一种甲磺酸乐伐替尼晶型X的制备方法
文献发布时间:2023-06-19 09:26:02
技术领域
本申请涉及药物晶型领域,具体涉及一种甲磺酸乐伐替尼晶型X及其制备方法。
背景技术
乐伐替尼(Lenvatinib),化学名为:4-[3-氯-4-(环丙基氨基羰基)氨基苯氧基]-7-甲氧基-6-喹啉甲酰胺,其结构式如(I)式所示。
乐伐替尼是由日本卫材株式会社研发的一种甲状腺癌药物。乐伐替尼是一种口服多受体酪氨酸激酶(RTK)抑制剂,其与其受体具有新颖的结合模式,除抑制参与肿瘤增殖的其他促血管生成和致癌信号通路相关RTK外,还能够选择性抑制血管内皮生长因子(VEGF)受体的激酶活性。2015年02月13日获得FDA批准,用于放射性碘难治性局部复发或转移的进展性分化型甲状腺癌患者的治疗,之后在欧盟和日本相继获批上市。
甲磺酸乐伐替尼,具有多晶型。专利CN100569753C、CN101337931B、CN101337932B和CN101337933B分别公开了乐伐替尼甲磺酸盐的晶型A、B、C、F(水合物)、I(醋酸合物)。结合其专利试验数据可知,晶型A、C相对晶型B、I更稳定。
基因毒性杂质是指能直接或间接损伤细胞DNA,产生致突变和致癌作用的物质。对乐伐替尼的结构式进行分析可知,该化合物容易与醇类溶剂发生醇解反应,生成醇解杂质;同时由于该化合物的成盐过程必须使用甲磺酸,而甲磺酸亦容易与醇类溶剂反应生成甲磺酸酯,甲磺酸酯是一类常见的基因毒性杂质。本申请在经过一系列的前期筛选试验后发现,晶型X仅在甲醇和纯水的溶剂体系中可以被制备,而甲醇与甲磺酸易生成甲磺酸酯(基毒杂质),甲醇与乐伐替尼游离碱在加热条件下易生成醇解杂质,从而容易导致产品的基毒杂质和醇解杂质超标。
现有技术中未公开晶型X,也未公开不使用醇类溶剂的制备晶型X的制备方法,因此,本领域仍然需要一种一种制备工艺简单,产品质量更优的晶型X的制备方法,而本申请符合这样的需求。
发明内容
本申请在不断地研究过程中,通过试验摸索,意外的获得了一种新的甲磺酸乐伐替尼晶型X的制备方法。该工艺可意外的将醇解杂质和基毒杂质降至检出限以下,同时保证最终产品的稳定性,并且可明显提高结晶收率。
具体的,本申请公开了甲磺酸乐伐替尼晶型X的一种制备方法,具体包含以下步骤:
(1)、将乐伐替尼于乙腈和纯化水中搅拌打浆,其中乐伐替尼与乙腈、纯化水的质量体积比为1g:(2-6ml):(2-6ml),搅拌打浆温度为20-30℃;
(2)、将醋酸、乙腈和甲磺酸的混合溶液滴加至步骤(1)的混悬液中,乐伐替尼与醋酸、乙腈、的质量体积比为1g:(1-3ml):(2-6ml),与甲磺酸的质量比为1g:(0.22-0.45g);
(3)、待步骤(2)的混悬液溶清后,加入晶型X的晶种的用量为乐伐替尼投料量的0-5%质量分数,然后将乙腈滴加至溶液中,乙腈用量与乐伐替尼质量体积比为(1-6ml):1g,保温温度为15-30℃,保温搅拌时间为12-24h,抽滤干燥得固体粉末;
其中,晶型的X射线粉末衍射具有如下所述的衍射角:22.84°±0.2°、20.16°±0.2°、9.46°±0.2°、14.52°±0.2°、24.86°±0.2°、18.92°±0.2°、26.82°±0.2°、8.68°±0.2°、10.32°±0.2°。
其中,步骤(1)中,乐伐替尼与乙腈、纯化水的质量体积比优选为1g:(2-4ml):(2-4ml),更优选1g:3ml:2ml,搅拌打浆温度优选为25-30℃,更优选为30℃。
