一种PML/RARA融合基因的检测方法
文献发布时间:2024-04-18 19:58:30
技术领域
本发明涉及基因检测领域,确切地说是一种PML/RARA融合基因的检测方法。
背景技术
急性早幼粒细胞白血病(APL)是急性髓性白血病(AML)的一个独特亚型,是一种高度侵袭性疾病,由于其特征性凝血功能障碍而表现为严重的出血综合征。APL的特点是染色体特异性重排t(15;17)(q22;q21),导致第17号染色体维甲酸受体α基因(RARA)与第15号染色体早幼粒细胞白血病基因(PML)融合,进而产生致癌性PML/RARA融合蛋白。病理上,PML基因通过调节细胞增殖和凋亡发挥肿瘤抑制作用,而RARA基因编码一种核受体,控制参与分化、凋亡和颗粒形成的基因的转录。PML/RARA融合基因产物可以破坏PML核小体(NBs),抑制RARA-RXR转录网络,从而阻碍DNA的损伤反应、衰老和细胞凋亡。这导致造血干细胞在早幼粒细胞阶段的阻滞和自噬和凋亡的损害。根据PML断点的位置(通常位于内含子6、外显子6或内含子3),可以产生不同的PML/RARA亚型,包括长型(bcr1)、变异型(bcr2)和短型(bcr3)。在所有APL阳性病例中,bcr1占比45-55%,表明其在APL中普遍存在。因此,PML/RARA基因组片段中bcr1亚型的鉴定和分类,对了解APL的特征起着至关重要的作用,对APL预后、风险分层和精准医疗更具有重要意义。
一般来说,检测PML/RARA融合基因常用的方法有染色体分析、荧光原位杂交(FISH)、流式细胞术(FCM)、实时定量逆转录聚合酶链反应(RT-PCR)等。尽管染色体分析可以识别单细胞结构和数量上的异常,但它是一个耗时和资源密集型的过程,灵敏度低,不适合快速诊断。RT-PCR被认为是APL遗传确认的金标准,因为它能够高度敏感地识别特定的PML/RARA亚型。然而,这种方法检测到的这些基因的表达水平可能并不能准确反映它们在所有APL细胞中的比例。尽管FISH分析可以通过定量治疗过程中所需的异常信号来确定携带融合基因的细胞比例,但其准确性受到固定过程的限制,并且在有限数量的体内样本中快速定量痕量生物标志物可能不那么有效。FCM只能在单细胞水平上提供有关细胞形状、大小和荧光特征的信息,它在准确定量痕量生物标志物方面也有类似的局限性,可能无法提供全面的遗传信息。此外,这些方法耗时,昂贵,技术要求高,通常需要一到两天的时间才能得到结果。APL的及时诊断对于实施靶向治疗和优化患者预后至关重要。考虑到全反式维甲酸(ATRA)和三氧化二砷(AsO
近年来,RNA引导的CRISPR/Cas(clustered regularly interspaced shortpalindromic repeats/CRISPR associated nucleases)系统因其高效、快速的精确核酸检测而备受关注。在已发现的与CRISPR相关的Cas蛋白中,Cas12a是一种2型V-A型CRISPR相关酶,在CRISPR RNA(crRNA)存在下特异性切割目标双链DNA(dsDNA)或单链DNA(ssDNA)。同时,Cas12a表现出反式切割活性,导致附近单链非靶向核酸被不加区分地随机切割。通过诱导荧光报告核酸(ssDNA-FQ)被非特异性的反式切割,这种切割活性被用于特异性靶基因的体外检测,进而导致荧光信号的增加。至关重要的是,由于其固有的低表达水平,核酸检测需要结合信号转导和核酸扩增技术。Cas12a已与核酸扩增方法(如聚合酶链反应(PCR))和等温扩增方法(如链位移扩增(SDA)、滚环扩增(RCA)和环介导等温扩增(LAMP)20-23)结合使用,以开发成本效益高、通用且高效的核酸检测系统和生物标志物诊断策略。特别是RCA,已成为一种严格且高度特异性的等温基因扩增方法。它需要引物和环状DNA模板之间的完全互补才能启动,这有利于单核苷酸多态性分析。因此,CRISPR/Cas12a技术与RCA技术的整合对于开发高效的多路等温核酸检测平台具有显著的优势。
