星型高反式1,4-结构含量的聚1,3-戊二烯弹性体及其制备方法以及应用
文献发布时间:2023-06-19 19:30:30
技术领域
本发明涉及高分子材料技术领域,具体地,涉及一种星型高反式1,4-结构含量的聚1,3-戊二烯弹性体及其制备方法以及应用。
背景技术
星型聚合物与线型聚合物相比,相同分子量下具备更低的扩散系数、熔体黏度和结晶度,且分子表面有更小的流体动力学体积以及官能度更高等独特的性质。其熔融黏度与总分子量无关而只取决于单臂分子量大小,这是与线型聚合物的最大区别。从工艺生产的角度而言,以星核类型的多官能度引发剂制备的星型聚合物在门尼黏度控制和凝胶自加速现象上要显著优于同工艺下的线型聚合物,这是由于星型聚合物的球形对称结构、较小的原子空间排列尺寸所带来的分子间弱相互作用。因为星核与各臂之间的化学键束缚作用,单位体积的星型聚合物具有的自由末端数要显著低于线型,这一特性也体现在硫化橡胶的链末端效应上:当整个大分子链作弹性运动时,末端受分子间网络状束缚越少,就越难以恢复原位点,能量则无法完全释放,造成滞后效应;例如溶聚丁苯和锂系聚丁二烯橡胶均需要通过硅锡偶联剂偶联后进一步提高抗湿滑性,优化滚动阻力。星型聚合物的这种紧密结构和支链式高片段密度也赋予了自身更高的力学强度和模量。
锂系合成橡胶和热塑性弹性体可通过(1)偶联法(2)多官能度引发剂法制备星型聚合物。偶联法工艺简单,在合成星型聚合物上很容易实现,偶联剂自身也比较廉价易得,核原子对弹性体的性能还有补强,如溶聚丁苯利用四氯化锡偶联后锡碳键易断裂,可有效促进炭黑等填料的分散,从而提高制品的抗湿滑性能和降低滚动阻力;利用硅氧烷等制备的星型或者线-星混合的SBS干胶用于沥青改性时的高温性能十分突出。但是偶联剂在使用过程中的偶联效率和偶联度对工艺要求较高,偶联效率低,或偶联度不足均会增加星型聚合物中低分子量线型单臂聚合物造成产品性能大幅下降;硅锡偶联剂还会与一些极性调节剂相排斥,使得偶联效果变差。同时偶联效果与活性链末端的位阻大小有很大关系,如聚异戊二烯链末端的甲基、聚苯乙烯的头尾位阻差异等,往往需要引入一小段丁二烯带帽或添加无规化试剂来得到足够高的偶联效率和偶联度。二乙烯基苯(DVB)作为偶联剂工艺难度大,DVB用量多则体系内副反应增多,随机性大会使得链之间交联和DVB齐聚物微凝胶核都会让溶液黏度急剧增大;DVB用量低偶联度不高,难以制备臂数稳定的星型聚合物。多官能度引发剂即多锂法制备星型聚合物则可有效解决上述因偶联效果不佳导致的单臂线型聚合物残余,以及复合极性调节剂存在下的低偶联效率等情况。以DVB为内核的多锂引发剂通过扩链后可有效解决自身的溶解性,同时与某些μ-型配体的极性调节剂兼容性更好,即可实现聚合物链的高效结构控制。CN101899135A采用DVB为核的多锂鳌合引发剂制备了星型高抗冲SBC,星型结构赋予材料高抗冲和透明性的同时,优化了材料的加工性能。
锂系聚二烯烃弹性体材料中,各类聚丁二烯产品均可制成支化度在4或更高的星型聚合物以提高物理性能,并解决窄分子量线型产品的冷流现象。如非流动型低顺式丁二烯橡胶LCBR和各种支化结构的乙烯基丁二烯橡胶。US 4482677将DVB在有少量Bd单体存在下由正丁基锂引发预聚合成支化度小于6的多官能引发剂用于高乙烯基聚丁二烯的制备,有效解决了线型同类产品熔体黏度(高剪切)和冷流(低剪切)性能差的弊端。
高反1,4-结构聚二烯烃橡胶具有低生热、高耐疲劳和耐磨性的优异的低温性能的同时,还有效平衡了轮胎的滚动阻力,是发展高性能节能轮胎的理想配胶。目前由不饱和烃单体制备高反1,4-结构橡胶的方法多见于丁二烯和异戊二烯单体均聚或者两者共聚,还没有出现关于星型结构的1,3-戊二烯的研究报道。阴离子聚合可实现星型高反式聚丁二烯的制备,但是高结晶度影响了该弹性体的加工性能,尤其是大幅提高了硫化过程的工艺成本。US4048418、US4148983、US5066754和CN103709295分别涉及铁系、钴系、钒系和稀土系催化剂制备线型和低支化度聚1,3-戊二烯的方法,有的尚不能有效实现聚合物微观结构的精确控制,分子量分布也普遍偏高PDI>2.5。US4482771通过阴离子聚合制备了线型的高丙烯基含量聚1,3-戊二烯,高Tg(Tg>﹣20℃)限制了在部分领域的应用,且无法有效克服1,3-戊二烯单体中戊炔和环戊二烯残余导致的聚合物分子量不高的问题。
