一种中压制备色谱快速提取莪术有效成分的方法
文献发布时间:2023-06-19 11:47:31
技术领域
本发明涉及中药制剂领域,特别是一种中压制备色谱快速提取莪术有效成分的方法。
背景技术
莪术来源于姜科植物蓬莪术、广西莪术和温郁金的根茎部位,其具有抗肿瘤、抗细菌、行气破血,消除积肿、缓解疼痛等效果。通常用于血液问题,呼吸不适,心脏疼痛,积食便秘,胃腹肿胀疼痛,淤血,痛经,癓瘕痞块,跌打损伤以及早期宫颈癌等症状,为临床常用药。莪术中主要成分可分为两大类,即挥发油(1.96%~2.5%)和姜黄素类。姜黄素是一种具有降血脂、抗氧化、抗炎和消除自由基等作用的酚类色素物,挥发油则包含莪术抗肿瘤、抗癌症的主要有效成分,这些成分为多种倍半萜类化合物和桉油精。莪术挥发油的有效成分中主要抗肿瘤活性的成分有莪术酮、吉马酮等化合物,这些化合物对肿瘤细胞均具有一定的抑制作用。而莪术挥发油中的莪术酮、吉马酮含量相对较高,一般被认为是莪术抗肿瘤、抗癌的最主要有效成分。
莪术酮又名莪术呋喃烯酮,是一种倍半萜类化合物,在莪术挥发油中的含量约为1%~2.5%,分子式为C
由于天然产物中一般具有的成分及杂质种类较多,分离纯化其中有效成分的纯度一般不高。现有技术从植物中提取莪术挥发油、以及从莪术挥发油中提取莪术酮、吉马酮的方法均存在提取纯度低、操作繁琐等缺点。因此,亟需寻求一种高效、便捷提取莪术酮、吉马酮的方法以满足中药治疗的需要。
发明内容
本发明要解决的技术问题为克服现有技术中提取莪术酮、吉马酮的方法均存在提取纯度低、操作繁琐等缺点的不足之处,提供一种中压制备色谱快速提取莪术有效成分的方法。
为了解决本发明的技术问题,所采取的技术方案为,一种中压制备色谱快速提取莪术有效成分的方法,所述有效成分为莪术酮和吉马酮,具体包括如下步骤:
S1、取含有莪术的植物根茎除去杂质后切片、烘干、粉碎,过筛得到粒径为40-100目的莪术粉;
S2、提取莪术粉中的莪术挥发油;
S3、将莪术挥发油用乙醇溶解后与硅胶混合均匀,然后装入中压制备色谱的硅胶色谱柱,使用洗脱剂进行梯度洗脱,洗脱剂的流速为10-40mL/min,压力为0.048-0.103Mpa,根据中压制备色谱检测器的紫外吸收信号所显示的莪术挥发油中各组分的吸收峰,分别收集各洗脱液,将含有相同成分的过柱洗脱液合并;
在合并的过柱洗脱液中加入乙醇至刚好溶解,再经薄层色谱法纯化分别得到莪术酮和吉马酮馏分,将两份馏分分别依次经过减压旋蒸浓缩、冷冻干燥,即得高纯莪术酮和吉马酮。
作为上述中压制备色谱快速提取莪术有效成分的方法进一步的改进:
优选的,所述提取莪术粉中的莪术挥发油的方法为回流加热法或者超声提取法。
优选的,所述回流加热法的具体步骤为:称取莪术粉置于洁净干燥圆底烧瓶中,按10-20mL/g的液料比加入乙醇,加入搅拌磁子,置于搭建好的回流装置中,温度80-100℃,加热回流8-16h,将提取液减压抽滤,滤液转移至旋转蒸发仪中浓缩至溶剂完全去除,得到褐色油状物即为莪术挥发油。
优选的,所述超声提取法的具体步骤为:称取莪术粉置于离心管中,加入提取剂至莪术粉的浓度为2mg/mL,置于超声清洗机中超声震动10-30分钟,离心管内呈浑浊混悬浮液状态,将离心管置于离心机内离心,上层清液即为莪术挥发油。
优选的,所述提取剂为乙醇、乙酸乙酯、N,N-二甲基甲酰胺即DMF、二甲基亚砜即DMSO、二氧六环中的任意一种。
优选的,所述提取剂为乙醇。
优选的,所述洗脱剂为石油醚-乙酸乙酯体系、正己烷-乙酸乙酯体系、石油醚-二氯甲烷体系中的一种。
优选的,所述洗脱剂为石油醚-乙酸乙酯体系,石油醚为A相、乙酸乙酯为B相,两者按体积比(100~81):(0~19)配制而成。
本发明相比现有技术的有益效果在于:
1)本发明通过提取和分离两步从莪术切片中制备了莪术酮和吉马酮。本发明在第一步粗提莪术挥发油时,对比了超声提取法和加热回流提取法提取莪术挥发油,检测莪术挥发油所含的主要物质为莪术酮和吉马酮;超声提取法中不同有机溶剂对莪术中有效成分的提取率存在差异;在加热回流提取法中,排除了加热回流法的实验溶剂的影响因素,方便了实验的操作,解决了实验废料排放的后处理问题,提升了实验效率,为以后莪术挥发油的工业化生产提供了一个经济环保的参考途径。然后以薄层色谱探索展开剂选择体系,选择展开剂体系为石油醚:乙酸乙酯=5:1,通过中压快速制备色谱除去杂质,薄层色谱板纯化得到莪术酮从莪术粉中的提取率约为0.2%,莪术酮的纯度为93.29%;吉马酮从莪术粉中的提取率提取率约为0.2%,吉马酮的纯度为98.35%。
第二步利用了中压快速制备色谱对莪术挥发油中有效成分进行纯化。中压快速制备色谱具有分离速度快、上样量大、制备毫克到数十克的样品所需时间较短,分离效率高、且分离纯化后的样品纯度也相对较高的特点。同时,其可自主填充分离柱的填料,增加了分离溶剂体系的可选择性。中压快速制备色谱在天然产物分离纯化工作中已经发展为一种备受欢迎的分离纯化方法。本研究首次将中压快速制备色谱运用于莪术中有效成分的分离纯化,并获得高纯度莪术酮和吉马酮,为生产医用级莪术酮和吉马酮提供了数据参考及高效环保的生产途径的参考。
附图说明
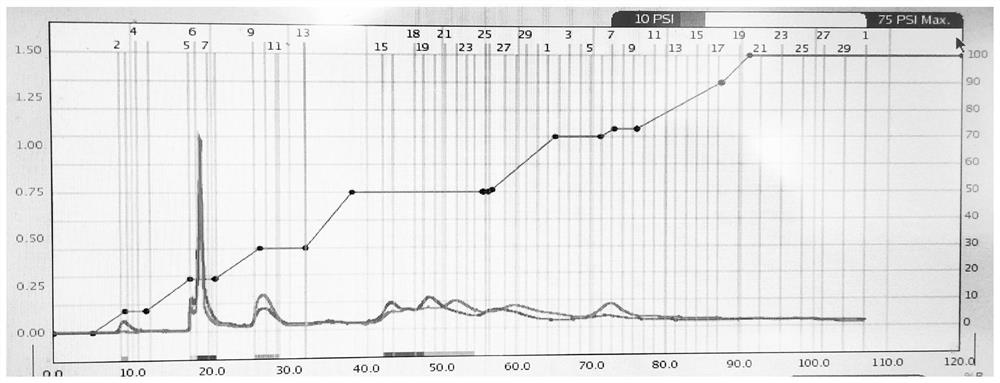
图1是中压制备色谱洗脱极性设置与出峰情况结果,横坐标为时间,单位为min;左边纵坐标为吸光强度,单位为Abs;右边纵坐标为B相百分比,单位为%;
图2(a)为实施例3经过薄层色谱法纯化后样品中莪术酮LCMS鉴定图、图2(b)为莪术酮1HNMR鉴定图、图2(c)为莪术酮HPLC鉴定图;
图3(a)为实施例3经过薄层色谱法纯化后样品中吉马酮LCMS鉴定图、图3(b)为吉马酮1HNMR鉴定图、图3(c)为吉马酮HPLC鉴定图。
具体实施方式
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合实施例,对本发明进行进一步详细说明,基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
提取莪术有效成分的原材料为莪术根茎切片采购于亳州市众益堂中药材销售有限公司,产自广西壮族自治区钦州市灵山县,属于广西莪术。取莪术根茎切片适量,用粉碎机粉碎后,粉末过40目筛后,收集并保存在塑封袋中备用。
莪术挥发油的提取实验中所用到的主要仪器情况见下表1。
表1 莪术挥发油的提取主要仪器基本情况一览表
莪术挥发油的提取实验中所用到的主要试剂情况见下表2。
表2 莪术挥发油的提取主要试剂基本情况一览表
实施例1莪术挥发油的超声提取法及结果
称取备用的莪术粉约2毫克,置于九个相同1.5mL的离心管中,分别加入不同有机溶剂,使液面至离心管1mL刻度处,做好编号C1~C9,置于超声清洗机中(温度20℃)超声震动10分钟。超声提取液送液相质谱联用仪检测后,提取到的主要物质为莪术酮和吉马酮。不同有机溶剂对这两种物质提取结果统计见下表3。
表3 编号、溶剂、超声提取结果一览表
由表3中超声提取莪术挥发油并对其有效成分进行检测的结果可知,不同有机溶剂对莪术中有效成分莪术酮和吉马酮的提取率存在差异,有些有机溶剂甚至无法提取出莪术粉中的吉马酮。
实施例2莪术挥发油的加热回流提取法及结果
称取备用的莪术粉30g,置于500mL洁净干燥圆底烧瓶中,按10mL/g的液料比,加入300mL乙醇,加入搅拌磁子,置于搭建好的回流装置中,温度85℃,加热回流过夜(约16h)。将过夜后的提取液减压抽滤,滤液转移至旋转蒸发仪中浓缩至溶剂完全去除,得到褐色油状物。称重后取适量上步所得褐色油状物于液质联用仪送样瓶中,加适量甲醇送液相质谱联用仪检测。以提取所得莪术挥发油量与莪术粉量的比计算得率。即:
将所得莪术挥发油转移到合适容器中,称重为1.8克莪术油,计算提取率为6.0%。超声提取液送液相质谱联用仪检测显示,以乙醇做提取剂时,回流加热法提取到的莪术中有效成分的种类比超声提取法提取到的多,但提取的莪术酮和吉马酮的量不如以超声提取法,测试莪术挥发油中莪术酮峰面积占比为15.215%、吉马酮峰面积占比为6.03%。
实施例3提取莪术挥发油的有效成分莪术酮和吉马酮
中压色谱快速制备实验中所用到的主要仪器情况见下表4。
表4 中压色谱快速制备主要仪器基本情况一览表
CHEETAH系统中压快速纯化制备色谱,其设计理念采用工业一体化,可实现实时在线监控,操作简单实用,更是大大缩短了样品的分离时间,实现样品分离的同时提升了产品价值。该制备色谱可标示出不同出峰物质,自动收集到不同试管中,且实现峰-管位置对应,方便实验过程中的实时处理,即在运行过程中,其洗脱梯度、流速、收集方式等可变因素可以任意自由更改改组合。溶剂输送泵为二元梯度,最多可进行四元切换。检测器采用双波长紫外检测器,其检测波长可在200~400nm范围内调整,吸光度为≤2AU范围。上样方式可选择液体上样或固体上样,收集方式支持峰接收、全收集、手动收集等方式的自由组合,且可实现没有紫外吸收或紫外吸收较弱的物质的快速制备纯化。
实验所用制备层析柱为天津博纳艾杰尔科技有限公司的ClaricepTM Flash中压纯化制备柱,包括预装柱80g(CS140080-0),其柱体积为160mL,直径为3.2cm,长21.0cm,填充基质为40-60μm硅胶,最大压力为180psi;非预装柱12g(CS140012-0),其柱体积为24mL,直径为21.4mm,长76mm,最大压力为180psi,所用拌样硅胶为100-200目,上海盛亚化工有限公司产品。
中压色谱快速制备实验中所用到的主要试剂情况见下表5。
表5 中压色谱快速制备实验主要试剂基本情况一览表
干法上样过柱,取加热回流提取的莪术挥发油1.8克置于100mL接收瓶中,加适量乙醇,使挥发油完全溶解在溶剂中,加约2g硅胶(硅胶量约为待吸附样品的一倍量),在旋转蒸发仪中浓缩,使溶剂完全去除,将已吸附硅胶上的样品装填至空白干净12g规格的硅胶柱中,用棉花或石英砂压实,制得装样品硅胶柱,准备上柱。
通过薄层色谱法确定过柱洗脱剂体系为石油醚-乙酸乙酯体系,石油醚为A相,乙酸乙酯为B相,先用B相溶剂冲洗管路约100mL后,换A相重复上一步骤,在中压制备色谱上装空白预装硅胶柱(80g),用A相溶剂将预装硅胶柱完全润湿后,在预装硅胶柱上方装样品硅胶柱,流速为35mL/min,压力为4.76-10.34KPa,体积分数100%石油醚洗脱7min后,逐渐增加乙酸乙酯进入洗脱体系,当有峰出现时,保持当前极性洗脱,峰转变为水平时,恢复极性增大趋势,重复上述操作。
中压制备色谱洗脱极性设置与出峰情况结果如图1所示。从图1可以看出,在B相体积分数为19%,即石油醚:乙酸乙酯体积比81:19极性下,出现一个在254nm波长下吸收较强的峰,收集在两个试管中。将两个试管中的溶液合并在一个干燥洁净100mL接收瓶中,在旋转蒸发仪中将溶剂旋干,转移到合适容器中,得到加热回流提取的莪术挥发油经经中压制备色谱过柱后收集的产物,称量约0.205g,取适量送LCMS检测。通过中压制备色谱获得主要产物只用20分钟左右,收率11.4%。
为了拿到高纯度的莪术酮和吉马酮,将中压制备色谱过柱后收集的产物进行薄层色谱法纯化。将收集到的0.205g产品,加适量乙醇溶剂溶解,用1.5mL滴管在200×200mm薄层硅胶板上距其一端2cm处划一条约0.5cm宽的样品带,因为一块200×200mm薄层硅胶板的上样量为0~100mg,为了保证分离纯度和效率,这里共用四块相同200×200mm薄层硅胶板上样。展开剂用石油醚:乙酸乙酯=5:1体系。
