一种具有硼酸酯结构的阿霉素前药及其合成方法
文献发布时间:2023-06-19 12:04:09
技术领域
本发明涉及抗癌药物缀合物设计制备技术领域,尤其涉及一种具有硼酸酯结构的阿霉素前药及其合成方法。
背景技术
在异常的氧化应激环境下,肿瘤细胞促使超氧阴离子自由基、羟基自由基以及过氧化氢的过度表达。这种活性氧通过线粒体、过氧化物酶体以及内质网等独特的氧代谢途径激发。通过减少线粒体内电子传递链中的氧分子,这些超氧阴离子自由基有规律地产生,暗示癌细胞具有显著的氧化潜能。因此,活性氧响应也常用于癌细胞中药物释放,使其具有更高的选择性。一系列包含活性氧响应聚合物,例如聚(丙烯硫醚)、含硒共聚物、聚硫醚缩酮以及含氧化不稳定基团(如硼酸酯、脯氨酸和硫代缩酮)已得到广泛可用于癌症治疗。修饰抗癌药物将活性氧反应基团引入前药中可降低细胞毒性,对提高非选择性药物的安全性具有重要意义。
硼酸酯基团对细胞中活性氧具有反应活性,硼酸酯中的硼原子含有一个空的p-轨道,易受亲核物质如H
有鉴于此,有必要提供一种具有硼酸酯结构的阿霉素前药及其合成方法,在苯硼酸酯结构上引入组氨酸、胆固醇、透明质酸等其他功能性分子,赋予阿霉素溶酶体逃逸、靶向等功能。
发明内容
本发明的目的在于克服传统技术中存在的上述问题,提供一种具有硼酸酯结构的阿霉素前药及其合成方法,在苯硼酸酯结构上引入组氨酸、胆固醇、透明质酸等其他功能性分子,赋予阿霉素溶酶体逃逸、靶向等功能。
为实现上述技术目的,达到上述技术效果,本发明是通过以下技术方案实现:
一种具有硼酸酯结构的阿霉素前药,其结构通式如式(I)所示:
式(I)中,R为功能性分子,所述功能性分子包括组氨酸、胆固醇、透明质酸中的至少一种。
一种阿霉素前药的合成方法,该合成方法包括如下步骤:
1)按一定配比将对羟甲基苯硼酸频哪醇酯、4-二甲氨基吡啶溶于无水四氢呋喃,冰浴条件下加到溶对硝基氯甲酸苯酯后在20-30℃下反应12-24h后用硅胶柱色谱分离纯化得到4-硝基苯4-(4,4,5,5-四甲基-1,3,2-二氧杂环-2-基)碳酸苄酯白色晶体;
2)按一定配比将4-硝基苯4-(4,4,5,5-四甲基-1,3,2-二氧杂环-2-基)碳酸苄酯、阿霉素、4-二甲氨基吡啶溶于N,N-二甲基甲酰胺中,在20-30℃下反应12-24h后加入到乙醚中,获得的红色固体溶于二甲基亚砜,加入1mol/L盐酸溶液,室温搅拌12-24h后加入到乙醚中,得到的红色固体经硅胶柱色谱分离纯化得到中间产物a;
3)按一定配比将功能性分子溶于一定溶剂后加入1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐、N-羟基硫代琥珀酰亚胺、三乙胺搅拌1-3h,加入5-10毫摩尔多巴胺加至上述溶液中,20-30℃反应12-24h后经柱层析得到中间产物b;
4)按一定配比中间产物b与中间产物a加入至N,N-二甲基甲酰胺,过程干燥,在20-30℃下反应12-24h,经柱层析得到目标产物。
进一步地,如上所述阿霉素前药的合成方法,步骤1)中,对羟甲基苯硼酸频哪醇酯、4-二甲氨基吡啶和溶对硝基氯甲酸苯酯的摩尔比为1:1-4:0.5-2。