发明内容
本发明的目的是为了克服现有技术存在的聚1,3-戊二烯的数均分子量较低的问题,以及现有技术中聚1,3-戊二烯的制备方法存在分子量分布过宽、偶联剂法效果不佳、臂数无法有效控制的缺陷问题,提供一种星型高反式1,4-结构含量的聚1,3-戊二烯弹性体及其制备方法以及应用,该制备方法简单,制备得到的星型高反式1,4-结构含量的聚1,3-戊二烯弹性体相比同类线型产品有更高的分子量,为1,3-戊二烯的高性能弹性体材料化提供了技术支持。
为了实现上述目的,本发明第一方面提供了一种星型高反式1,4-结构含量的聚1,3-戊二烯弹性体,其中,所述弹性体具有式(1)所示的结构表达式:
(PPD—PB)n—A,式(1);
其中,PPD为聚1,3-戊二烯嵌段,PB为聚丁二烯嵌段,n为多官能度锂引发剂mLi的官能度,n为3-50,A为mLi的有机残基;
其中,所述弹性体中的反式1,4-结构含量为65-95摩尔%,顺式1,4-结构含量为0-20摩尔%,1,2-结构含量为5-20摩尔%,3,4-结构含量为0-10摩尔%。
本发明第二方面提供了一种星型高反式1,4-结构的聚1,3-戊二烯的制备方法,其中,所述的制备方法包括:
(1)将丁二烯单体与第一非质子溶剂在反应器中混合预热,得到混合溶液;
(2)将所述混合溶液与催化剂接触进行陈化处理;其中,所述催化剂包括主催化剂以及可选的助催化剂,其中,所述主催化剂包括有机钡盐或有机钠盐,所述助催化剂为烷基铝;
(3)将经步骤(2)后的产物与第一极性调节剂和多官能度有机锂引发剂mLi混合进行引发聚合反应至丁二烯单体全部转化;
(4)将1,3-戊二烯单体加入经步骤(3)后的体系中混合引发聚合反应后终止反应,得到星型高反式1,4-结构含量的聚1,3-戊二烯弹性体(PPD—PB)n—A;
其中,PPD为聚1,3-戊二烯嵌段,PB为聚丁二烯嵌段,n为多官能度锂引发剂mLi的官能度,n为3-50,A为mLi的有机残基。
本发明第三方面提供了一种由前述所述的制备方法制备得到的星型高反式1,4-结构含量的聚1,3-戊二烯弹性体。
本发明第四方面提供了一种前述所述的星型高反式1,4-结构含量的聚1,3-戊二烯弹性体作为配胶在轮胎胎面和/或胎侧中的应用。
通过上述技术方案,本发明的技术方案具有如下优点:
(1)采用本发明的制备方法,各组分原料易得,反应速率快,转化率高。
(2)本发明制备得到的星型高反式1,4-结构含量的聚1,3-戊二烯弹性体的臂数可控,能够有效解决1.3-戊二烯单体中ppm级戊炔和环戊二烯残余导致的聚合物分子量不高的问题。
附图说明
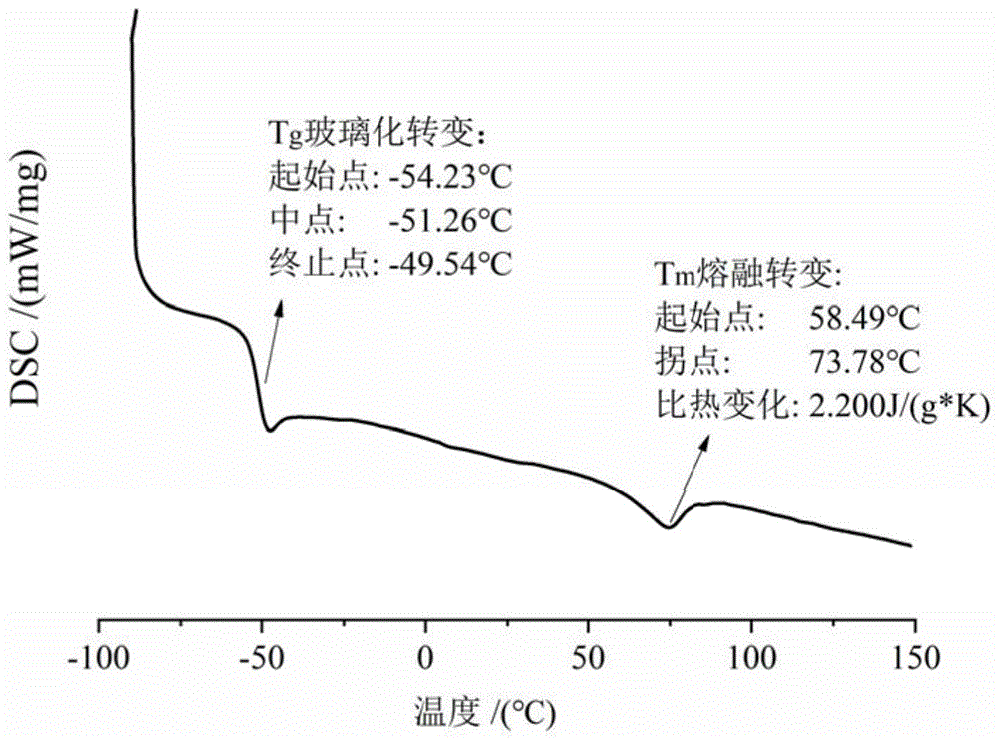
图1为本发明实施例1制备的星型高反式1,4-结构含量的聚1,3-戊二烯弹性体的DSC曲线图;
图2为本发明实施例1制备的星型高反式1,4-结构含量的聚1,3-戊二烯弹性体的GPC曲线图。
具体实施方式
在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。
本发明第一方面提供了一种星型高反式1,4-结构含量的聚1,3-戊二烯弹性体,其中,所述弹性体具有式(1)所示的结构表达式:
(PPD—PB)n—A,式(1);
其中,PPD为聚1,3-戊二烯嵌段,PB为聚丁二烯嵌段,n为多官能度锂引发剂mLi的官能度,n为3-50,A为mLi的有机残基;
其中,所述弹性体中的反式1,4-结构含量为65-95摩尔%,顺式1,4-结构含量为0-20摩尔%,1,2-结构含量为5-20摩尔%,3,4-结构含量为0-10摩尔%。