通常,在涉及Cas12a蛋白、crRNA和ssDNA-FQ的反应中,它们的相互作用依赖于反应物之间的随机接触。根据碰撞理论,增加局部反应物浓度会促进分子频繁碰撞,从而提高反应效率和动力学。然而,以往的研究忽略了这一点。分子间G四链体(inter-G-quadruplex)是一种独特的DNA序列,具有四链体结构,在分子生物学、生物医学和分析化学中显示出巨大的潜力。在这种结构中,四个鸟嘌呤碱基(G碱基)通过Hoogsteen氢键连接形成一个鸟嘌呤四聚体,然后与两个或更多其他鸟嘌呤四聚体堆叠,形成一个G四链体。受其结构特性的启发,分子间G四链体可以作为一个支架来限制反应物,增加CRISPR/Cas12a蛋白的局部浓度,从而提高反应效率和动力学,以便及时诊断疾病和生物医学研究。
基于以上考虑,本研究利用分子间G四链体的结构功能所产生空间约束效应加速激活CRISPR/Cas12a系统,并将该平台应用于PML/RARA融合基因bcr1异构体的超灵敏检测。我们使用RCA作为分析工具来扩增目标基因,并引入一个缺口位点来产生具有多个串联G束的ssDNA。扩增的ssDNA通过Hoogsteen氢键结合,形成分子间G四链体,有利于捕获crRNA并与Cas12蛋白结合。分子间G四链体的空间约束效应极大地增强了CRISPR/Cas12a系统的局部浓度,使得荧光基团和猝灭基团修饰的ssDNA-FQ能够快速、非特异性地切割。据我们所知,这是第一次尝试利用分子间G四链体来创造空间限制效果以加速激活CRISPR/Cas12a系统。我们的结果表明,通过遵循这种方法,目标基因可以精准快速的被定性和定量。
发明内容
本发明的目的在于提供一种PML/RARA融合基因的检测方法。
上述目的通过以下方案实现:
一种PML/RARA融合基因的检测方法,其特征在于,包括以下步骤:
(1)crRNA的制备
通过体外转录合成crRNA;
(2)将4μL双蒸馏水ddH2O、2μL的2μM锁式探针PP、2μL的2μM bcr1、1μL浓度为5U/μL的T4 DNA连接酶和1μL 10×T4连接缓冲液在15-17℃下孵育25-35分钟,进行结环反应,形成环型探针PP;
(3)将2μM引物2μL、浓度为5U/μL的Klenow片段聚合酶1μL、10×NEBuffer 2反应缓冲液2μL、浓度为10U/μL的Nt.BbvCI 1μL、10×CutSmart 2μL、25mM的dNTPs2μL混合均匀,配制成10μL的混合物,再将10μL的环型探针PP作为DNA模板加入到混合物中,进行RCA扩增,将得到的20μL的扩增混合物混36-38℃孵育1-1.5小时,在64-66℃下10-12分钟使酶灭活;
(3)将8μL 1μM Cas12a、8μL 1μM crRNA、5μL 10×reaction Buffer、1μL RNaseInhibitor 40U/μL的混合物在24-26℃下孵育10-12分钟,然后将22μL的混合物与2μL的10μM ssDNA-FQ和6μL的ddH
(4)最后,将步骤(3)所得50μL处理后得样品加入150μL的ddH
所述的一种PML/RARA融合基因的检测方法,其特征在于:在基因序列表内,所述的所有单链DNA的最终浓度都是根据200μL溶液的总体积来确定的,在生物传感方面,使用523nm处的荧光强度来评估靶目标物存在和不存在时的检测性能。
所述的一种PML/RARA融合基因的检测方法,其特征在于:
本实验使用PerkinElmer FL8500荧光分光光度计Waltham,USA进行所有荧光测量;仪器设置为:激发波长440nm,狭缝宽度10/10nm,扫描速度240nm/min,记录范围460-650nm,PMT探测器电压650V。
所述的一种PML/RARA融合基因的检测方法,其特征在于:电泳实验采用Servicebio PW-600电泳分析仪,凝胶成像照片采用ChampGel 7000凝胶成像系统,缓冲液的pH值使用LeiCi pH-3e台式pH计测量,此外,Bio-Rad T100热循环系统用于控制恒定的反应温度。
