(br,1H),5.88-5.99(m,1H)
第3工序
以与参考例1的第7、8工序相同的方式进行反应,而获得化合物16。
第4工序
利用Waters制造的SFC30系统(Daicel制造的CHIRALPAK IB,液化碳酸-甲醇)将化合物16(870mg,2.41mmol)进行手性分割,而获得化合物i2(270mg,产率31%)。
分析条件
<Waters制造的SFC30系统(SPRC4·5N406)>
色谱柱:CHIRALPAK IB/SFC(5μm、i.d.250x4.6 mm)(DAICEL)
流速:8.0mL/分;UV检测波长:254nm
背压:100bar
流动相:[A]液化碳酸、[B]甲醇
在保持5%溶剂[B]1分钟后,以6分钟进行5%-40%溶剂[B]的线性梯度。其后保持40%溶剂[B]2分钟后,保持5%溶剂[B]1分钟。
洗脱时间:7.3分钟
参考例3
[化学式36]
第1工序
于冰浴冷却下向化合物17(4.00g,16.3mmol)的二氯甲烷(40mL)溶液滴加草酰氯(1.56mL,17.9mmol)及DMF(0.013mL,0.162mmol),一边上升至室温一边搅拌5小时。减压蒸馏除去反应液,使所获得的残渣溶解于二氯甲烷(40mL)中,于冰浴冷却下添加2,2,2-三氟乙醇(2.44g,24.4mmol)、三乙胺(4.50mL,32.5mmol)、4-(二甲基氨基)吡啶(99.0mg,0.812mmol),一边上升至室温一边搅拌1小时。减压蒸馏除去反应液,向所获得的残渣添加1mol/L盐酸水溶液,利用乙酸乙酯进行萃取。利用1mol/L盐酸水溶液、饱和食盐水洗涤有机层后,利用无水硫酸镁进行干燥,而获得化合物18(5.33g,产率100%)。
1H-NMR(CDCl3)δ:4.64(q,J=8.2Hz,2H),5.38(s,2H),6.49(d,J=5.6Hz,1H),7.30-7.38(m,3H),7.43-7.49(m,2H),7.75(d,J=5.6Hz,1H).
第2~3工序
以与参考例1的第5、6工序相同的方式进行反应而获得化合物20。
1H-NMR(CDCl3)δ:4.55(q,J=8.3Hz,2H),5.18(s,2H),5.29(s,2H),6.37(d,J=7.8Hz,1H),7.30-7.42(m,6H).
第4、5工序
以与参考例1的第7工序相同的方式进行反应而获得化合物23。
LC/MS(ESI):m/z=342.1[M+H]
第六工序
向化合物23(820mg,2.40mmol)的二氯甲烷(16.5mL)溶液添加Boc2O(0.837mL,3.60mmol)、三乙胺(0.499mL,3.60mmol)及4-(二甲基氨基)吡啶(44.0mg,0.360mmol)并于室温下搅拌3.5小时。向反应液添加1mol/L盐酸水溶液,利用乙酸乙酯进行萃取。利用1mol/L盐酸水溶液、饱和食盐水洗涤有机层后,利用无水硫酸钠进行干燥,减压蒸馏除去溶剂。通过硅胶柱色谱法(氯仿-甲醇)对所获得的残渣进行纯化而获得化合物24(593mg,产率56%)及化合物i3(170mg,产率16%)。
化合物24:LC/MS(ESI):m/z=441.9[M+H]
第七工序
使化合物24(547mg,1.24mmol)溶解于乙酸(5.5mL)中,于80℃下搅拌5小时。将反应液的溶剂减压蒸馏除去,通过硅胶柱色谱法(氯仿-甲醇)对所获得的残渣进行纯化而获得化合物i3(454mg,产率100%)。
1H-NMR(CDCl3)δ:1.46(d,J=6.4Hz,3H),3.45(dd,J=10.5,10.5Hz,1H),3.55(dd,J=11.7,4.3Hz,1H),3.92(dd,J=11.7,3.6Hz,1H),3.95-4.01(m,2H),4.76(dq,J=13.9,4.3Hz,1H),5.19(d,J=10.2Hz,1H),5.22(d,J=10.2Hz,1H),5.36(d,J=12.9Hz,1H),6.28(d,J=7.8Hz,1H),7.25(d,J=7.8Hz,1H),7.28-7.36(m,3H),7.56-7.61(m,2H).
