一种高纯度枸橼酸托法替布的制备方法
文献发布时间:2023-06-19 09:24:30
技术领域
本发明涉及药物合成领域,更具体地,涉及一种高纯度枸橼酸托法替布的制备方法。
背景技术
枸橼酸托法替布(Tofacitinib citrate),又名枸橼酸托法替尼,是口服有效的蛋白酪氨酸激酶(JAK)抑制剂,由美国Pfizer公司研发。2012年11月6日FDA批准其片剂上市,用于对氨甲喋呤治疗应答不充分或不耐受的中至重度活动性类风湿关节炎成人患者。截止目前,枸橼酸托法替布已获得全球80多个国家批准上市,于2017年3月10日获得CFDA批准上市许可。
枸橼酸托法替布(CAS:540737-29-9;化学名称:(3R,4R)-4-甲基-3-(甲基-1H-吡咯并[2,3-d]嘧啶-4-氨基)-β-氧-1-哌啶丙腈,2-羟基-1,2,3-丙烷三羧酸(1:1))结构如下式所示:
目前,关于枸橼酸托法替布的合成方法已有多篇文献报道,原研Pfizer公司在CN101233138A中公开了以下合成路线:
其中,化合物1(2,4-二氯-7H-吡咯[2,3-d]嘧啶)与化合物2((3R,4R)-N,4-二甲基-1-(苯基甲基)-3-哌啶胺)发生取代反应生成化合物3,化合物3经氢化还原得到化合物4,化合物4与氰基乙酸乙酯发生亲核取代反应、再与枸橼酸成盐一锅化反应制备得到枸橼酸托法替布。然而,在该合成路线中,制得的终产品枸橼酸托法替布存在较多的杂质,最大单杂高达0.28%,且产品颜色偏黄,不能满足药用要求。
因此,亟需开发一种高纯度、且颜色符合药用要求的枸橼酸托法替布的制备方法。
发明内容
本发明的目的是克服上述现有技术中的缺陷和不足,提供一种高纯度枸橼酸托法替布的制备方法。
本发明上述目的通过以下技术方案实现:
步骤S1.氰基乙酸经特戊酰氯活化后,与N-[(3R,4R)-4-甲基哌啶-3-基]-N-甲基-7H-吡咯并[2,3-d]嘧啶-4-胺在有机溶剂中发生取代反应,得到反应液;
步骤S2.向S1制得的反应液中加酸性水溶液调至pH值为1~3,分液,收集水相;
步骤S3.将S2中水相调至pH值为7~8,用二氯甲烷萃取,收集有机相,浓缩;
步骤S4.向S3制得的浓缩物中加入醇水混合溶剂,析晶得托法替布;
步骤S5.将S4制得的托法替布与枸橼酸成盐得到高纯度枸橼酸托法替布。
本发明将S1中的反应液中主要成分为托法替布,随后加入酸性水溶液将反应液中的托法替布与酸结合生成可溶性盐,此时溶液分为水相和有机相两层,其中大部分杂质溶解在有机相中,再通过分液可达到杂质分离效果;进一步地往含有托法替布可溶性盐的水相中加入pH调节剂,再加入二氯甲烷将托法替布萃取出来,浓缩后将所得的浓缩物,即托法替布油状物,经醇水混合溶剂析晶、过滤、干燥等工序进一步纯化,可获得化学纯度不低于99.5%,最大单杂低至0.03%以下的托法替布精品,最后再与枸橼酸成盐,可获得符合药用要求的高纯度的枸橼酸托法替布。
优选地,步骤S2中所述酸性水溶液为盐酸、稀硫酸、氢溴酸中的一种或几种。
更优选地,所述酸性水溶液为盐酸。
更优选地,所述盐酸的浓度为0.5~5mol/L。
更优选地,所述盐酸的浓度为1~2mol/L。
优选地,步骤S4中所述醇水混合溶剂为甲醇和水的混合溶剂或乙醇和水的混合溶剂。
更优选地,所述醇水混合溶剂为甲醇和水的混合溶剂。
更优选地,所述甲醇和水的体积比为2:1~1:5。
优选地,步骤S3中所述pH值调节剂为碳酸钠、碳酸氢钠、碳酸钾、氢氧化钠、氨水中的一种或几种。
优选地,步骤S3中所述的萃取次数为1~3次。具体为将S3中萃取得到的水相用二氯甲烷再萃取,得到的有机相与S3中的有机相合并,循环1~3次,再进行浓缩。
与现有技术相比,本发明具有如下有益效果:
(1)本发明本发明两步法所制得的枸橼酸托法替布的总收率可达74.4%,所制得的枸橼酸托法替布的纯度高达99.81%,最大单杂可低至0.03%以下。与现有技术相比,采用本发明的制备方法制得的枸橼酸托法替布收率上升了约7.1%,纯度也得到了有效提升。
(2)本发明制得的终产品枸橼酸托法替布的颜色具有明显改善,完全达到药用标准。
(3)本发明的枸橼酸托法替布的制备方法,合成路线简单,反应条件易控易操作,产品总收率高,纯度高,杂质少,终产品稳定,利于工业化生产。
附图说明
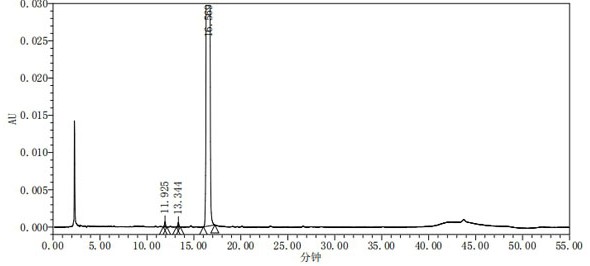
图1是实施例1制得的枸橼酸托法替布的HPLC纯度检测色谱图。
具体实施方式
为了更清楚、完整的描述本发明的技术方案,以下通过具体实施例进一步详细说明本发明,应当理解,此处所描述的具体实施例仅用于解释本发明,并不用于限定本发明,可以在本发明权利限定的范围内进行各种改变。
实施例1
一种高纯度枸橼酸托法替布的制备方法,包括以下步骤:
步骤S1.往100L搪玻璃反应罐中依次加入37.89kg二氯甲烷、3.48kg氰基乙酸和4.15kg三乙胺,然后通入氮气保护,降温至-10~0℃。往反应釜中滴加4.62kg特戊酰氯。控温-10~0℃反应2h。反应完毕,往反应釜中滴加25.26kg二氯甲烷和3.35kg(N-[(3R,4R)-4-甲基哌啶-3-基]-N-甲基-7H-吡咯并[2,3-d]嘧啶-4-胺)的混合溶液,滴加完后,继续保温反应1.5h,取样监控反应进程,反应完毕后得到反应液;
步骤S2.向S1制得的反应液中加1mol/L盐酸49.98L调至pH值为1~3,搅拌均匀后静置分液,萃取,收集水相;
步骤S3.向S2中的水相中搅拌下滴加饱和碳酸钠溶液约19.41kg调pH值至7~8,加入55kg二氯甲烷,静置,萃取,收集有机相,将得到的水相再次用二氯甲烷萃取,合并两次有机相,浓缩;
步骤S4.向S3制得的浓缩物中加入加入甲醇18.31kg、纯化水35.42kg,升温至40-50℃搅拌1h后,降温至10-25℃搅拌1h,过滤,用纯化水12.4kg洗涤滤饼,所得固体于55~65℃减压干燥后,得到白色固体托法替布3.15kg,收率73.9%;
步骤S5.将14.75kg丙酮、5.19kg纯化水和2.95kg S4制得的托法替布3.10kg依次加入到50L玻璃反应釜中,体系升温至50~55℃;往反应釜中加入2.02kg枸橼酸与8.08kg水溶液,将体系保温50~55℃搅拌1h,然后降温至0~10℃,保温0~10℃析晶1h,过滤,滤饼用2.90kg丙酮淋洗滤饼,所得固体于55~65℃干燥后,得到白色固体4.72kg,即为枸橼酸托法替布,收率为94.4%,采用HPLC进行纯度检测,结果如附图1所示,制得的枸橼酸托法替布的纯度为99.87%,最大单杂为0.03%;
实施例2
一种高纯度枸橼酸托法替布的制备方法,与实施例1基本相同,其区别在于,仅改变酸性水溶液的种类和浓度,考察制备终产品枸橼酸托法替布收率、纯度及杂质情况,见表1(本申请中的终产品均指枸橼酸托法替布)。
表1
实施例3
一种高纯度枸橼酸托法替布的制备方法,与实施例1基本相同,其区别在于,仅改变碱性溶液种类,考察制备终产品枸橼酸托法替布收率、纯度及杂质情况,见表2。
表2
对比例1
参照专利CN101233138A实施例13的方法制得枸橼酸托法替布。
表3
对比表1~表3可知,相比于采用其他制备方法得到的枸橼酸托法替布,本发明所制得的枸橼酸托法替布具备更高的纯度,最大单杂显著降低,且产品成色也完全能够达到药用要求。
以上实施例使用具体实施方式及试验,对本发明作了详尽的描述,但在本发明基础上,可以对之作一些不偏离本发明主旨的修改或改进,这对本领域技术人员而言是显而易见的。因此,这些不偏离本发明主旨基础上所做的修改或改进,均属于本发明要求保护的范围。
- 一种枸橼酸托法替布中间体及枸橼酸托法替布的制备方法
- 一种枸橼酸托法替布肠溶缓释微丸及其制备方法