所述的一种PML/RARA融合基因的检测方法,其特征在于:
步骤(1)的crRNA的制备方法为:为了制备crRNA,在Tap DNA聚合酶的参与下,以短T7引物T7-crRNA-F和长T7引物T7-crRNA-R为底物生成crRNA模板DNA;DNA模板在94-96℃时进行热变性,然后逐渐冷却到环境温度退火;随后,在50μL的转录系统中,将200ng模板DNA与5μL的1μM的T7 RNA聚合酶、10μL的5×T7转录缓冲液和2μL的100mM NTPs结合,加入RNaseInhibitor 40U/μL,36-38℃孵育8-10小时,然后,加入4μL体积浓度为1U/μL的DNase I和10×6μL体积的Reaction Buffer,然后将所得产物保存在-80℃左右备用即得。
所述的一种PML/RARA融合基因的检测方法,其特征在于:
本方法荧光检测中当靶目标bcr1不存在的情况下检测不到荧光信号,靶目标bcr1存在的情况下才有信号。
所述的一种PML/RARA融合基因的检测方法,其特征在于:
bcr1浓度在1fM-100pM范围内,523nm波长处的峰值荧光值F
所述的一种PML/RARA融合基因的检测方法,其特征在于:
bcr1浓度在10nM-250nM范围内,523nm波长处的峰值荧光值F
所述的一种PML/RARA融合基因的检测方法,其特征在于:
对于靶目标bcr1具有极高的特异性,可以准确快速地区分野生型和其他典型PML/RARA亚型,如bcr2,bcr3,PML-L,PML-S,PML-V。
所述的一种PML/RARA融合基因的检测方法,其特征在于:
在含有1%人血清的环境中,bcr1浓度在50fM-50nM范围内,523nm波长处的峰值荧光值F
本发明的有益效果为:
(1)为了证明分子间G四链体在融合基因检测中的空间约束效应,我们设计了一个切割后触发类似物(NTA)和一系列切割触发变体(NTV),包括NTV1、NTV2和NTV3,它们被替换了不同数量的G碱基。这些变体被用来强调基于分子间G四链体的空间限制对Cas12a切割效率的增强。硫黄素T(ThT)的荧光可以通过其诱导、稳定和结合特性有效地指示分子间G四链体的存在。为了验证分子间G四链体的存在,将含有5μL 10μM NTA或NTV的反应混合物(200μL)与含有1μM ThT的Tris缓冲液(50mM,pH 7.4,100mM NaCl)195μL在37℃下孵育1小时。随后,测量了激发波长为430nm和发射波长为496nm的荧光强度。此外,在体外Cas12a酶切ssDNA-FQ的整个过程中,采用Bio-Rad CFX Connect real-time PCR系统(California,USA)进行实时荧光监测。这种监测使我们能够基于分子间G四链体的空间限制施加的空间约束来评估其对Cas12a活性的加速。
(2)PML/RARA基因组片段的三种亚型的准确检测和分类对于预测急性早幼粒细胞白血病(APL)的疾病进展、风险分层和给予精确的药物治疗至关重要。在这项研究中,我们开发了一种高度特异性的核酸检测平台,能够在等温条件下定量PML-RARA三种主要亚型中的长型。该平台整合了CRISPR/Cas12a核酸酶方法和滚环扩增(RCA)技术的优势。值得注意的是,RCA辅助的CRISPR/Cas12a反式切割系统通过利用了分子间G四链体结构的空间限制效应。这种创新设计有效地提高了CRISPR/Cas12a的局部浓度,从而加快了其对荧光报告核酸的切割效率,使PML/RARA融合基因表达的荧光光谱的高效率检测成为可能。通过从人血清样本中检测PML/RARA融合基因,验证了该检测平台在APL病例筛查、诊断和预后方面的可靠性和实际应用潜力。我们的研究结果为检测融合基因提供了一种简单、省时、高灵敏度的方法,为APL的快速诊断提供了一种有希望的方法,可以很容易地应用于临床诊断。
附图说明
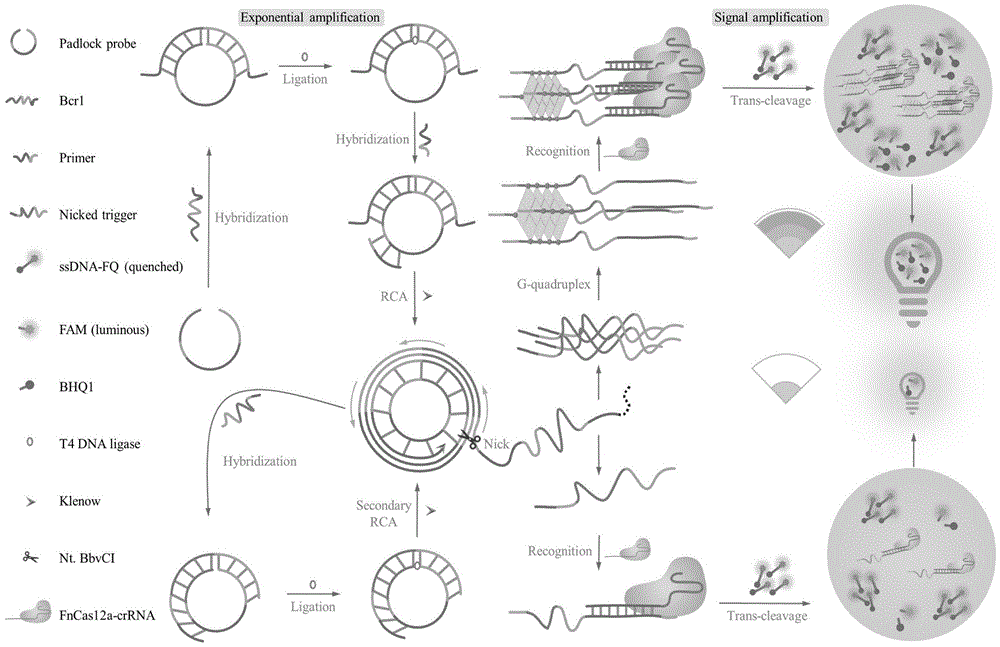
图1为:融合基因检测原理图;