本发明的发明人意外发现:采用多官能度有机锂引发剂,在主催化剂(有机钡盐或有机钠盐)与可选的助催化剂(烷基铝)的催化下,经极性调节剂调节引发1,3-戊二烯阴离子聚合,得到了星型高反1,4-结构含量的聚1,3-戊二烯弹性体,而非线性聚1,3-戊二烯;另外,本发明的方法的多官能度有机锂引发剂与有机钡盐或有机钠盐合成简单,原料易得,有机钡盐或有机钠盐还可通过水蒸汽凝聚回收;另外,本发明提供了一种解决目前市场上的1,3-戊二烯单体中炔烃和环戊二烯残余量大于20ppm时导致的聚1,3-戊二烯分子量较低的办法,得到的星型高反1,4-结构含量的聚1,3-戊二烯弹性体相比同类线型产品有更高的分子量,为1,3-戊二烯的高性能弹性体材料化提供了技术支持。
根据本发明,所述弹性体的数均分子量为5×10
根据本发明,所述弹性体的分子量分布PDI(PDI=重均分子量Mw/数均分子量Mn)的值为1.05-2.5,优选为1.05-1.8。
根据本发明,以所述弹性体的总重量为基准,所述聚1,3-戊二烯嵌段的含量为50-95质量%,所述聚丁二烯嵌段的含量为2-50质量%;优选地,以所述弹性体的总重量为基准,所述聚1,3-戊二烯嵌段的含量为91.5-95质量%,所述聚丁二烯嵌段的含量为4.2-8.5质量%。
根据本发明,所述聚1,3-戊二烯嵌段与所述聚丁二烯嵌段的质量比为(1-22):1,优选为(10-22):1。
根据本发明,所述弹性体中反式1,4-结构含量为70-95摩尔%,优选为82-88摩尔%。
根据本发明,优选地,所述弹性体中顺式1,4-结构含量为0-5摩尔%,1,2-结构含量为5-15摩尔%,3,4-结构含量为0-3摩尔%的单元。
根据本发明,所述弹性体的玻璃化温度T
根据本发明,所述弹性体的臂数由多官能度锂引发剂mLi的官能度n决定,n为3-50,优选为3-10。在本发明中,“臂数”是指每个与mLi的有机残基A连接的PPD-PB嵌段共聚物的数目。
根据本发明,采用溶液聚合的方法,通过多锂引发剂制备含有高反1,4-结构含量的星型高分子量的聚1,3-戊二烯共聚物。
如前所述,本发明第二方面提供了一种星型高反式1,4-结构含量的聚1,3-戊二烯弹性体的制备方法,其中,所述的制备方法包括:
(1)将丁二烯单体与第一非质子溶剂在反应器中混合预热,得到混合溶液;
(2)将所述混合溶液与催化剂接触进行陈化处理;其中,所述催化剂包括主催化剂以及可选的助催化剂,其中,所述主催化剂包括有机钡盐或有机钠盐,所述助催化剂为烷基铝;
(3)将经步骤(2)后的产物与第一极性调节剂和多官能度有机锂引发剂mLi混合进行引发聚合反应至丁二烯单体全部转化;
(4)将1,3-戊二烯单体加入经步骤(3)后的体系中混合引发聚合反应后终止反应,得到星型高反式1,4-结构含量的聚1,3-戊二烯弹性体(PPD—PB)n—A;
其中,PPD为聚1,3-戊二烯嵌段,PB为聚丁二烯嵌段,n为多官能度锂引发剂mLi的官能度,n为3-50,A为mLi的有机残基。
根据本发明,所述多官能度锂引发剂mLi的用量由单臂的聚1,3-戊二烯/聚丁二烯嵌段共聚物PPD-PB决定,单臂数均分子量设计值为1×10
根据本发明,所述多官能度有机锂引发剂mLi的制备方法包括:将二乙烯基苯、不饱和烃、极性调节剂按比例与非质子溶剂混合后,加入烷基锂进行加成反应得到的;
其中,二乙烯基苯、不饱和烃、第二极性调节剂与烷基锂的用量的摩尔比为(0.1-10):(1-100):(1-100):1;
其中,烷基锂加成反应的条件包括:温度为-20-60℃,时间为0.5-12h。
根据本发明,所述不饱和烃选自丁二烯、苯乙烯、4-叔丁基苯乙烯、1,1-二苯基乙烯、异戊二烯和1,3-戊二烯中的一种或多种;丁二烯、异戊二烯、1,3-戊二烯属于共轭二烯烃,苯乙烯类衍生物实际上是大π键与双键进行π与双共轭。
根据本发明,所述烷基锂选自甲基锂、正丁基锂、仲丁基锂、叔丁基锂和苯基锂中的一种或多种。
根据本发明的一种优选的具体实施方式,本发明所用的多官能度有机锂引发剂mLi合成方法如下:在高纯氩气氛围的手套箱内,按配比向高温烘烤过的聚合瓶中加入100克环己烷、4克丁二烯、1.4克三乙胺、1.8克二乙烯基苯,均匀混合后逐步加入浓度为1.6mol/L的正丁基锂环己烷溶液9.3毫升,于50℃下反应2小时后用吉尔曼双滴定确定浓度备用,该方法制备的多锂引发剂呈紫红色。