其中,步骤(2)中,乐伐替尼与醋酸、乙腈、的质量体积比优选为1g:(1-2ml):(2-4ml),更优选1g:1ml:2ml,与甲磺酸的质量比优选为1g:(0.-0.35g),更优选为1g:0.27g。
其中,步骤(3)中,加入晶型X的晶种的用量优选为乐伐替尼投料量的2-5%质量分数,更优选3%,乙腈用量与乐伐替尼质量体积比优选为(1-3ml):1g,更优选2ml:1g,保温温度优选为20-25℃,更优选25℃,保温搅拌时间优选为12-18h,更优选12h。
更优选的,本申请的制备方法,包含以下步骤:
(1)将乐伐替尼于乙腈和纯化水中搅拌打浆,其中乐伐替尼与乙腈用量比例为1g:3ml,乐伐替尼与纯化水用量比例为1g:2ml,搅拌打浆温度为30℃;
(2)将醋酸、乙腈和甲磺酸的混合溶液滴加至步骤(1)的混悬液中,乐伐替尼与醋酸用量比例为1g:1ml,乐伐替尼与乙腈用量比例为1g:2ml,乐伐替尼与甲磺酸用量比例为1g:0.27g;
(3)待步骤(2)的混悬液溶清后,加入晶型X的晶种,晶种用量为乐伐替尼投料量的3%质量分数,然后将乙腈滴加至溶液中,乙腈用量与乐伐替尼用量比例为2ml:1g,滴加完毕后在25℃下保温搅拌12h,抽滤干燥得固体粉末;
更优选的,本申请甲磺酸乐伐替尼晶型X的制备方法,包含以下步骤:
将10.0g乐伐替尼分散于20ml纯化水和30ml乙腈中,于30℃搅拌打浆形成悬浮液;然后将2.7g甲磺酸、10ml醋酸和20ml乙腈的混合溶液在滴加至悬浮液中;待滴加完毕并且混悬液溶清后加入3%的晶种X,然后将20ml乙腈滴加至悬浮液中,滴加完毕后继续25℃搅拌12h,抽滤干燥得固体粉末8.93g。
或者,更优选的,本申请甲磺酸乐伐替尼晶型X的制备方法,包含以下步骤:
将10.0g乐伐替尼分散于30ml纯化水和30ml乙腈中,于30℃搅拌打浆形成悬浮液;然后将2.7g甲磺酸、10ml醋酸和30ml乙腈的混合溶液滴加至悬浮液中;待滴加完毕并且混悬液溶清后加入3%的晶种X,然后将50ml乙腈滴加至悬浮液中,滴加完毕后继续搅拌25℃搅拌12h,抽滤干燥得固体粉末8.53g。采用本申请制备的甲磺酸乐伐替尼晶型X具有以下优点:
1、在目前晶型X的制备中,必须采用甲醇和纯水的溶剂体系,而该工艺会导致醇解杂质和基毒杂质(甲磺酸酯)的生成,导致产品中的醇解杂质和基毒杂质明显高于化学药物杂质研究的技术指导原则中单杂(小于0.10%)和EMEA《基因毒性杂质限度指南》中基毒杂质小于1.5μg/d(根据剂量24mg/d可换算为62.5ppm)的限度,不符合产品质量要求。而本申请制备,可以避免使用醇类溶剂和加热步骤,既能保证所得产品为晶型X,同时可降低能耗并提高收率,反应条件温和、操作简单,最关键的是所得产品无醇解杂质,且基毒杂质无增加风险,产品质量更优,更符合原料药产品质量要求。
2、通过本申请晶型X制备过程中的各关键参数控制,本申请制备方法能够显著提高晶型X的收率,工艺重现性好,工业化生产条件可控,有利于大规模产业化。
附图说明
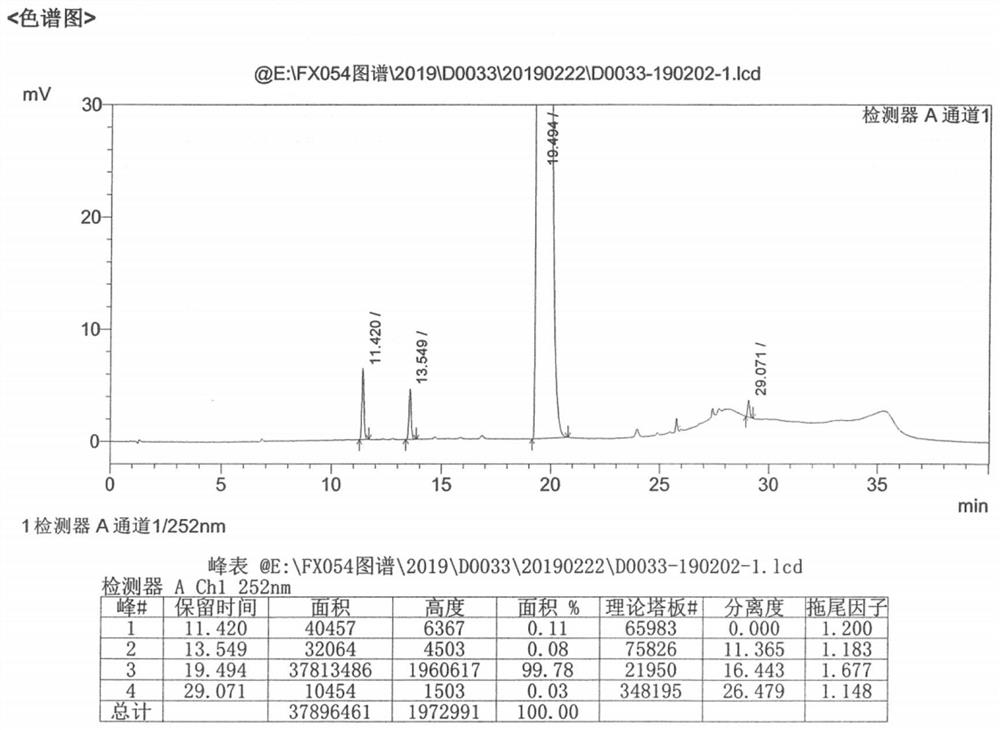
图1实施例1甲磺酸乐伐替尼晶型X的HPLC图谱
图2实施例1甲磺酸乐伐替尼晶型X的XRPD图谱
具体实施方式