图2为:验证检测方法可行性的荧光光谱响应图;ssDNA-FQ(a)、ssDNA-FQ+PP(b)、ssDNA-FQ+PP+bcr1(c)、ssDNA-FQ+PP+Klenow(d)、ssDNA-FQ+PP+bcr1+Klenow(e)、ssDNA-FQ+PP+Klenow+Nt.BbvCI(f)和ssDNA-FQ+PP+bcr1+Klenow+Nt.BbvCI(g)与T4连接酶和Cas12a-crRNA在引物和dNTPs存在下孵育。实验条件:[PP]=[bcr1]=[引物]=20nM;[ssDNA-FQ]=100nM;[Cas12a-crRNA]=80nM;[T4连接酶]=[Klenow]=25U/mL;[Nt.BbvCI]=50U/mL;[dNTPs]=250μM;
图3为:证明信号扩增(RCA)成功实现的PAGE凝胶电泳图;DNA标记物的非变性PAGE分析,bcr1(a)、PP(B)、PP+bcr1+T4连接酶(c)、PP+bcr1+T4连接酶+Klenow+dNTPs(d)和PP+bcr1+T4连接酶+Klenow+dNTPs+Nt.BbvCI(e)。实验条件:[PP]=[bcr1]=[引物]=20nM;[ssDNA-FQ]=100nM;[Cas12a-crRNA]=80nM;[T4连接酶]=[Klenow]=25U/mL;[Nt.BbvCI]=50U/mL;[dNTPs]=250μM;
图4为:分子间G四链体对增强CRISPR/Cas12a切割效率的空间约束效应的描述图;
图5为:RCA扩增后所产生的触发器结合ThT后产生的荧光光谱响应图;ThT(a)、PP+ThT(B)、bcr1+ThT(c)、PP+bcr1+T4连接酶+引物+Klenow+dNTPs+ThT(d)和PP+bcr1+T4连接酶+引物+Klenow+dNTPs+Nt.BbvCI+ThT(e)的荧光测定。实验条件:[PP]=[bcr1]=[primer]=20nM;[T4连接酶]=[Klenow]=25U/mL;[Nt.BbvCI]=50U/mL;[dNTPs]=250μM;[ThT]=1μM。
图6为:切割后触发类似物(NTA)和一系列切割触发变体(NTV),包括NTV1、NTV2和NTV3结合ThT后产生的荧光光谱响应图;仅ThT(a)、NTA+ThT(b)、NTV1+ThT(C)、NTV2+ThT(d)和NTV3+ThT(e)的荧光测量。实验条件:[NTA]=[NTV1]=[NTV2]=[NTV3]=250nM;[ThT]=1μM。
图7为:CRISPR/Cas12a系统在切割后触发类似物(NTA)和一系列切割触发变体(NTV),包括NTV1、NTV2和NTV3的激活下切割ssDNA-FQ的效率(实时荧光强度的记录);仅Cas12a-crRNA+ssDNA-FQ(a),Cas12a-crRNA+NTA+ssDNA-FQ(b),Cas12a-crRNA+NTV1+ssDNA-FQ(c),Cas12a-crRNA+NTV2+ssDNA-FQ(d)and Cas12a-crRNA+NTV3+ssDNA-FQ(e);实验条件:[NTA]=[NTV1]=[NTV2]=[NTV3]=1μM;[ssDNA-FQ]=4μM;[Cas12a-crRNA]=100nM.
验证分子间G四链体的形成增强了CRISPR/Cas12a切割效率;Cas12a-crRNA+NTV2(D)和Cas12a-crRNA+NTV3(e)对ssDNA-FQ切割的时间依赖性荧光响应。[NTA]=[NTV1]=[NTV2]=[NTV3]=1μM;[ssDNA-FQ]=4μM;[Cas12a-crRNA]=100nM;
图8为:生物传感器对bcr1在1fM-250nM不同浓度下的荧光发射光谱图。插图放大了bcr1在低浓度范围内(1fM至100pM)的荧光光谱;实验条件:[PP]=[Primer]=20nM;[ssDNA-FQ]=100nM;[Cas12a-crRNA]=80nM;[T4 Ligase]=[Klenow]=25U/mL;[Nt.BbvCI]=50U/mL;[dNTPs]=250μM.