制备催化剂时,有机钡盐BAD、BHT均可来自商业购买,或按照已有公开的常见合成路线制备,以甲苯为溶剂配置成溶液使用。正丁基锂、叔丁基锂由巴陵石化公司提供。
根据本发明,本发明的催化-引发反应体系是在无水无氧条件下,使用的催化剂由主催化剂和可选的助催化剂组成,所述助催化剂与所述主催化剂的用量的摩尔比为(0-100):1,优选为(0-4):1。
根据本发明,所述主催化剂为可用于锂系制备反式聚不饱和烃的有机钡盐或有机钠盐;其中,所述有机钡盐选自烷氧基钡盐、环烷氧基钡盐、醇醚钡盐、醇胺钡盐、酚钡盐和羧酸钡盐中的一种或多种;优选地,所述有机钡盐选自薄荷醇钡、百里酚钡(BHT)、二乙二醇单乙醚钡(BAD)、吗啉乙醇钡、十二烷基苯磺酸钡、四氢糠酸钡和四氢糠醇钡中的一种或多种;更优选地,所述有机钡盐为百里酚钡和/或二乙二醇单乙醚钡。
根据本发明,所述有机钠盐为烷氧基钠盐和/或磺酸钠盐;优选地,所述有机钠盐选自十二烷基硫酸钠(SDS)、对甲苯磺酸钠、十二烷基苯磺酸钠(SDBS)、四氢糠醇钠和叔戊醇钠的一种或多种,优选为十二烷基苯磺酸钠和十二烷基硫酸钠。
根据本发明,所述助催化剂为烷基铝,其中,所述烷基铝选自三甲基铝(TMA)、三乙基铝(TEA)、三丙基铝、三异丁基铝(TIBA)、三异丙基铝、三辛基铝和甲基铝氧烷中的一种或多种,优选为三甲基铝、三乙基铝和三异丁基铝。
根据本发明,所述非质子溶剂选自环己烷、正己烷、正戊烷、正庚烷、苯、加氢油和抽提油中的一种或多种,优选为环己烷;其中,苯包括甲苯、乙苯和二甲苯。
根据本发明,所述极性调节剂选自四氢呋喃(THF)、二氧六环、三乙胺、双四氢糠丙烷、N,N-二甲基四氢糠胺、四氢糠醇乙基醚、四氢糠醇丁基醚、五甲基二乙基三胺、二哌啶乙烷、三苯基膦和二硫化碳中的一种或多种,优选为四氢呋喃、四氢糠醇乙基醚和二硫化碳中的一种或多种。
根据本发明,所述催化剂与所述多官能度有机锂引发剂mLi的用量的摩尔比为(0.01-100):1,优选为(0.5-1.25):1。
根据本发明,所述第一极性调节剂与所述多官能度有机锂引发剂mLi的用量的摩尔比为(0-100):1,优选为(0-20):1。
根据本发明,丁二烯、1,3-戊二烯单体与所述第一非质子溶剂的用量的质量比为(1-50):(50-100):1000。
根据本发明,所述混合预热的条件包括:温度为25-80℃,时间为0.5-3h;
根据本发明,所述陈化处理的条件包括:温度为25-60℃,时间为10min-5h;
根据本发明,所述聚合反应的条件包括:温度为0-100℃,时间为1-24h。
根据本发明,聚合结束后加入0.5-3mL含防老化剂和抗氧剂的水溶液或醇溶液终止反应,过量甲醇沉淀后在40-60℃下真空干燥24-72小时。
在本发明中,防老剂选自防老化剂264和/或防老化剂2264。
在本发明中,抗氧剂选自抗氧剂1010、抗氧剂168和抗氧剂1076中的一种或多种。
根据本发明,所述终止反应中所使用的终止剂一般选自水、甲醇、乙醇和异丙醇中的任意一种,优选为异丙醇。
根据本发明,所使用的1,3-戊二烯单体选自顺式1,3-戊二烯(Zp)、反式1,3-戊二烯(Ep)、任意比例的混合异构1,3-戊二烯(Pd)、2-甲基-1,3-戊二烯(Mpd)、2,3-二甲基-1,3-戊二烯(Dmpd);优选为顺式1,3-戊二烯(Zp)、反式1,3-戊二烯(Ep)和特定比例的混合异构1,3-戊二烯(Pd,E/Z=65/35)。
根据本发明的一种优选的具体实施方式,一种星型高反式1,4-结构含量的聚1,3-戊二烯弹性体的制备方法包括:
(1)将丁二烯单体与第一非质子类溶剂配置成混合溶液,加入烘烤冲氮除氧处理过的聚合瓶中;聚合瓶25-80℃预热0.5-3小时;
(2)预热后,加入催化剂陈化10-300分钟;
(3)陈化结束经过破杂后加入第一极性调节剂和多官能度有机锂引发剂mLi引发第一段聚丁二烯聚合,聚合温度为0-100℃,聚合时间为1-24小时;
(4)聚合结束后加入1,3-戊二烯单体,该段聚合温度为0-100℃,聚合时间为1-24小时;反应完毕向最终得到的聚合母液内加入0.5-3mL含防老化剂和抗氧剂的醇溶液终止反应,过量甲醇沉淀后在40℃下真空干燥24小时,得到星型的含高反式1,4-结构含量的聚1,3-戊二烯弹性体;