爬板共出现四条带,在紫外分析仪中将四条带用铅笔轻轻画出轮廓(注意避免将边沿线画在圈内,因为边沿线可能存在产品交叉),每条带刮1cm长,刮下来的硅胶转移至1.5mL离心管中,加适量乙酸乙酯,超声震荡,使硅胶上的物质全部溶解在离心管中,离心机中离心,分别得到含有有效成分莪术酮和吉马酮的上层清液,取适量上层清液送LCMS检测。
称量得莪术酮70mg,从莪术挥发油中提取率为34.1%,从莪术粉中的提取率为0.2%;吉马酮68mg,从莪术挥发油中提取率为33.2%,从莪术粉中的提取率为0.2%。
莪术酮鉴定结果见图2(a)莪术酮LCMS鉴定图、图2(b)莪术酮1HNMR鉴定图、图2(c)莪术酮HPLC鉴定图。
图2(a)为莪术酮的LCMS鉴定图,谱图中只有一个较高峰,即保留时间(RT)=3.688min时,质核比为231.0,而莪术酮的分子量为230.13,根据EM+1,即[M+H]
图2(b)为莪术酮的核磁氢谱鉴定图,溶剂为氘带氯仿,解析谱图得,氘带氯仿的溶剂峰的化学位移为7.262,定标为7.270,其余莪术酮化学结构上氢的化学位移分别为:1HNMR(400MHz,CHLOROFORM-d)δ7.10(s,1H),5.82(d,J=10.8,17.6Hz,1H),5.01(s,1H),4.99(d,J=1.6Hz,1H),4.95(d,J=5.2Hz,1H),4.76(s,1H),3.02(s,1H),2.95-2.89(m,1H),2.83-2.76(m,1H),2.19(d,J=1.2Hz,3H),1.84(s,3H),1.19(s,3H)。根据莪术酮的化学结构,莪术酮的结构有3个-CH
图2(c)为莪术酮的HPLC鉴定图,主要鉴定所制备的莪术酮的纯度,从谱图可以看出,在254nm波长下,莪术酮的保留时间为5.18min,纯度为93.29%,提取率约为0.2%。
吉马酮鉴定结果见图3(a)吉马酮LCMS鉴定图、图3(b)吉马酮1HNMR鉴定图、图3(c)吉马酮HPLC鉴定图。
图3(a)为吉马酮的LCMS鉴定图,谱图中只有一个较高峰,即RT=4.250min时,,质核比为219.0,而吉马酮的分子量为218.12,根据EM+1,即[M+H]+,吉马酮的MS值则应为219,因此确定该保留时间下出峰为吉马酮的峰。
图3(b)为吉马酮的核磁氢谱鉴定图,溶剂为氘带氯仿,解析谱图得,氘带氯仿的溶剂峰出在7.262,定标为7.270,其余吉马酮化学结构上氢的化学位移为:1HNMR(400MHz,CHLOROFORM-d)δ4.99(d,J=12.0Hz,1H),4.72(d,J=9.2Hz,1H),3.50(d,J=4.8Hz,1H),3.42(d,J=10.4Hz,1H),3.01-2.92(m,2H),2.90-2.83(m,1H),2.44-2.31(m,1H),2.20-2.15(m,1H),2.09(s,1H),1.78(s,3H),1.73(s,3H),1.64(s,3H),1.45(s,3H)。根据吉马酮的化学结构,吉马酮的结构有4个-CH3,-CH3上的H的化学位移分别为1.78、1.73、1.64、1.45;4个-CH2,-CH2上的H的化学位移分别为3.50、3.42、3.01-2.92、2.90-2.83、2.44-2.31、2.20-2.15、2.09;2个-H,其化学位移分别为4.99、4.72,与文献对比后确定鉴定的化合物为吉马酮。
图3(c)为吉马酮的HPLC鉴定图,主要鉴定所制备的吉马酮的纯度,从谱图可以看出,在254nm波长下,吉马酮的保留时间为5.87min,纯度为98.35%,提取率约为0.2%。
本领域的技术人员应理解,以上所述仅为本发明的若干个具体实施方式,而不是全部实施例。应当指出,对于本领域的普通技术人员来说,还可以做出许多变形和改进,所有未超出权利要求所述的变形或改进均应视为本发明的保护范围。
- 一种中压制备色谱快速提取莪术有效成分的方法
- 中压制备色谱且纯化高纯吴茱萸碱和吴茱萸次碱的方法