图9为:生物传感器在不同浓度Klenow下的荧光发射光谱图。插图中的条形图为不同浓度Klenow下的F/F
图11为:生物传感器在不同浓度Cas12a-crRNA下的荧光发射光谱图。插图中的条形图为不同浓度Cas12a-crRNA下的F/F
图12为:生物传感器在不同酶促反应时间下的荧光发射光谱图。插图中的条形图为不同酶促反应时间下的F/F
图13为:生物传感器在不同ssDNA-FQ裂解反应时间下的荧光发射光谱图。插图中的条形图为不同ssDNA-FQ裂解反应时间下的F/F
图14为:生物传感器在不同缓冲体系下的荧光发射光谱图。插图中的条形图为不同缓冲体系下的F/F
图15为:从1fM到250nm,523nm处峰值荧光强度与bcr1浓度的关系示意图;误差条从至少三次重复测试中获得。实验条件:[PP]=[Primer]=20nM;[ssDNA-FQ]=100nM;[Cas12a-crRNA]=80nM;[T4 Ligase]=[Klenow]=25U/mL;[Nt.BbvCI]=50U/mL;[dNTPs]=250μM.
图16为:从1fM到100pM,523nm处峰值荧光强度与靶目标bcr1浓度的对数呈线性相关示意图及方程式;误差条从至少三次重复测试中获得。实验条件:[PP]=[Primer]=20nM;[ssDNA-FQ]=100nM;[Cas12a-crRNA]=80nM;[T4 Ligase]=[Klenow]=25U/mL;[Nt.BbvCI]=50U/mL;[dNTPs]=250μM.
图17为:在10nM-250nM范围内,523nm处峰值荧光强度与靶目标bcr1浓度的对数呈线性相关示意图及方程式;误差条从至少三次重复测试中获得。实验条件:[PP]=[Primer]=20nM;[ssDNA-FQ]=100nM;[Cas12a-crRNA]=80nM;[T4 Ligase]=[Klenow]=25U/mL;[Nt.BbvCI]=50U/mL;[dNTPs]=250μM.
图18为:生物传感器响应靶目标bcr1和非靶目标(Bcr2,Bcr3,PML-L,PML-S,PML-V)的荧光强度对比示意图;误差条从至少三次重复测试中获得。实验条件:[PP]=[Bcr1]=[Bcr2]=[Bcr3]=[PML-L]=[PML-S]=[PML-V]=[Primer]=20nM;[ssDNA-FQ]=100nM;
[Cas12a-crRNA]=80nM;[T4 Ligase]=[Klenow]=25U/mL;[Nt.BbvCI]=50U/mL;[dNTPs]=250μM.
图19为:生物传感器响应血清中50fM-50nM不同浓度融合基因bcr1的荧光发射光谱;实验条件:[PP]=[bcr1]=[Primer]=20nM;[ssDNA-FQ]=100nM;[Cas12a-crRNA]=80nM;[T4 Ligase]=[Klenow]=25U/mL;[Nt.BbvCI]=50U/mL;[dNTPs]=250μM.
图20为:从50fM到50nm,523nm处峰值荧光强度与血清中bcr1浓度的关系示意图;从50fM到50pM,523nm处峰值荧光强度与血清中靶目标bcr1浓度的对数呈线性相关示意图及方程式;误差条从至少三次重复测试中获得。实验条件:[PP]=[bcr1]=[Primer]=20nM;[ssDNA-FQ]=100nM;[Cas12a-crRNA]=80nM;[T4 Ligase]=[Klenow]=25U/mL;[Nt.BbvCI]=50U/mL;[dNTPs]=250μM.