实施例1
[化学式37]
第1工序
使化合物i1(1100g,3360mmol)及7,8-二氟-6,11-二氢二苯并噻庚因-11-醇(977g,3697mmol)悬浮于50wt%T3P的乙酸乙酯溶液(3208g,5041mmol)及乙酸乙酯(1.1L)中。于室温下向反应液添加甲磺酸(436ml,6721mmol),于70℃下搅拌5小时30分钟。于冰浴冷却下向反应液添加水,于室温下搅拌1小时后,添加THF,利用乙酸乙酯进行萃取。利用水及8%碳酸氢钠水溶液洗涤有机层,利用无水硫酸钠进行干燥,减压蒸馏除去溶剂。使所获得的残渣溶解于THF(5.5L)中,添加碳酸钾(790g,5713mmol),升温至50℃,滴加苄基溴(240ml,2016mmol),于60℃下搅拌8小时30分钟。于冰浴冷却下向反应液滴加2mol/L盐酸水溶液,于室温下搅拌10分钟,利用乙酸乙酯进行萃取。利用水及8%碳酸氢钠水溶液洗涤有机层,利用无水硫酸镁进行干燥。添加活性碳(Norit SX-2,240g),进行硅藻土过滤,于减压下将滤液蒸馏去除。向所获得的残渣添加乙酸乙酯及己烷,使固体析出后,进行滤取,由此获得化合物25(1019g,1776mmol,产率53%)。
第2工序
于室温下向化合物25(1200g,2092mmol)的DMA(3.6L)溶液添加氯化锂(443g,10.5mol),于80℃下搅拌3小时。于冰浴冷却下向反应液添加丙酮(1.2L)、0.5mol/L盐酸水溶液(6.0L)及水(2.4L)并搅拌1小时。滤取所析出的固体。使所获得的固体溶解于氯仿中,添加异丙醚,使固体析出后,进行滤取,由此获得化合物III-2(950g,1965mmol,产率94%)。
实施例2
[化学式38]
第1工序
使化合物i1(400mg,1.22mmol)及6,11-二氢二苯并噻庚因-11-醇(418mg,1.83mmol)溶解于50%T3P乙酸乙酯溶液(7.27mL,12.2mmol)中,于封管条件下以110℃搅拌1.5小时。向反应液添加水,利用乙酸乙酯进行萃取。利用饱和食盐水洗涤有机层后,利用无水硫酸钠进行干燥,减压蒸馏除去溶剂。通过硅胶柱色谱法(氯仿-甲醇及乙酸乙酯-甲醇)对所获得的残渣进行纯化而获得化合物26(316mg,产率47%)。
1H-NMR(CDCl3)δ:2.86(dd,J=11.4,11.4Hz,1H),3.26-3.40(m,2H),3.55(d,J=13.4Hz,1H),3.70(d,J=10.4Hz,1H),3.86(d,J=10.4Hz,1H),4.48(d,J=9.5Hz,1H),4.66(d,J=13.4Hz,1H),5.20(s,1H),5.43-5.50(m,2H),5.63(d,J=10.9Hz,1H),5.79(d,J=7.8Hz,1H),6.40(d,J=7.7Hz,1H),6.62-6.69(m,1H),7.02-7.07(m,3H),7.18(d,J=7.4Hz,1H),7.27-7.44(m,6H),7.60-7.66(m,2H).
第2工序
以与实施例1的第2工序相同的方式进行反应而获得化合物III-1。
1H-NMR(CDCl3)δ:2.98(dd,J=13.0,12.3Hz,1H),3.46(dd,J=13.1,10.0Hz,1H),3.55-3.63(m,2H),3.79(d,J=11.4Hz,1H),3.96(d,J=11.0Hz,1H),4.62-4.66(m,2H),5.26(s,1H),5.52(d,J=13.4Hz,1H),5.75(d,J=7.7Hz,1H),6.70(d,J=7.7Hz,1H),6.79-6.85(m,1H),7.05-7.12(m,3H),7.23(d,J=7.4Hz,1H),7.30(t,J=7.3Hz,1H),7.36(d,J=7.4Hz,1H),7.44(t,J=7.4Hz,1H).
实施例3
[化学式39]
第1工序
使化合物27(290mg,0.880mmol)与化合物i1(240mg,0.733mmol)溶解于50%T3P乙酸乙酯溶液(2.4mL)中,于封管条件下以100℃搅拌1.5小时。向反应液添加水,利用乙酸乙酯进行萃取。利用饱和食盐水洗涤有机层,利用无水硫酸镁进行干燥后,减压蒸馏除去溶剂。通过硅胶柱色谱法(氯仿-乙酸乙酯-甲醇)对所获得的残渣进行纯化而获得化合物28(106mg,产率24%)。
1H-NMR(CDCl3)δ:2.37(s,3H),2.94-3.03(m,1H),3.15-3.23(m,1H),3.28(t,J=10.4Hz,1H),3.58(d,J=13.2Hz,1H),3.66(dd,J=3.2Hz,11.6Hz,1H),3.84(dd,J=2.8Hz,10.8Hz,1H),4.40-4.52(m,2H),5.49(t,J=13.6Hz,2H),5.60(d,J=10.4Hz,2H),5.78(d,J=7.6Hz,1H),6.41(d,J=7.2Hz,1H),6.66-6.71(m,1H),6.98-7.12(m,4H),7.21(d,J=7.6Hz,1H),7.30-7.42(m,4H),7.56-7.61(m,2H).
第2工序
向化合物28(100mg,0.168mmol)的甲醇(1mL)溶液添加2mol/L氢氧化钠水溶液(252μL,0.504mmol)并于室温下搅拌1小时。向反应液添加2mol/L盐酸(0.3mL),利用氯仿进行萃取后,减压蒸馏除去溶剂。使所获得的残渣溶解于DMA(1.0mL)中,添加氯化锂(35.6mg,0.839mmol),于100℃下搅拌15小时。通过反相硅胶柱色谱法(乙腈-水)对反应液进行纯化而获得化合物III-24(20mg,产率26%)。
1H-NMR(CDCl3)δ:3.09(t,J=11.2Hz,1H),3.40-3.58(m,3H),3.76(d,J=10.8Hz,1H),3.91(d,J=10.8Hz,1H),4.66(d,J=13.2Hz,1H),4.73(d,J=9.6Hz,1H),5.50(d,J=13.6Hz,1H),5.79(d,J=6.8Hz,1H),6.25(s,1H),6.61-6.70(m,2H),6.79(d,J=6.8Hz,1H),6.93-7.08(m,3H),7.10-7.19(m,2H).