进一步地,如上所述阿霉素前药的合成方法,步骤2)中,4-硝基苯4-(4,4,5,5-四甲基-1,3,2-二氧杂环-2-基)碳酸苄酯、阿霉素和4-二甲氨基吡啶的摩尔比为1:1-5:3-15。
进一步地,如上所述阿霉素前药的合成方法,步骤3)中,功能性分子、1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐、N-羟基硫代琥珀酰亚胺、三乙胺和多巴胺的摩尔比为1:1-2.5:0.5-2.5:1-2:1-2。
进一步地,如上所述阿霉素前药的合成方法,步骤3)中,功能性分子为组氨酸时,该组氨酸采用叔丁氧羰基保护氨基,获得氨基双保护组氨酸。
进一步地,如上所述阿霉素前药的合成方法,步骤3)中,功能性分子为胆固醇时,将胆固醇与琥珀酸酐反应进行羧基化处理,获得胆固醇琥珀酸单酯。
进一步地,如上所述阿霉素前药的合成方法,步骤4)中,中间产物a和中间产物b的摩尔比为1:1-5:。
本发明的有益效果是:
本发明提供的阿霉素前药结构设计科学合理,其结构中含有易于被活性氧氧化的硼酸酯基团,在活性氧存在下发生断裂,释放阿霉素。并且结构中的组氨酸、胆固醇等功能性分子,也使其具有溶酶体逃逸与匹配载药体系等功能。
当然,实施本发明的任一产品并不一定需要同时达到以上的所有优点。
附图说明
为了更清楚地说明本发明实施例的技术方案,下面将对实施例描述所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
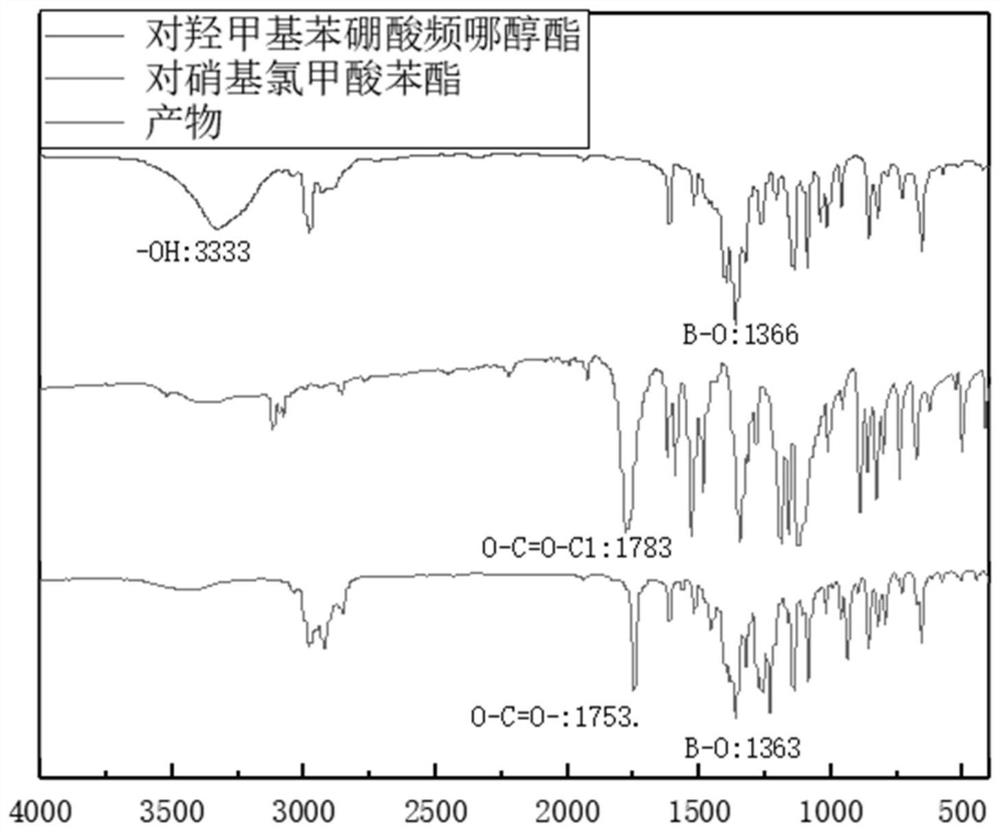
图1为本发明实施例1中白色晶体红外谱图;
图2为本发明实施例1中氨基双保护的组氨酸红外光谱;
图3为本发明实施例2中胆固醇琥珀酸单酯红外谱图;
图4为本发明实施例2中产物红外谱图。
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其它实施例,都属于本发明保护的范围。
一种具有硼酸酯结构的阿霉素前药,其特征在于,其结构通式如式(I)所示:
式(I)中,R为功能性分子,所述功能性分子包括组氨酸、胆固醇、透明质酸中的至少一种。
一种阿霉素前药的合成方法,该合成方法包括如下步骤:
1)按一定配比将对羟甲基苯硼酸频哪醇酯、4-二甲氨基吡啶溶于无水四氢呋喃,冰浴条件下加到溶对硝基氯甲酸苯酯后在20-30℃下反应12-24h后用硅胶柱色谱分离纯化得到4-硝基苯4-(4,4,5,5-四甲基-1,3,2-二氧杂环-2-基)碳酸苄酯白色晶体;
2)按一定配比将4-硝基苯4-(4,4,5,5-四甲基-1,3,2-二氧杂环-2-基)碳酸苄酯、阿霉素、4-二甲氨基吡啶溶于N,N-二甲基甲酰胺中,在20-30℃下反应12-24h后加入到乙醚中,获得的红色固体溶于二甲基亚砜,再加入1mol/L盐酸溶液,室温搅拌12-24h加入到乙醚中,得到的红色固体经硅胶柱色谱分离纯化得到中间产物a;