具体实施方式
1、检测方法
1.1材料和方法
T4 DNA连接酶(5U/μL)和10×T4连接缓冲液(400mM Tris,pH 7.8,100mM MgCl
Table S1:本研究中所用到的单链DNA的序列见基因序列表
1.2仪器
本实验使用PerkinElmer FL8500荧光分光光度计(Waltham,USA)进行所有荧光测量。仪器设置为:激发波长440nm,狭缝宽度10/10nm,扫描速度240nm/min,记录范围460-650nm,PMT探测器电压650V。电泳实验采用Servicebio PW-600电泳分析仪(中国武汉),凝胶成像照片采用ChampGel 7000凝胶成像系统(中国北京)。缓冲液的pH值使用LeiCi pH-3e台式pH计(中国上海)测量。此外,Bio-Rad T100热循环系统(Hercules,USA)用于控制恒定的反应温度。
1.3聚丙烯酰胺凝胶电泳(PAGE)分析
将10μL含有不同组分的由100×SYBR Green I染色的样品滴装入新鲜制备的12%native-PAGE凝胶的电泳加样孔中。DNA电泳分析在1×TBE缓冲液中进行,在80V恒定电位下进行90分钟。随后,将凝胶直接暴露在紫外线下,并使用凝胶成像系统拍摄图像。
1.4crRNA的制备
通过体外转录合成crRNA。为了制备crRNA,在Tap DNA聚合酶的参与下,以短T7引物(T7-crRNA-f)和长T7引物(T7-crRNA-r)为底物生成crRNA模板DNA。DNA模板在95℃时进行热变性,然后逐渐冷却到环境温度退火。随后,在50μL的转录系统中,将200ng模板DNA与5μL的1μM的T7 RNA聚合酶、10μL的5×T7转录缓冲液和2μL的100mM NTPs结合。加入RNaseInhibitor(40U/μL),37℃孵育9小时。然后,加入4μL体积的DNase I(1U/μL)和10×6μL体积的Reaction Buffer,以促进多余DNA模板的消化。然后将所得产物保存在-80℃以备将来使用。
1.5PML-RARA融合基因检测
为了进行bcr1分析,将4μL双蒸馏水(ddH
1.6分子间G四链体的空间约束效应表征
为了证明分子间G四链体在融合基因检测中的空间约束效应,我们设计了一个切割后触发类似物(NTA)和一系列切割触发变体(NTV),包括NTV1、NTV2和NTV3,它们被替换了不同数量的G碱基。这些变体被用来强调基于分子间G四链体的空间限制对Cas12a切割效率的增强。硫黄素T(ThT)的荧光可以通过其诱导、稳定和结合特性有效地指示分子间G四链体的存在。为了验证分子间G四链体的存在,将含有5μL 10μM NTA或NTV的反应混合物(200μL)与含有1μM ThT的Tris缓冲液(50mM,pH 7.4,100mM NaCl)195μL在37℃下孵育1小时。随后,测量了激发波长为430nm和发射波长为496nm的荧光强度。此外,在体外Cas12a酶切ssDNA-FQ的整个过程中,采用Bio-Rad CFX Connect real-time PCR系统(California,USA)进行实时荧光监测。这种监测使我们能够基于分子内G四链体的空间限制施加的空间约束来评估其对Cas12a活性的加速。
3.结果与讨论
3.1PML/RARA融合基因检测原理
为了检测bcr1,我们设计了一种名为PP的探针,通过序列分析确定PP探针的末端区域与靶基因互补,而中间部分包含限制性内切酶Nt.BbvCI的识别序列GCTGAGG,以及引物结合序列TGAGGTTTTTTT。为了简化探针设计和减少相互碱基识别的干扰,探针的其余区域由富T序列组成。当靶基因bcr1存在时,如图1所示,PP探针形成一个环,由T4 DNA连接酶连接。随后,引物与PP结合,使用Kelnow DNA聚合酶进行RCA扩增并诱导靶基因的位移。由于探针中存在限制性内切酶的识别位点,在滚环过程中形成的外周核酸链被Nt.BbvCI切割,在PP模板上诱导了新一轮聚合酶介导的复制。已经产生的DNA链,称为切割后触发器,从PP环上剥离。值得注意的是,移位的靶基因和切割后触发器都可以反复用于引起下一轮PP结环和随后的RCA扩增。有趣的是,含有大量G碱基的切割后触发器可以通过G碱基之间的Hoogsteen氢键形成G四链体结构。这些G四链体结构是由鸟嘌呤四聚体相互堆叠而形成的。基于此,G四链体结合crRNA,激活CRISPR/Cas12a的反式切割活性。G四链体独特的空间限制效应,限制了crRNA和Cas12蛋白,允许CRISPR/Cas12a系统的局部浓度显著增加。因此,ssDNA-FQs的信号报告探针可以被CRISPR/Cas12a系统高效、快速地切割,从而释放出强烈的荧光信号,指示被检测目标基因的浓度。在靶基因缺失的情况下,虽然引物仍能与PP探针结合并启动扩增、切割反应,但这种条件下的单链产物不能与crRNA结合,无法激活CRISPR/Cas12a系统。这有效地控制了背景信号。基于这两种情况的对比分析,我们认为我们建立的通过靶基因诱导的RCA诱导分子间G四链体并与CRISPR/Cas12a结合的方法可以用于bcr1的检测。如图1所示。