依据与上述实施例相同的方法,使用市售化合物或参考例所示的中间化合物而合成以下的实施例化合物。
[表1]
[表2]
[表3]
[表4]
[表5]
[表6]
[表7]
[表8]
[表9]
实施例4
[化学式40]
向化合物III-2(1.00g,2.07mmol)的DMA(5ml)的悬浮液添加氯甲基碳酸甲酯(0.483g,3.10mmol)及碳酸钾(0.572g,4.14mmol)、碘化钾(0.343g,2.07mmol),升温至50℃并搅拌6小时。进一步向反应液添加DMA(1ml)并搅拌6小时。将反应液冷却至室温,添加DMA(6ml),于50℃下搅拌5分钟,进行过滤。于冰浴冷却下向所获得的滤液滴加1mol/L盐酸水(10ml)及水(4ml)并搅拌1小时。滤取所析出的固体,于60℃下进行3小时减压干燥,而获得化合物II-6(1.10g,1.93mmol,产率93%)。
1H-NMR(DMSO-D6)δ:2.91-2.98(1H,m),3.24-3.31(1H,m),3.44(1H,t,J=10.4Hz),3.69(1H,dd,J=11.5,2.8Hz),3.73(3H,s),4.00(1H,dd,J=10.8,2.9Hz),4.06(1H,d,J=14.3Hz),4.40(1H,d,J=11.8Hz),4.45(1H,dd,J=9.9,2.9Hz),5.42(1H,dd,J=14.4,1.8Hz),5.67(1H,d,J=6.5Hz),5.72-5.75(3H,m),6.83-6.87(1H,m),7.01(1H,d,J=6.9Hz),7.09(1H,dd,J=8.0,1.1Hz),7.14-7.18(1H,m),7.23(1H,d,J=7.8Hz),7.37-7.44(2H,m).
实施例5
[化学式41]
第1工序
在氮气氛围中且于0℃下向氯甲酸氯甲酯(300mg,2.33mmol)与化合物30(330mg,2.79mmol)的二氯甲烷(6.0mL)溶液添加吡啶(207μL,2.56mmol),并于0℃下搅拌30分钟,升温至室温,进一步搅拌1小时。向反应液添加2mol/L盐酸,利用二氯甲烷进行萃取。利用饱和食盐水洗涤有机层,利用无水硫酸镁进行干燥后,减压蒸馏除去溶剂,而获得化合物31(440mg,产率90%)。
1H-NMR(CDCl3)δ:1.65(s,6H),3.77(s,3H),5.71(s,2H).
第2工序
使化合物III-2(300mg,0.62mmol)、碳酸钾(172mg,1.24mmol)、碘化钾(103mg,0.62mmol)与化合物31(261mg,1.24mmol)溶解于DMA(3.0mL)中,于80℃下搅拌3小时。向反应液添加2mol/L盐酸,利用乙酸乙酯进行萃取。利用饱和食盐水洗涤有机层,利用无水硫酸镁进行干燥后,减压蒸馏除去溶剂。通过硅胶柱色谱法(氯仿-甲醇)对所获得的残渣进行纯化,而获得化合物II-61(350mg,产率86%)。
1H-NMR(CDCl3)δ:1.63(s,3H),1.67(s,3H),2.86-2.93(m,1H),3.38-3.61(m,2H),3.68-3.78(m,4H),3.90-3.96(m,1H),4.06(d,J=14.0Hz,1H),4.51(dd,J=2.0Hz,9.6Hz,1H),4.65(d,J=12.4Hz,1H),5.21(d,J=14.4Hz,1H),5.36(s,1H),5.80-5.95(m,3H),6.85-6.92(m,2H),7.03-7.22(m,5H).
实施例6
[化学式42]
向化合物III-2(90mg,0.186mmol)的二氯甲烷(2mL)溶液添加乙酸酐(0.053mL,0.558mmol)、三乙胺(0.077mL,0.558mmol)、催化剂量的DMAP,并于室温下搅拌2小时。将反应液的溶剂减压蒸馏除去,通过硅胶柱色谱法(氯仿-甲醇)对所获得的残渣进行纯化。向所获得的溶液添加醚,使固体析出后,进行滤取,由此获得化合物II-4(71mg,73%)。
1H-NMR(CDCl3)δ:2.46(s,3H),2.88-2.99(m,1H),3.35-3.50(m,1H),3.60-3.65(m,1H),3.75-3.83(m,1H),3.90-4.00(m,1H),4.05(d,J=14.0Hz,1H),4.52-4.57(m,1H),4.60-4.70(m,1H),5.24-5.34(m,1H),5.35(s,1H),5.88(d,J=7.6Hz,1H),6.85-6.82(m,1H),6.90-7.05(m,2H),7.06-7.20(m,4H)
LC/MS(ESI):m/z=526.2[M+H]
实施例7
[化学式43]
第1工序
在氮气氛围中且于0℃下向三光气(300mg,2.54mmol)的二氯甲烷(6.0mL)溶液添加吡啶(257μL,3.17mmol)并搅拌15分钟。其后,向反应液添加化合物30(377mg,1.27mmol)的二氯甲烷(1.0mL)溶液,于0℃下搅拌15分钟后,升温至室温,进一步搅拌15分钟。于减压下将反应液浓缩后,添加乙酸乙酯(4.0mL),进行过滤。于减压下将滤液蒸馏去除,而获得化合物32(380mg)。
第2工序
于0℃下向化合物III-2(350mg,0.724mmol)的二氯甲烷(3.5mL)溶液添加化合物32(196mg,1.09mmol)、三乙胺(301μL,2.17mmol),并于0℃下搅拌30分钟。向反应液添加2mol/L盐酸,利用二氯甲烷进行萃取。利用饱和食盐水洗涤有机层,利用无水硫酸镁进行干燥后,减压蒸馏除去溶剂。通过硅胶柱色谱法(氯仿-甲醇)对所获得的残渣进行纯化,而获得化合物II-65(380mg,产率84%)。
1H-NMR(CDCl3)δ:1.73(s,3H),1.77(s,3H),2.90-2.99(m,1H),3.37-3.43(m,1H),3.57(t,J=8.8Hz,1H),3.76(dd,J=2.8Hz,12.0Hz,1H),3.81(s,3H),3.94(dd,J=2.8Hz,10.8Hz,1H),4.05(d,J=14.0Hz,1H),4.55(dd,J=2.8Hz,9.6Hz,1H),4.65(d,J=12.0Hz,1H),5.28(d,J=12.0Hz,1H),5.34(s,1H),5.89(d,J=8.0Hz,1H),6.86-6.95(m,2H),7.03-7.15(m,5H).