其中,所述的催化剂由摩尔比为(0-100):1的主催化剂、助催化剂两组分组成,主催化剂为有机钡盐化合物;助催化剂为烷基铝;所述多官能度有机锂引发剂mLi为二乙烯基苯DVB、不饱和烃与烷基锂反应后得到的多鳌合型有机锂引发剂,R为碳原子数10-100的烃基,官能度n范围为3-50;所述催化剂与多官能度有机锂引发剂mLi的摩尔比为(0.01-100):1;所述第一极性调节剂与多官能度有机锂引发剂mLi的摩尔比为(0-100):1,丁二烯、1,3-戊二烯单体与所述第一非质子溶剂的用量的质量比为(1-50):(50-100):1000。
本发明第三方面提供了一种由前述所述的制备方法制备得到的星型高反式1,4-结构含量的聚1,3-戊二烯弹性体。
本发明第四方面提供了一种前述所述的星型高反式1,4-结构含量的聚1,3-戊二烯弹性体作为配胶在轮胎胎面和/或胎侧中的应用。
以下将通过实施例对本发明进行详细描述。
合成的聚合物的微观结构采用美国Varian公司生产的型号为INOVA-400的核磁仪进行测定,频率为400MHz,以四甲基硅烷(TMS)为内标,溶剂为氘代氯仿。
红外分析使用岛津公司生产的型号为IRAffinity-1的红外仪进行测定,以CS
分子量和分子量分布采用英国PL公司生产的型号为GPC-220的凝胶渗透色谱仪测定,淋洗液为THF,流速为1.0mL/min,测试温度为40℃。
玻璃化转变温度的测定采用德国耐驰仪器有限公司生产的DSC200F3差示量热扫描仪进行。一次升温25℃~100℃,10℃/min,保温10min;降温100℃~(-100)℃,5℃/min;二次升温(-100)~150℃,5℃/min;吹扫气:氮气,50mL/min;保护气:氮气,50mL/min。
本发明中聚合反应所使用的1,3-戊二烯单体、苯乙烯、丁二烯、二乙烯基苯和极性调节剂均经过严格的除水氧操作,水氧含量达到阴离子聚合要求。
实施例1
本实施例在于说明采用本发明的方法制备的星型高反式1,4-结构聚1,3-戊二烯弹性体。
(1)在高纯氩气氛围的手套箱内,将2.9g环己烷与0.085g的丁二烯加入高温烘烤后的聚合瓶,50℃下预热1小时;
(2)预热后,加入含百里酚钡(BHT)0.21mmol的甲苯溶液(BHT浓度为0.55mol/L)、含0.42mmol三甲基铝(TMA)的己烷溶液(TMA浓度为0.1mol/L)后在50℃下静置陈化30分钟;
(3)再加入3.4mmol四氢呋喃,混合溶液经过破杂处理后加入浓度为0.1mol/L的多锂引发剂mLi0.85mmol,于80℃下反应2小时至丁二烯全部转化;
(4)加入由15.6g环己烷和1.7g聚合级1,3-戊二烯配置成的混合溶液,聚1,3-戊二烯的单臂分子量设计值为20,000g/mol,第二段继续在80℃下反应4小时,待单体全部转化后加入2mL含1%的防老化剂264和1%抗氧剂1010的异丙醇-甲苯溶液终止反应,过量甲醇沉淀后,反复用甲醇层析4~5次,在40℃下真空干燥24小时得到星型高反式1,4-结构含量的聚1,3-戊二烯弹性体;其各微观结构含量、实测数均分子量Mn和分布PDI如表1中编号1所示。
该聚合物的DSC曲线和GPC曲线分别如附图1、附图2所示,从图1能够看出:受“冻住”的大分子链队的无定形区随着慢慢升温至-54℃后开始“解冻”,温度达到-49℃后可以自由运动,即玻璃态向高弹态转变;但由于聚合物的反式-1,4-结构含量大于80%,聚合物链比较规整,高弹态下存在的分子链中较为规整的部分定向排列形成部分结晶区,随着温度进一步升高出现了结晶区的融化,因为晶型结构不一存在多晶形态,从58℃出现一个较宽的熔融峰。图中还可看出聚合物的玻璃化转变温度为-51℃,结晶的熔融温度为74℃;
从图2能够看出:星型聚1,3-戊二烯弹性体为单峰正态分布,没有出现明显的肩峰和双峰,本发明所使用的mLi制备得到的聚合物臂数可控。
重量法求得丁二烯和1,3-戊二烯的总单体转化率99%,
实施例2