3.2bcr1生物传感可行性论证
为了验证基于扩增传感系统检测目标融合基因的可行性,图2给出了不同样品的荧光光谱。在曲线a中,表示只有5个核苷酸双标记的ssDNA-FQ、Primer、T4 Ligase和dNTPs的溶液系统,由于ssDNA-FQ上的荧光猝灭,仅能观察到低荧光背景信号。当系统中不存在Klenow时(曲线b和c),无论bcr1是否存在,都观察到与仅含ssDNA-FQ的系统相似的低荧光信号。同样,在没有bcr1的情况下,Klenow的参与并没有导致荧光信号的改善(曲线d)。然而,当bcr1与PP、Klenow和Cas12a-crRNA复合物一起呈现时(曲线e),可以观察到高荧光强度。这是因为在靶基因诱导的RCA过程中,单链RCA产物可以与Cas12a-crRNA复合物结合,激活Cas12a的侧切活性。最重要的是,当系统中同时存在Klenow和Nt.BbvCI检测bcr1时,观察到最大的荧光信号(曲线g),同时背景信号仍然被很好地抑制(曲线f)。曲线g与f的信号差异较大,说明本工作对bcr1检测是可行的。
3.3分子间G四链体的形成及其空间约束效应的论证
已知ThT可以结合G四链体间或G四链体内结构,从而产生荧光发射。对切割后触发器的序列分析表明,切割后触发器不能形成分子间G四链体结构。因此,如果ThT与切割后触发器孵育导致荧光增强,如图4所示,则为分子间G四链体结构的形成提供了强有力的证据。为了验证这一假设,在图5中,我们首先测试了ThT的固有荧光(曲线a),将ThT与PP混合(曲线b),将ThT与bcr1混合(曲线c)。正如预期的那样,它们都没有明显的荧光响应。此外,当我们将PP与bcr1混合并生成长RCA产物时,如曲线d所示,该成分对ThT的荧光信号没有明显的增加。然而,当我们加入Nt.BbvCI来启动RCA切割反应时,我们惊喜地观察到一个显著的荧光信号,如曲线e所示。这可能是由于聚合酶和核酸内切酶的共同作用,导致产生了更多的切割后触发器。这些切割后触发器可以相互作用并形成预期的分子间G四链体结构。为了进一步支持我们的假设,我们设计了额外的切割后触发器变体,NTV1,NTV2和NTV3,与原始切割后触发器相比,它们分别减少了2个,4个和8个“G”碱基。从图6的检测结果可以看出,随着“G”碱基数量减少,被“A”取代,导致G束减少,荧光信号也随之减少。这一比较数据突出了优化“G”碱基数量对于形成分子间G四链体结构的重要性。最后,在crRNA和Cas12a蛋白存在的情况下,我们彻底检查了切割后触发器、NTV1、NTV2和NTV3对ssDNA-FQ切割的反应动力学。如图7所示,当crRNA和Cas12a蛋白未被激活时,与ssDNA-FQ结合后没有荧光释放(曲线a)。相反,当切割后触发器与crRNA和Cas12a蛋白相互作用,导致CRISPR/Cas12a系统快速激活时,反应体系荧光信号快速增加(曲线b)。然而,NTV1、NTV2和NTV3与crRNA和Cas12a蛋白结合时,也引起荧光信号释放。但反应速率明显较慢。减缓效应与缺失“G”碱基的数量呈正相关。因此,基于上述数据,我们证明了在反应过程中分子间G四链体结构的形成,并且分子间G四链体结构表现出明显的空间约束效应,提高了反应动力学和效率。图7中ThT(a)、PP+ThT(B)、bcr1+ThT(c)、PP+bcr1+T4连接酶+引物+Klenow+dNTPs+ThT(d)和PP+bcr1+T4连接酶+引物+Klenow+dNTPs+Nt.BbvCI+ThT(e)的荧光测定。[PP]=[bcr1]=[primer]=20nM;[T4连接酶]=[Klenow]=25U/mL;[Nt.BbvCI]=50U/mL;[dNTPs]=250μM;[ThT]=1μM。(C)仅ThT(a)、NTA+ThT(b)、NTV1+ThT(C)、NTV2+ThT(d)和NTV3+ThT(e)的荧光测量。[NTA]=[NTV1]=[NTV2]=[NTV3]=250nM;[ThT]=1μM。(D)仅Cas12a-crRNA(a)、Cas12a-crRNA+NTA(b)、Cas12a-crRNA+NTV1(c)、Cas12a-crRNA+NTV2(D)和Cas12a-crRNA+NTV3(e)对ssDNA-FQ切割的时间依赖性荧光响应。[NTA]=[NTV1]=[NTV2]=[NTV3]=1μM;[ssDNA-FQ]=4μM;[Cas12a-crRNA]=100nM。
2.4分析性能调查