实施例8
[化学式44]
于冰浴冷却下向化合物33(276mg,0.402mmol)的THF(1mL)溶液添加乙酸(121mg,2.01mmol)、1mol/L TBAF THF溶液(1.21mL,1.21mmol),并于室温下搅拌4小时。将反应液的溶剂减压蒸馏除去,通过硅胶柱色谱法(乙酸乙酯-甲醇)对所获得的残渣进行纯化,而获得化合物II-129(179mg,产率78%)。
LC/MS(ESI):m/z=572.0[M+H]
实施例9
[化学式45]
于室温下向化合物III-2(300mg,0.62mmol)的DMF(4mL)溶液添加碳酸钾(258mg,1.8mmol)、4-(氯甲基)苯基乙酸酯(344mg,1.87mmol)、碘化钠(139mg,1.87mmol),并于65℃下搅拌1小时。向反应溶液添加水,利用乙酸乙酯进行萃取。利用水洗涤有机层,利用硫酸钠进行干燥,减压蒸馏除去溶剂。通过硅胶柱色谱法(乙酸乙酯-甲醇)对所获得的残渣进行纯化,而获得化合物II-115(120mg,产率31%)。
LC/MS(ESI):m/z=631.95[M+H]
实施例10
[化学式46]
于室温下向化合物III-2(150mg,0.31mmol)的二氯甲烷(2mL)溶液添加聚合物担载三苯基膦3mmol/g(310mg,0.93mmol)、吡啶-4-基甲醇(68mg,0.62mmol)、DEAD 40%甲苯溶液(270mg,0.62mmol),并于室温下搅拌30分钟。通过氨基柱色谱法(乙酸乙酯-甲醇)对反应溶液进行纯化,而获得化合物II-143(63mg,产率35%)。
LC/MS(ESI):m/z=575.00[M+H]
实施例11
[化学式47]
向化合物III-2(65mg,0.134mmol)的吡啶(0.8mL)溶液添加二甲氨基甲酰氯(21.7mg,0.202mmol),并于80℃下搅拌整夜。向反应液添加1mol/L盐酸,利用乙酸乙酯进行萃取。利用饱和食盐水洗涤有机层,利用无水硫酸镁进行干燥后,减压蒸馏除去溶剂。利用乙酸乙酯-己烷对所获得的残渣进行固体化而获得化合物II-27(65mg,产率87%)。
1H-NMR(CDCl3)δ:2.89(t,J=11.2Hz,1H),2.99(s,1H),3.01(s,3H),3.18-3.26(m,4H),3.45(t,J=10.8Hz,1H),3.59(t,J=10.8Hz,1H),3.70-3.80(m,1H),3.90-3.98(m,1H),4.03(d,J=13.6Hz,1H),4.50-4.70(m,2H),5.21-5.35(m,2H),5.82(d,J=7.6Hz,1H),6.91(t,J=7.6Hz,1H),7.00-7.20(m,6H).
实施例12
[化学式48]
向二氯磷酸乙酯(135mg,0.829mmol)的二氯甲烷(3mL)溶液添加L-缬氨酸甲酯盐酸盐(139mg,0.829mmol),于-78℃下滴加三乙胺(168mg,1.66mmol)的二氯甲烷(2mL)溶液。将反应液于室温下搅拌1小时后,添加化合物III-2(200mg,0.414mmol)与三乙胺(126mg,1.25mmol),于相同温度下搅拌6小时。将反应液进行浓缩,通过硅胶柱色谱法(乙酸乙酯-甲醇)进行纯化,而获得化合物II-55(112mg,产率38%)。
LC/MS(ESI):m/z=705.05[M+H]
实施例13
[化学式49]
于-78℃下向二氯磷酸乙酯(202mg,1.24mmol)的二氯甲烷(3mL)溶液滴加三乙胺(126mg,1.24mmol)与乙醇酸甲酯(112mg,1.24mmol)的二氯甲烷(2mL)混合溶液。将反应液于室温下搅拌2小时后,添加化合物III-2(200mg,0.414mmol)与三乙胺(126mg,1.25mmol),并于相同温度下搅拌1小时。将反应液进行浓缩,通过硅胶柱色谱法(乙酸乙酯-甲醇)进行纯化,而获得化合物II-57(143mg,产率52%)。
LC/MS(ESI):m/z=664.00[M+H]
实施例14
[化学式50]
于-78℃下向磷酰氯(1.53g,10mmol)的二氯甲烷(10mL)溶液滴加三乙胺(2.12g,20.95mmol)与乙醇酸甲酯(1.89mg,21mmol)的二氯甲烷(5mL)混合溶液。将反应液于室温下搅拌2小时。于室温下向反应液(2mL)添加化合物III-2(200mg,0.414mmol)与三乙胺(126mg,1.25mmol),并于相同温度下搅拌1小时。将反应液进行浓缩,通过硅胶柱色谱法(乙酸乙酯-甲醇)进行纯化,而获得化合物II-58(166mg,产率57%)。
LC/MS(ESI):m/z=707.90[M+H]
按照与上述实施例相同的方法,使用市售化合物而合成以下的实施例化合物。
[表10]
[表11]
[表12]
[表13]
[表14]
[表15]
[表16]
[表17]
[表18]
[表19]
[表20]
[表21]
[表22]
[表23]
[表24]
[表25]
[表26]
[表27]
[表28]
[表29]
[表30]
[表31]
[表32]
[表33]
[表34]
[表35]
[表36]
[表37]
[表38]
[表39]