3)按一定配比将功能性分子溶于一定溶剂加入1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐、N-羟基硫代琥珀酰亚胺、三乙胺搅拌1-3h,加入5-10毫摩尔多巴胺加至上述溶液中,20-30℃反应12-24h,经柱层析得到中间产物b;
4)按一定配比将中间产物b与中间产物a加入至N,N-二甲基甲酰胺,过程干燥,在20-30℃下反应12-24h,经柱层析得到目标产物。
以下为制备过程中使用的部分试剂代号:
EDCI 1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐
NHS N-羟基硫代琥珀酰亚胺
本发明具体实施例如下:
实施例1
本实施例中功能性分子为氨基双保护的组氨酸,氨基双保护的组氨酸制备:将5毫摩尔组氨酸溶于水与二氧六环的混合溶液,冰浴条件滴加7.5毫摩尔二碳酸二叔丁酯后,缓慢滴加5毫摩尔K
本实施例中阿霉素前药的合成方法包括如下步骤:
1)1毫摩尔对羟甲基苯硼酸频哪醇酯、1.5毫摩尔4-二甲氨基吡啶溶于无水四氢呋喃,冰浴条件下滴加1毫摩尔溶对硝基氯甲酸苯酯,滴加完毕25℃反应12h,反应结束后用硅胶柱色谱分离纯化得到4-硝基苯4-(4,4,5,5-四甲基-1,3,2-二氧杂环-2-基)碳酸苄酯白色晶体。4-硝基苯4-(4,4,5,5-四甲基-1,3,2-二氧杂环-2-基)碳酸苄酯红外谱图见图2;
2)0.1毫摩尔4-硝基苯4-(4,4,5,5-四甲基-1,3,2-二氧杂环-2-基)碳酸苄酯、0.1毫摩尔阿霉素、0.15毫摩尔4-二甲氨基吡啶溶于N,N-二甲基甲酰胺中,25℃反应12h,反应结束将反应液逐滴加入到乙醚中,将得到红色固体溶于二甲基亚砜,加入1mol/L盐酸溶液,室温搅拌12-24h,逐滴加入到乙醚中,得到红色固体,经硅胶柱色谱分离纯化得到中间产物。
3)5毫摩尔双保护组氨酸,6毫摩尔1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐、6毫摩尔N-羟基硫代琥珀酰亚胺、5-10毫摩尔三乙胺溶于水中,室温下搅拌1h。加入5毫摩尔多巴胺加至上述溶液中,25℃反应24h,经柱层析得到产物。将产物溶于10%三氟乙酸水溶液,室温搅拌2-4h,经柱层析得到产物。
4)将0.1毫摩尔步骤2)产物与0.1毫摩尔步骤3)产物及无水硫酸镁加入至N,N-二甲基甲酰胺,25℃反应24h后逐滴加入乙醚中,得到产物。
实施例2
本实施例中功能性分子为胆固醇琥珀酸单酯,胆固醇琥珀酸单酯制备:2毫摩尔胆固醇、4毫摩尔琥珀酸酐、2毫摩尔4-二甲氨基吡啶溶于二氯甲烷,65℃回流过夜,旋干溶剂后,用丙酮结晶得到胆固醇琥珀酸单酯。胆固醇琥珀酸单酯红外谱图见图3。
本实施例中阿霉素前药的合成方法包括如下步骤:
1)1毫摩尔对羟甲基苯硼酸频哪醇酯、2毫摩尔4-二甲氨基吡啶溶于无水四氢呋喃,冰浴条件下加至1.5毫摩尔溶对硝基氯甲酸苯酯,滴加完毕25℃反应12h,反应结束后用硅胶柱色谱分离纯化得到4-硝基苯4-(4,4,5,5-四甲基-1,3,2-二氧杂环-2-基)碳酸苄酯白色晶体。4-硝基苯4-(4,4,5,5-四甲基-1,3,2-二氧杂环-2-基)碳酸苄酯红外谱图见图1;