在最佳实验条件下(图8、9、10、11、12、13),我们通过测量不同目标浓度样品的荧光强度来评估检测系统的灵敏度。如图14所示,荧光强度与靶目标浓度呈正相关,范围从1fM到250nM。附图为低浓度(0至100pM)下记录的bcr1荧光光谱。荧光强度与不同浓度靶基因的动态响应关系如图15所示。可见,随着bcr1浓度的逐渐增加,荧光响应明显增强。为了准确描述荧光峰响应与靶浓度的关系,图16和图17分别显示了523nm波长处的荧光峰值(F
插图放大了bcr1在低浓度范围内(1fM至100pM)的荧光光谱;从1fM到250nm,523nm处峰值荧光强度与bcr1浓度的关系;从1fM到100pM,峰值荧光强度与靶浓度的对数呈线性相关;在10nM-250nM范围内,峰值荧光强度与靶浓度的对数呈线性相关。误差条从至少三次重复测试中获得。实验条件:[PP]=[bcr1]=[引物]=20nM;[ssDNA-FQ]=100nM;[Cas12a-crRNA]=80nM;[T4连接酶]=[Klenow]=25U/mL;[Nt.BbvCI]=50U/mL;[dNTPs]=250μM。
2.5特异性研究
特异性是准确检测bcr1的关键。为了评估所提出的生物传感系统的特异性,我们通过检测正常基因外显子的野生型DNA片段和可能存在于APL患者中的PML/RARA的其他典型亚型来比较传感信号。为了实现这一目标,我们人为设计了一系列融合基因片段(Bcr2,Bcr3,PML-L,PML-S和PML-V),这些片段专门用于激活生物传感系统以靶向操纵bcr1。空白样品,不包含任何目标分析物,作为对照。如图18所示,改变碱基的存在阻碍了挂锁探针介导的连接过程中的特异性识别,从而阻碍了信号放大。当我们将目标bcr1(F
2.6实际样本分析
众所周知,人类血清中含有多种核酸降解酶,这些酶可能会影响基于核酸的传感策略的准确性。因此,我们评估了传感系统在检测人血清中PML/RARA融合基因时的荧光响应。通过将已知浓度的bcr1加入稀释100倍的人血清中制备模拟样品。在加入bcr1之前,将1%人血清加热至65℃10分钟,使脱氧核糖核酸酶失活。如图19所示,在1%的人血清中,随着融合基因浓度的增加,传感系统的荧光强度不断增加。传感器系统的荧光强度与不同浓度靶基因的动态响应关系如图20所示。值得注意的是,即使在复杂的人血清环境中,在50fM-50nM范围内,该检测系统仍然显示出荧光强度与bcr1靶基因浓度的对数之间具有很强的线性相关性。回归曲线为F
3.结论
总之,本研究结合基于CRISPR/Cas12a核酸酶的系统和RCA技术的优势,提出了一种检测PML/RARA基因组片段的新方法。RCA辅助的切割反应导致大量切割后触发器的产生,分子间G四链体结构的利用有效地提高了CRISPR/Cas12a的局部浓度,从而加速了报告核酸的切割效率。PML/RARA融合基因的长异构体作为本研究的目标分析物,该方法达到了令人印象深刻的1fM的检测限。此外,在1fM至100pM和10nM至250nM的浓度范围内,观察到目标浓度的对数与荧光强度之间的精确线性相关。与其他DNA检测方法相比,即使在复杂的样品环境中,该方法也具有飞摩尔灵敏度。本研究提出的方法为进一步开发强大的CRISPR/Cas传感器系统提供了巨大的潜力,并为急性早幼粒细胞白血病(APL)的诊断提供了快速和适应性的范例。
实验条件优化。为了提高分析性能,我们优化了反应缓冲溶液,调整了Klenow、Nt.BbvCI和Cas12a-crRNA的浓度,微调了酶促反应时间和ssDNA-FQ裂解时间,以保持系统中较高的信号放大能力。从图15的实验结果可以看出,最优缓冲溶液为缓冲液f。从图16、S17和S18可以看出,荧光信号强度最初随着Klenow、Nt.BbvCI和Cas12a-crRNA浓度的增加而增加,然后达到平台。如图所示的柱状图表明,信号噪声比最初上升,随后下降,可能是由于高浓度的工具酶对反应进展的抑制作用。因此,后续实验以25U/mL的Klenow、50U/mL的Nt.BbvCI和25nM的Cas12a-crRNA为最佳反应浓度。此外,在本实验中,酶促反应的持续时间对信号强度和信噪比都有显著影响。荧光信号强度在酶促反应时间的5-60分钟范围内呈先上升后稳定的趋势,而信噪比则呈上升后下降的趋势,如图S19所示。因此,60分钟的酶促反应时间被认为是最佳的。如图20所示,当ssDNA-FQ切割时间为0.5-3小时时,体系的荧光强度迅速增加,2小时后荧光强度缓慢增加。为了优化效率,我们选择剪切时间为2小时为最适合CRISPR/Cas12a的剪切时间。在对关键实验参数进行优化后,选择最优实验条件进行后续实验。
- 一种检测PML-RARA融合基因PML B2区域突变的引物及检测方法
- 一种检测PML-RARA融合基因的LAMP引物组、试剂盒及检测方法