本发明的化合物和/或本发明的化合物的母化合物对于由流行性感冒病毒诱发的症状和/或疾病是有用的。例如,对于伴随着发热、恶寒、头痛、肌肉痛、全身疲劳感等的类似感冒症状;或者咽喉痛、鼻涕、鼻塞、咳嗽、痰等气管炎症状;腹痛、呕吐、腹泻等胃肠症状;以及伴随着急性脑病、肺炎等二次感染的并发症的治疗和/或预防、症状改善是有效的。
本发明的化合物是前药,因此具有口服吸收性高;表现出良好的生物利用率;表现出良好的清除率;肺移行性高等优点,因此可成为优异的医药品。
本发明的化合物的母化合物具有对帽结构依赖性核酸内切酶的抑制活性高、且由于是病毒特异性酶因此选择性高等的效果,因此可成为副作用得以减轻的医药品。
进一步,本发明的化合物和/或本发明的化合物的母化合物具有代谢稳定性高;溶解度高;口服吸收性高;表现出良好的生物利用率;表现出良好的清除率;肺移行性高;半衰期长;非蛋白质结合率高;hERG通道抑制低;CYP抑制低;显现CPE(CytoPathic Effect,细胞变性效果)抑制效果;和/或在光毒性试验、Ames试验、遗传毒性试验中显示阴性;或不具有肝障碍等毒性等优点。因此,本发明的化合物可成为优异的医药品。
本发明的化合物和/或本发明的化合物的母化合物可以以口服或非口服的方式进行给药。在口服给药的情况下,本发明化合物可以以通常的制剂、例如片剂、粉剂、颗粒剂、胶囊剂等固体剂;水剂;油性悬浮剂;或者糖浆剂或酏剂等液剂中的任意剂型使用。在非口服给药的情况下,本发明的化合物可以以水性或油性悬浮注射剂、滴鼻液的形式使用。在其制备时,可任意地使用惯用的赋形剂、结合剂、润滑剂、水性溶剂、油性溶剂、乳化剂、悬浮化剂、保存剂、稳定剂等。本发明的药物组合物是通过将治疗有效量的本发明化合物与药学上可接受的载体或稀释剂一并组合(例如进行混合)而制造。
本发明的化合物的给药量根据给药方法、患者的年龄、体重、状态及疾病的种类而不同,但通常在口服给药的情况下,对成人每1天将约0.05mg~3000mg、优选为约0.1mg~1000mg根据需要而进行分割给药即可。另外,在非口服给药的情况下,对成人每1天给药约0.01mg~1000mg、优选为给药约0.05mg~500mg。
试验例1:帽依赖性核酸内切酶(CEN)抑制活性的测定
1)底物的制备
购买对5'末端的G进行了2磷酸化修饰,且对2'位的羟基进行了甲氧基化修饰,将自5'末端起第6位的U进行了Cy3标记,并将3'末端进行了BHQ2标记的30merRNA(5'-pp-[m2'-O]GAA UAU(-Cy3)GCA UCA CUA GUA AGC UUU GCU CUA-BHQ2-3':Japan BioServices公司制造),使用EPICENTRE公司制造的ScriptCap系统而加成帽结构(产物为m7G[5']-ppp-[5'][m2'-O]GAA UAU(-Cy3)GCA UCA CUA GUA AGC UUU GCU CUA(-BHQ2)-3')。利用改性聚丙烯酰胺凝胶电泳法对其进行分离、纯化,将其用作为底物。
2)酶的制备
RNP(ribonucleoprotein,核糖核蛋白)依据常规方法由病毒粒子制备而成(参考文献:VIROLOGY(1976)73,p327-338OLGA M.ROCHOVANSKY)。具体而言,将A/WSN/33病毒1x10
3)酶反应
向聚丙烯制的384孔盘分注酶反应液(组成:53mM三(羟甲基)氨基甲烷-盐酸盐(pH值7.8)、1mM MgCl
3)抑制率(IC
将反应已停止的溶液以85℃加热5分钟,于冰上急冷2分钟后,利用ABI PRIZM3730遗传分析仪(Genetic Analyzer)进行分析。通过解析软体ABI Genemapper而定量帽依赖性核酸内切酶产物的波峰,将PC、NC的荧光强度分别设为0%抑制、100%抑制而求出受检化合物的CEN反应抑制率(%)后,使用曲线拟合软体(XLfit2.0:Model 205(IDBS公司制造)等)而求出IC
试验例2:CPE抑制效果确认试验
<材料>
·2%FCS(Fetal Calf Serum,胎牛血清)E-MEM(Eagle's Minimum EssentialMedium,伊格尔最低必需培养基)(向MEM(Minimum Essential Medium,最低必需培养基)(Invitrogen)添加康霉素及FCS而进行调整)
·0.5%BSA(Bovine Serum Albumin,牛血清白蛋白)E-MEM(向MEM(MinimumEssential Medium,最低必需培养基)(Invitrogen)添加康霉素及BSA而进行调整)
·HBSS(Hanks'Balanced Salt Solution,汉克斯平衡盐溶液)
·MDBK(Mardin-Darby Bovine Kidney,马达氏牛肾)细胞
利用2%FCS E-MEM调整至适当细胞数(3×10
·MDCK(Madin-Darby Canine Kidney,马达氏犬肾)细胞
利用HBSS清洗2次后,利用0.5%BSA E-MEM调整至适当细胞数(5×10
·胰蛋白酶溶液
使来自猪胰的胰蛋白酶(Trypsin from porcine pancreas)(SIGMA)溶解于PBS(-)中,利用0.45μM的过滤器进行过滤。
·EnVision(PerkinElmer)
·WST-8套组(Kishida Chemical)