本实施例在于说明采用本发明的方法制备的星型高反式1,4-结构聚1,3-戊二烯弹性体。
按照与实施例1相同的方法制备聚1,3-戊二烯弹性体,所不同之处在于:仅以“混合异构1,3-戊二烯(Pd,E/Z=65/35)”替代了“顺式1,3-戊二烯单体”进行星型高反式1,4-结构含量的聚1,3-戊二烯弹性体的制备;其聚合工艺,聚合物各微观结构含量、实测数均分子量Mn和分布PDI如表1中编号2所示。
重量法求得丁二烯和1,3-戊二烯的总单体转化率97%,产物的玻璃化转变温度为-47℃,结晶的熔融温度为70℃。
实施例3
本实施例在于说明采用本发明的方法制备的星型高反式1,4-结构聚1,3-戊二烯弹性体。
按照与实施例1相同的方法制备聚1,3-戊二烯弹性体,所不同之处在于:仅以“反式1,3-戊二烯单体(Ep)”替代了“顺式1,3-戊二烯单体(Zp)”进行星型高反式1,4-结构含量的聚1,3-戊二烯弹性体的制备;其聚合工艺,聚合物各微观结构含量、实测数均分子量Mn和分布PDI如表1中编号3所示。
重量法求得丁二烯和1,3-戊二烯的总单体转化率94%,产物的玻璃化转变温度为-42℃,未观察到聚合物的熔融峰。
实施例4
本实施例在于说明采用本发明的方法制备的星型高反式1,4-结构聚1,3-戊二烯弹性体。
按照与实施例1相同的方法制备聚1,3-戊二烯弹性体,所不同之处在于:仅以“二乙二醇单乙醚钡(BAD)”替代了“百里酚钡(BHT)”进行星型高反式1,4-结构含量的聚1,3-戊二烯弹性体的制备,有机钡盐与TMA的配比没有变化,其聚合工艺,聚合物各微观结构含量、实测数均分子量Mn和分布PDI如表1中编号4所示。
重量法求得丁二烯和1,3-戊二烯的总单体转化率92%,产物的玻璃化转变温度为-49℃,结晶的熔融温度为71℃。
实施例5
本实施例在于说明采用本发明的方法制备的星型高反式1,4-结构聚1,3-戊二烯弹性体。
按照与实施例1相同的方法制备聚1,3-戊二烯弹性体,所不同之处在于:仅以“三乙基铝(TEA)”替代了“三甲基铝”进行星型高反式1,4-结构聚1,3-戊二烯的制备,BHT与TEA的配比没有变化,其聚合工艺,聚合物各微观结构含量、实测数均分子量Mn和分布PDI如表1中编号5所示。
重量法求得丁二烯和1,3-戊二烯的总单体转化率95%,产物的玻璃化转变温度为-51℃,结晶的熔融温度为72℃。
实施例6
本实施例在于说明采用本发明的方法制备的星型高反式1,4-结构聚1,3-戊二烯弹性体。
按照与实施例1相同的方法制备聚1,3-戊二烯弹性体,所不同之处在于:仅改变TMA的配比,具体地:
将“含0.42mmol三甲基铝(TMA)的己烷溶液(TMA浓度为0.1mol/L)”替换为“含0.85mmol三甲基铝(TMA)的己烷溶液(TMA浓度为0.1mol/L)”;
制备了星型高反式1,4-结构聚1,3-戊二烯,其聚合工艺,聚合物各微观结构含量、实测数均分子量Mn和分布PDI如表1中编号6所示。
重量法求得丁二烯和1,3-戊二烯的总单体转化率92%,产物的玻璃化转变温度为-54℃,结晶的熔融温度为79℃。
实施例7
本实施例在于说明采用本发明的方法制备的星型高反式1,4-结构聚1,3-戊二烯弹性体。
按照与实施例1相同的方法制备聚1,3-戊二烯弹性体,所不同之处在于:仅改变BHT与TMA和多锂引发剂的配比,具体地:
将“含百里酚钡(BHT)0.21mmol的甲苯溶液(BHT浓度为0.55mol/L)”替换为“含百里酚钡(BHT)0.15mmol的甲苯溶液(BHT浓度为0.55mol/L)”;
将“含0.42mmol三甲基铝(TMA)的己烷溶液(TMA浓度为0.1mol/L)”替换为“含0.30mmol三甲基铝(TMA)的己烷溶液(TMA浓度为0.1mol/L)”;
制备了星型高反式1,4-结构聚1,3-戊二烯,其聚合工艺,聚合物各微观结构含量、实测数均分子量Mn和分布PDI如表1中编号7所示。
重量法求得丁二烯和1,3-戊二烯的总单体转化率98%,产物的玻璃化转变温度为-48℃,结晶的熔融温度为63℃。
实施例8
本实施例在于说明采用本发明的方法制备的星型高反式1,4-结构聚1,3-戊二烯弹性体。