2)0.1毫摩尔4-硝基苯4-(4,4,5,5-四甲基-1,3,2-二氧杂环-2-基)碳酸苄酯、0.1毫摩尔阿霉素、0.15毫摩尔4-二甲氨基吡啶溶于N,N-二甲基甲酰胺中,25℃反应12h,反应结束将反应液逐滴加入到乙醚中,将得到红色固体溶于二甲基亚砜,加入1M盐酸溶液,室温搅拌12-24小时,再加入到乙醚中,得到红色固体,经硅胶柱色谱分离纯化得到中间产物。
3)1毫摩尔胆固醇琥珀酸单酯、1.2毫摩尔(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐、1.2毫摩尔N-羟基硫代琥珀酰亚胺溶于二氯甲烷与N,N-二甲基甲酰胺混合溶剂,加入室温下活化,加入1毫摩尔多巴胺,25度反应24h。反应结束后,分离纯化得到产物。产物红外谱图见图4。
4)将0.1毫摩尔步骤2)产物与0.2毫摩尔步骤3)产物和无水硫酸镁加入至N,N-二甲基甲酰胺,25℃反应24h后逐滴加入乙醚中,得到产物。
实施例3
本实施例中功能性分子为透明质。
本实施例中阿霉素前药的合成方法包括如下步骤:
1)1毫摩尔对羟甲基苯硼酸频哪醇酯、3毫摩尔4-二甲氨基吡啶溶于无水四氢呋喃,冰浴条件下加至1.5毫摩尔溶对硝基氯甲酸苯酯,滴加完毕25℃反应12h,反应结束后用硅胶柱色谱分离纯化得到4-硝基苯4-(4,4,5,5-四甲基-1,3,2-二氧杂环-2-基)碳酸苄酯白色晶体。4-硝基苯4-(4,4,5,5-四甲基-1,3,2-二氧杂环-2-基)碳酸苄酯红外谱图见图1;
2)0.1毫摩尔4-硝基苯4-(4,4,5,5-四甲基-1,3,2-二氧杂环-2-基)碳酸苄酯、0.1毫摩尔阿霉素、0.15毫摩尔4-二甲氨基吡啶溶于N,N-二甲基甲酰胺中,25℃反应12h,反应结束将反应液逐滴加入到乙醚中,得到红色固体,溶于二甲基亚砜,加入1M盐酸溶液,室温搅拌12-24小时,再加入到乙醚中,得到红色固体,经硅胶柱色谱分离纯化得到中间产物。
3)2毫摩尔透明质酸、2.4毫摩尔(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐、2.4毫摩尔N-羟基硫代琥珀酰亚胺溶于水,室温下活化,加入1毫摩尔多巴胺,25℃反应24h。反应结束后分离纯化得到产物。
4)将0.1毫摩尔步骤2)产物与0.35毫摩尔步骤4)产物和无水硫酸镁加入至N,N-二甲基甲酰胺,25℃反应24h后逐滴加入乙醚中,得到产物。
以上公开的本发明优选实施例只是用于帮助阐述本发明。优选实施例并没有详尽叙述所有的细节,也不限制该发明仅为具体实施方式。显然,根据本说明书的内容,可作很多的修改和变化。本说明书选取并具体描述这些实施例,是为了更好地解释本发明的原理和实际应用,从而使所属技术领域技术人员能很好地理解和利用本发明。本发明仅受权利要求书及其全部范围和等效物的限制。
- 一种具有硼酸酯结构的阿霉素前药及其合成方法
- 一种具有还原性响应的阿霉素聚前药纳米胶束及其制备方法和应用