·10%SDS(sodium dodecyl sulfate,十二烷基硫酸钠)溶液
<操作程序>
·受检试样的稀释、分注
在使用MDBK细胞时,使用2%FCS E-MEM作为培养液,在使用MDCK细胞时,使用0.5%BSA E-MEM作为培养液。以下,关于病毒、细胞、受检试样的稀释,使用了相同的培养液。
预先将受检试样利用培养液稀释为适度的浓度,于96孔盘制作2~5倍阶梯性稀释系列(50μL/孔)。制作抗Flu活性测定用、细胞毒性测定用的2片。对各药剂实施三重测定。
在使用MDCK细胞时,仅在抗Flu活性测定用中,将胰蛋白酶以最终浓度达到3ug/mL的方式添加于细胞中。
·流行性感冒病毒的稀释、分注
预先将流行性感冒病毒利用培养液稀释为适当的浓度,向添加有受检试样的96孔盘以50μL/孔进行分注。向细胞毒性测定用的孔盘以50μL/孔分注培养液。
·细胞的稀释、分注
向添加有受检试样的96孔盘以100μL/孔分注已调整至适当细胞数的细胞。
利用孔盘搅拌器进行混合,于CO
·WST-8的分注
用肉眼在显微镜下对培养了3天的96孔盘进行观察,确认细胞的形态、结晶的有无等。以不吸取细胞的方式自孔盘去除上清液。
将WST-8套组利用培养液进行10倍稀释,向各孔以100μL/孔分注该WST-8溶液。利用孔盘搅拌器进行混合后,于CO
关于抗Flu活性测定用孔盘,在培养后,向各孔以10μL/孔分注10%SDS溶液,使病毒失活。
·吸光度的测定
对混合过的96孔盘利用EnVision于450nm/620nm的2种波长下测定吸光度。
<各测定项目值的算出>
基于如下述的计算式,使用Microsoft Excel或具有同等计算处理能力的程序而算出。
·50%流行性感冒感染细胞死亡抑制浓度(EC50)的算出
EC
Z=(50%-High%)/(High%-Low%)×{log(High conc.)-log(Low conc.)}+log(High conc.)
关于本发明化合物的母化合物,将试验例1及试验例2的测定结果示于表39。
[表40]
[表41]
根据以上的结果,母化合物由于表现出高的帽依赖性核酸内切酶(CEN)抑制活性、和/或高的CPE抑制效果,因此可成为作为由于感染流行性感冒病毒而诱发的症状和/或疾病的治疗和/或预防剂而有用的药物。
以下记载本发明化合物的生物试验例。
试验例3:CYP抑制试验
使用市售的混合人肝微粒体,以作为人类主要CYP5分子种类(CYP1A2、2C9、2C19、2D6、3A4)的典型底物代谢反应的7-乙氧基试卤灵的O-脱乙基化(CYP1A2)、甲苯磺丁脲的甲基-氢氧化(CYP2C9)、美芬妥英的4'-氢氧化(CYP2C19)、右美沙芬的O脱甲基化(CYP2D6)、特芬那定的氢氧化(CYP3A4)为指标,对各代谢物生成量被本发明化合物抑制的程度进行评价。
反应条件如下所述:底物、0.5μmol/L乙氧基试卤灵(CYP1A2)、100μmol/L甲苯磺丁脲(CYP2C9)、50μmol/L S-美芬妥英(CYP2C19)、5μmol/L右美沙芬(CYP2D6)、1μmol/L特芬那定(CYP3A4);反应时间、15分钟;反应温度、37℃;酶、混合人肝微粒体0.2mg蛋白质/mL;本发明化合物浓度1、5、10、20μmol/L(4点)。
在96孔盘中,作为反应溶液,向50mmol/L Hepes缓冲液中以上述组成添加各5种底物、人肝微粒体、本发明化合物,且添加作为辅酶的NADPH(nicotinamide adeninedinucleotide phosphate,烟酰胺腺嘌呤二核苷磷酸盐),引发了作为指标的代谢反应。于37℃下反应15分钟后,添加甲醇/乙腈=1/1(V/V)溶液,由此使反应停止。以3000rpm离心15分钟后,利用荧光多标计数仪定量离心上清液中的试卤灵(CYP1A2代谢物),利用LC/MS/MS(Liquid Chromatography-Mass Spectrometry-Mass Spectrometry,液相色谱-质谱联用)定量甲苯磺丁脲氢氧化物(CYP2C9代谢物)、美芬妥英4'氢氧化物(CYP2C19代谢物)、右啡烷(CYP2D6代谢物)、特芬那定醇体(CYP3A4代谢物)。
将反应体系中仅添加溶解有本发明化合物的作为溶剂的DMSO所得者设为对照组(100%),算出添加至溶剂中的本发明化合物的各浓度下的残存活性(%),使用浓度与抑制率,通过利用罗吉斯模型的逆推而算出IC
(结果)
化合物No.III-2:5种>20μmol/L
试验例4:BA(bioavailability,生物利用率)试验
口服吸收性的研究试验材料与方法
(1)使用动物:使用小鼠或SD大鼠。
(2)饲养条件:使小鼠或SD大鼠自由摄取固体饲料及杀菌自来水。
(3)给药量、分组的设定:以特定的给药量进行口服给药、静脉内给药。以下述方式设定组。(每种化合物给药量有变更)
口服给药1~30mg/kg(n=2~3)
静脉内给药0.5~10mg/kg(n=2~3)
(4)给药液的制备:口服给药是以溶液或悬浮液的形式进行给药。静脉内给药是在可溶化后进行给药。
(5)给药方法:口服给药在通过口喂管强制性地给药至胃内。静脉内给药是通过附带注射针的注射器自尾静脉进行给药。