按照与实施例1相同的方法制备聚1,3-戊二烯弹性体,所不同之处在于:没有添加TMA;
制备了星型高反式1,4-结构聚1,3-戊二烯,其聚合工艺,聚合物各微观结构含量、实测数均分子量Mn和分布PDI如表1中编号8所示。
重量法求得丁二烯和1,3-戊二烯的总单体转化率95%,产物的玻璃化转变温度为-51℃,结晶的熔融温度为71℃。
实施例9
本实施例在于说明采用本发明的方法制备的星型高反式1,4-结构聚1,3-戊二烯弹性体。
按照与实施例1相同的方法制备聚1,3-戊二烯弹性体,所不同之处在于:仅改变第一段反应中丁二烯的用量,具体地:
将“步骤(1)中,0.085g的丁二烯”替换为“0.177g的丁二烯”;
制备了星型高反式1,4-结构聚1,3-戊二烯,其聚合工艺,聚合物各微观结构含量、实测数均分子量Mn和分布PDI如表1中编号9所示。
重量法求得丁二烯和1,3-戊二烯的总单体转化率99%,产物的玻璃化转变温度为-55℃,结晶的熔融温度为70℃。
实施例10
本实施例在于说明采用本发明的方法制备的星型高反式1,4-结构聚1,3-戊二烯弹性体。
按照与实施例1相同的方法制备聚1,3-戊二烯弹性体,所不同之处在于:仅改变BHT、TMA和多锂引发剂的用量,具体地:
将“步骤(2)中,含百里酚钡(BHT)0.21mmol的甲苯溶液(BHT浓度为0.55mol/L)”替换为“含百里酚钡(BHT)0.10mmol的甲苯溶液(BHT浓度为0.55mol/L)”;
将“步骤(2)中,含0.42mmol三甲基铝(TMA)的己烷溶液(TMA浓度为0.1mol/L)”替换为“含0.21mmol三甲基铝(TMA)的己烷溶液(TMA浓度为0.1mol/L)”;
将“步骤(3)中,浓度为0.1mol/L的多锂引发剂mLi0.85mmol”替换为“浓度为0.1mol/L的多锂引发剂mLi0.43mmol”;
制备了星型高反式1,4-结构聚1,3-戊二烯,其聚合工艺,聚合物各微观结构含量、实测数均分子量Mn和分布PDI如表1中编号10所示。
重量法求得丁二烯和1,3-戊二烯的总单体转化率97%,产物的玻璃化转变温度为-52℃,结晶的熔融温度为72℃。
实施例11
本实施例在于说明采用本发明的方法制备的星型高反式1,4-结构聚1,3-戊二烯弹性体。
按照与实施例1相同的方法制备聚1,3-戊二烯弹性体,所不同之处在于:仅改变催化剂的类型和极性调节剂的用量,具体地:
将“步骤(2)中,含百里酚钡(BHT)0.21mmol的甲苯溶液(BHT浓度为0.55mol/L)、含0.42mmol三甲基铝(TMA)的己烷溶液(TMA浓度为0.1mol/L)”替换为“含0.17mmol十二烷基苯磺酸钠(SDBS)的甲苯溶液1.7mL(SDBS浓度为0.1mol/L)”,没有添加三甲基铝(TMA);
将“步骤(3)中,加入3.4mmolTHF”替换为“加入17mmolTHF”;
制备了星型高反式1,4-结构聚1,3-戊二烯,其聚合工艺,聚合物各微观结构含量、实测数均分子量Mn和分布PDI如表1中编号11所示。
重量法求得丁二烯和1,3-戊二烯的总单体转化率98%,产物的玻璃化转变温度为-47℃,未观察到聚合物的熔融峰。
实施例12
本实施例在于说明采用本发明的方法制备的星型高反式1,4-结构聚1,3-戊二烯弹性体。
按照与实施例1相同的方法制备聚1,3-戊二烯弹性体,所不同之处在于:仅改变极性调节剂THF的用量,具体地,
将“在步骤(3)中,加入3.4mmolTHF”替换为“加入0.17mmolTHF”;
制备了星型高反式1,4-结构聚1,3-戊二烯,其聚合工艺,聚合物各微观结构含量、实测数均分子量Mn和分布PDI如表1中编号12所示。
重量法求得丁二烯和1,3-戊二烯的总单体转化率99%,产物的玻璃化转变温度为-55℃,结晶的熔融温度为67℃。
对比例1
按照与实施例1相同的方法制备聚1,3-戊二烯弹性体,所不同之处在于:将“步骤(3)中多锂引发剂”替换为“正丁基锂”;
制备了线性的聚1,3-戊二烯弹性体,其聚合工艺,聚合物各微观结构含量、实测数均分子量和分布如表1中编号P1所示。