(6)评价项目:经时采血,使用LC/MS/MS对血浆中本发明化合物浓度进行测定。
(7)统计解析:关于血浆中本发明化合物浓度变化,使用非线性最小二乘法程式WinNonlin(注册商标)算出血浆中浓度-时间曲线下面积(AUC),根据口服给药组与静脉内给药组的AUC算出本发明化合物的生物利用率(BA)。
(结果)
化合物No.II-6:14.9%
化合物No.III-2:4.2%
根据以上的结果,前药的生物利用率较母化合物提高。
因此,本发明化合物可成为口服吸收性优异、作为由于感染流行性感冒病毒而诱发的症状和/或疾病的治疗和/或预防剂有用的药物。
试验例5:代谢稳定性试验
使市售的混合人肝微粒体与本发明化合物反应一定时间,通过反应样品与未反应样品的比较而算出残存率,对本发明化合物被肝代谢的程度进行评价。
于包含人肝微粒体0.5mg蛋白质/mL的0.2mL的缓冲液(50mmol/L Tris-HCl pH值7.4、150mmol/L氯化钾、10mmol/L氯化镁)中,于1mmol/L NADPH存在下于37℃下反应0分钟或30分钟(氧化反应)。反应后,向甲醇/乙腈=1/1(v/v)溶液的100μL添加反应液50μL并进行混合,以3000rpm进行15分钟离心。利用LC/MS/MS对该离心上清液中的本发明化合物进行定量,将反应0分钟时的化合物量设为100%而计算反应后的本发明化合物的残存量。需要说明的是,水解反应体是于NADPH非存在下进行反应,葡糖醛酸结合反应体是代替NADPH而于5mmol/L UDP-葡糖醛酸的存在下进行反应,以后实施相同的操作。
(结果)表示化合物浓度2μmol/L下的氧化代谢中的残存率。
化合物No.III-2:90.1%
试验例6:CYP3A4荧光MBI试验
CYP3A4荧光MBI试验是对基于代谢反应的本发明化合物的CYP3A4抑制的增强进行考察的试验。通过CYP3A4酶(大肠杆菌表达酶)而使7-苄氧基三氟甲基香豆素(7-BFC)脱苄基化,生成发出荧光的代谢物7-羟基三氟甲基香豆素(7-HFC)。以7-HFC生成反应为指标而评价CYP3A4抑制。
反应条件如下所述:底物,5.6μmol/L 7-BFC;预反应时间,0分钟或30分钟;反应时间,15分钟;反应温度,25℃(室温);CYP3A4含量(大肠杆菌表达酶),预反应时为62.5pmol/mL,反应时为6.25pmol/mL(10倍稀释时);本发明化合物浓度0.625、1.25、2.5、5、10、20μmol/L(6点)。
在96孔盘上,向K-Pi缓冲液(pH值7.4)中将酶、本发明化合物溶液以上述预反应的组成进行添加以作为预反应液,且以经底物与K-Pi缓冲液1/10稀释的方式将预反应液的一部分转移至另一96孔盘,添加作为辅酶的NADPH,而引发作为指标的反应(无预反应),反应给定时间后,添加乙腈/0.5mol/L Tris(三羟基氨基甲烷)=4/1(V/V),由此使反应停止。另外,也向剩余的预反应液添加NADPH而引发预反应(有预反应),预反应给定时间后,以利用底物与K-Pi缓冲液1/10稀释的方式将一部分转移至另一孔盘,引发作为指标的反应。反应给定时间后,添加乙腈/0.5mol/L Tris(三羟基氨基甲烷)=4/1(V/V),由此使反应停止。针对各进行过指标反应的孔盘,利用荧光读板仪测定作为代谢物的7-HFC的荧光值。(Ex=420nm,Em=535nm)
将在反应体系中仅添加有作为溶解有本发明化合物的溶剂的DMSO者设为对照组(100%),算出以各浓度添加本发明化合物时的残存活性(%),使用浓度与抑制率,通过利用罗吉斯模型的逆推而算出IC
(结果)
化合物No.III-2:(-)
试验例7:安氏突变试验(Fluctuation Ames Test)
对本发明化合物的诱突变性进行评价。
将冷冻保存的鼠伤寒沙门氏杆菌(Salmonella typhimurium TA98株、TA100株)20μL接种至10mL液体营养培养基(2.5%Oxoid nutrient broth No.2),于37℃下进行10小时振荡预培养。关于TA98株,系将9mL的菌液进行离心(2000×g,10分钟)而去除培养液。使菌悬浮于9mL的Micro F缓冲液(K
(结果)
化合物No.III-2:(-)
试验例8:hERG试验
以本发明化合物的心电图QT间隔延长危险性评价为目的,使用表达出humanether-a-go-go related gene(hERG)通道的HEK293细胞,而研究本发明化合物对于心室再极化过程中发挥重要作用的延迟整流K
使用全自动膜片钳系统(PatchXpress 7000A,AxonInstruments Inc.),并通过全细胞膜片钳法,将细胞保持为-80mV的膜电位后,记录赋予了+40mV的去极化刺激2秒钟,进一步赋予了-50mV的再极化刺激2秒钟时所诱发的I
(结果)表示化合物浓度0.3~10μmol/L下的抑制率。
化合物No.III-2:7.9%
试验例9:溶解性试验
本发明化合物的溶解度是在添加1%DMSO的条件下确定的。利用DMSO制备10mmol/L化合物溶液,将本发明化合物溶液2μL分别添加至JP-1液(向氯化钠2.0g、盐酸7.0mL添加水而制成1000mL)、JP-2液(向使磷酸二氢钾3.40g及无水磷酸氢二钠3.55g溶解于水中而制成1000mL者1容量添加水1容量)198μL中。于室温下振荡1小时后,对混液进行过滤。利用甲醇/水=1/1(V/V)对各滤液进行10倍稀释,通过绝对校准曲线法,使用LC/MS测定滤液中浓度。
(结果)
化合物No.III-2:42.2μmol/L
试验例10:粉末溶解度试验
向适当的容器添加适量的本发明化合物,向各容器添加JP-1液(向氯化钠2.0g、盐酸7.0mL添加水而制成1000mL)、JP-2液(向pH值6.8的磷酸盐缓冲液500mL添加水500mL)、20mmol/L牛胆酸钠(TCA)/JP-2液(向TCA1.08g添加JP-2液而制成100mL)各200μL。于试验液添加后全部溶解的情况下,适当追加本发明化合物。进行密闭并于37℃下振荡1小时后进行过滤,向各滤液100μL添加甲醇100μL而进行2倍稀释。稀释倍率系视需要进行变更。确认没有气泡及析出物,进行密闭并进行振荡。通过绝对校准曲线法,使用HPLC定量本发明化合物。
(结果)
化合物No.III-2:JP-1液;7.1μg/mL、JP-2液4.4μg/mL、20mmol/L TCA/JP-2液16.1μg/mL
试验例11Ames试验
使用沙氏杆菌(Salmonella typhimurium)TA98、TA100、TA1535、TA1537及大肠杆菌(Escherichia coli)WP2uvrA作为试验菌株,于利用预培养法的非代谢活化条件下及代谢活化条件下实施Ames试验,调查本发明的化合物有无基因突变诱发性。
(结果)
化合物No.III-2:(-)
试验例12光溶血试验
使本发明化合物以目标浓度溶解,于微量盘上,与自脱纤维绵羊血制备之0.1~0.0008%浓度的红血球浮游液(2.5v/v%)进行混合,使用紫外线荧光灯(GL20SE灯,三共电气及FL20S-BLB灯,Panasonic),进行UVA及UVB区域中的光照射(10J/cm
(结果)
化合物No.III-2:(-)
图1及2表示针对将作为母化合物的化合物III-2进行前药化而成的化合物II-6,测定在非断食下向大鼠口服给药后的化合物III-2及II-6的血浆中浓度变化而获得的结果。
另外,关于化合物II-6,由于全部血浆样品中的浓度为定量下限以下,因此可知将作为母化合物的化合物III-2进行前药化而成的化合物II-6是在给药后在体内迅速地转化为化合物III-2(参照图2)。
根据这些试验结果判明如下情况:经前药化而成的化合物在口服给药后被吸收至体内而在血中迅速地转化为母化合物。因此,本发明的化合物可成为作为由于感染流行性感冒病毒而诱发的症状和/或疾病的治疗和/或预防剂而有用的药物。
试验例13静脉内给药试验
静脉内给药试验的研究实验材料与方法
(1)使用动物:使用SD大鼠。
(2)饲养条件:使SD大鼠自由摄取固体饲料及杀菌自来水。
(3)给药量、分组的设定:根据特定的给药量向静脉内给药。以下述方式设定组。(每种化合物给药量有变更)
静脉内给药0.5~1mg/kg(n=2~3)
(4)给药液的制备:静脉内给药是在可溶化后进行给药。
(5)给药方法:静脉内给药是通过附带注射针的注射器自尾静脉进行给药。
(6)评价项目:经时采血,使用LC/MS/MS对血浆中本发明化合物浓度进行测定。
(7)统计解析:关于血浆中本发明化合物浓度变化,使用非线性最小二乘法程式WinNonlin(注册商标)算出总体清除率(CLtot)及消失半衰期(t1/2,z)。
(结果)
化合物No.III-2:
CLtot:16.4mL/min/kg
t1/2,z:3.4小时
根据以上的结果判明,化合物III-2系总体清除率较低,半衰期较长的化合物。
因此,本发明化合物可成为持续性优异、作为由于感染流行性感冒病毒而诱发的症状和/或疾病的治疗和/或预防剂而有用的药物。
制剂例
以下所示的制剂例仅仅是例示,完全不限定发明的范围。
制剂例1:片剂
将本发明化合物、乳糖及硬脂酸钙进行混合,进行破碎造粒并进行干燥,制成适当尺寸的颗粒剂。接着添加硬脂酸钙,进行压缩成形而制成片剂。
制剂例2:胶囊剂
将本发明化合物、乳糖及硬脂酸钙均匀地进行混合,制成粉末或细粒状而制作粉剂。将其填充于胶囊容器中而制成胶囊剂。
制剂例3:颗粒剂
将本发明化合物、乳糖及硬脂酸钙均匀地进行混合,压缩成型后,进行粉碎、整粒,进行筛别而制成适当尺寸的颗粒剂。
制剂例4:口腔崩解片
将本发明化合物及结晶纤维素进行混合,于造粒后进行压片而制成口腔崩解片。
制剂例5:干糖浆
将本发明化合物及乳糖进行混合,进行粉碎、整粒、筛别而制成适当尺寸的干糖浆。
制剂例6:注射剂
将本发明化合物及磷酸缓冲液进行混合而制成注射剂。
制剂例7:点滴剂
将本发明化合物及磷酸缓冲液进行混合而制成点滴剂。
制剂例8:吸入剂
将本发明化合物及乳糖进行混合并细微地粉碎,由此制成吸入剂。
制剂例9:软膏剂
将本发明化合物及凡士林进行混合而制成软膏剂。
制剂例10:贴剂
将本发明化合物及黏着石膏等基剂进行混合而制成贴剂。
工业实用性
本发明的化合物在吸收至体内后具有帽依赖性核酸内切酶(CEN)抑制活性。本发明的化合物可成为作为由于感染流行性感冒病毒而诱发的症状和/或疾病的治疗和/或预防剂而有用的药物。
- 经取代的多环性吡啶酮衍生物及其前药
- 经取代的多环性吡啶酮衍生物及其前药