重量法求得丁二烯和1,3-戊二烯的总单体转化率99%,产物的玻璃化转变温度为-39℃,未观察到聚合物的熔融峰。
对比例2
按照与实施例1相同的方法制备聚1,3-戊二烯弹性体,所不同之处在于:将“步骤(2)中的BHT和TMA”替换“0.17mmol叔丁醇钠(t-BuONa)”;
制备了聚1,3-戊二烯弹性体,其聚合工艺,聚合物各微观结构含量、实测数均分子量和分布如表1中编号P2所示。
重量法求得丁二烯和1,3-戊二烯的总单体转化率57%,产物的玻璃化转变温度为-44℃,未观察到聚合物的熔融峰。
对比例3
按照与实施例1相同的方法制备聚1,3-戊二烯弹性体,所不同之处在于:
将“在步骤(3)中,加入浓度为0.1mol/L的多锂引发剂mLi0.85mmol”替换为“加入浓度为0.14mol/L的多锂引发剂mLi-cons0.85mmol”;
另外,该对比例所使用的多官能度锂引发剂mLi-cons的制备方法包括:
在高纯氩气氛围的手套箱内,按配比向高温烘烤过的聚合瓶中加入100克环己烷、4克丁二烯、0.13克三乙胺、1.8克二乙烯基苯,均匀混合后逐步加入浓度为1.6mol/L的正丁基锂环己烷溶液9.3毫升,于50℃下反应2小时后用双滴定定浓备用,该对比例降低了制备mLi第二极性调节剂的用量,该配比下得到的多官能度锂引发剂命名为mLi-cons,浓度为0.14mol/L。
制备了聚1,3-戊二烯弹性体,其聚合工艺,聚合物各微观结构含量、实测数均分子量和分布如表1中编号P3所示。
重量法求得丁二烯和1,3-戊二烯的总单体转化率为99%,产物的玻璃化转变温度为-41℃,未观察到聚合物的熔融峰。
对比例4
按照与实施例1相同的方法制备聚1,3-戊二烯弹性体,所不同之处在于:将“将“步骤(2)中,含百里酚钡(BHT)0.21mmol的甲苯溶液(BHT浓度为0.55mol/L)、含0.42mmol三甲基铝(TMA)的己烷溶液(TMA浓度为0.1mol/L)”替换为“含百里酚钡(BHT)0.21mmol的甲苯溶液(BHT浓度为0.55mol/L)、含0.11mmol氯化二乙基铝(Et
制备了线性的聚1,3-戊二烯弹性体,其聚合工艺,聚合物各微观结构含量、实测数均分子量和分布如表1中编号P4所示。
重量法求得丁二烯和1,3-戊二烯的总单体转化率67%,产物的玻璃化转变温度为-43℃,未观察到聚合物的熔融峰。
表1
/>
注:1,2-%包括聚1,3-戊二烯结构单元中的反式1,2-和顺式1,2-结构含量。
总是,本发明中采用mLi这种多官能度有机锂引发剂,能够得到星型聚1,3-戊二烯。
对比例1使用的正丁基锂制备的聚1,3戊二烯为线型,只能得到线型聚合物;说明:如果不采用mLi而采用其他的烷基锂,则无法得到星型聚合物。
对比例2采用叔丁醇钠(t-BuONa)作为催化剂替代了有机钡和烷基铝,由于没有采用本发明限定的催化剂,由于t-BuONa无法提供如BHT与TMA配合形成的钡-铝二配物的大位阻效应,也不能像SDBS和极性调节剂形成新的活性中心,使得该星型聚合物中的反式1,4-结构的占比大大降低,1,3-戊二烯单体也无法完全转化。
对比例3由于降低了制备mLi过程中使用的第二极性调节剂三乙胺的用量,n-BuLi优先进攻DVB上的双键,丁二烯起不到增溶扩链的效果,使mLi-cons的臂数无法精确控制,结果得到的产物发生交联,可溶DMF的聚合物部分数均分子量较低,近似于线型聚合物,弹性体中的反式1,4-结构含量也明显低于实施例1-12。
对比例4采用氯化二乙基铝(Et
以上详细描述了本发明的优选实施方式,但是,本发明并不限于此。在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,包括各个技术特征以任何其它的合适方式进行组合,这些简单变型和组合同样应当视为本发明所公开的内容,均属于本发明的保护范围。
- 一种辐射交联反式-1,4-聚异戊二烯材料的制备方法及反式-1,4-聚异戊二烯材料
- 一种辐射交联反式-1,4-聚异戊二烯材料的制备方法及反式-1,4-聚异戊二烯材料