以下将结合实施例对本申请作进一步的详细描述,本申请的实施例仅用于说明本申请的技术方案,并非限制本申请的实质和范围。
本申请中所用到的缩写的解释如下:
XRPD:X射线粉末衍射
HPLC:高效液相色谱
本申请所述的X射线粉末衍射(XRPD)的测定是采用辽宁丹东浩元DX-2700X粉末衍射仪进行采集,具体参数如下表:
HPLC谱图的测定采用安捷伦Agilent1260DAD型液相色谱仪进行。本申请中,乐伐替尼HPLC纯度用以下方法进行:
(1)以十八烷基硅烷键合硅胶为填充剂的色谱柱
(2)检测器:紫外检测器(波长252nm和205nm)
(3)流速:每分钟1ml
(4)运行时间:从溶剂峰开始计时,约为乐伐替尼峰保留时间的2.7倍
(5)供试品溶液:取本品12.5mg,精密称定,加溶剂溶解稀释制成每1ml约含0.5mg的溶液,作为供试品溶液
(6)进样量:10μl,通过自动积分法测定供试品溶液,按峰面积计算供试品中乐伐替尼的纯度。
具体实施方式:筛选试验
为了避免醇解杂质和甲磺酸酯杂质对产品质量构成影响,避免醇类溶剂的使用,本申请发明人在获得技术方案的过程中对步骤(1)所使用的溶剂种类及其组合进行了大量的筛选,同时对终产品的晶型和有关物质进行了数据统计,结果如下表1所示。
表1不同溶剂体系制备晶型X的实验结果
*备注:起始原料为游离碱,其醇解杂质未检出,甲磺酸酯杂质为32ppm
由上表1可知,晶型X并不容易获得,且仅在甲醇和水的溶剂体系以及乙腈、醋酸和水的溶剂体系中可以制备得到。但是甲醇和水的溶剂体系所得晶型X其醇解杂质及甲磺酸酯的基毒杂质均远超限度。
实施例1:甲磺酸乐伐替尼晶型X的制备
将10.0g乐伐替尼分散于20ml纯化水和30ml乙腈中,于30℃搅拌打浆形成悬浮液;然后将2.7g甲磺酸、10ml醋酸和20ml乙腈的混合溶液在滴加至悬浮液中;待滴加完毕并且混悬液溶清后加入3%的晶种X,然后将20ml乙腈滴加至悬浮液中,滴加完毕后继续25℃搅拌12h,抽滤干燥得固体粉末8.93g。收率89.3%。HPLC纯度为99.78%,醇解杂质未检出,基毒杂质未增加。其粉末衍射数据如图2所示。
其
使用Cu-ka射线进行X射线粉末测定,其图谱具有下表所示衍射角和晶面间距:
表1
2θ衍射角的误差为±0.2°。
实施例2:甲磺酸乐伐替尼晶型X的制备
将10.0g乐伐替尼分散于30ml纯化水和30ml乙腈中,于30℃搅拌打浆形成悬浮液;然后将2.7g甲磺酸、10ml醋酸和30ml乙腈的混合溶液滴加至悬浮液中;待滴加完毕并且混悬液溶清后加入3%的晶种X,然后将50ml乙腈滴加至悬浮液中,滴加完毕后继续25℃搅拌12h,抽滤干燥得固体粉末8.53g,收率85.3%。HPLC纯度为99.77%,醇解杂质未检出,基毒杂质未增加。其粉末衍射数据与实施例1数据保持基本一致。
实施例3:甲磺酸乐伐替尼晶型X的制备
将10.0g乐伐替尼分散于20ml纯化水和30ml乙腈中,于30℃搅拌打浆形成悬浮液;然后将2.7g甲磺酸、10ml醋酸和20ml乙腈的混合溶液滴加至悬浮液中;待滴加完毕并且混悬液溶清后加入3%的晶种X,然后将20ml乙腈滴加至悬浮液中,滴加完毕后继续20℃搅拌12h,抽滤干燥得固体粉末9.35g。,收率93.5%。HPLC纯度为99.75%,醇解杂质未检出,基毒杂质未增加。其粉末衍射数据与实施例1数据保持基本一致。
实施例4:甲磺酸乐伐替尼晶型X的制备
将10.0g乐伐替尼分散于30ml纯化水和30ml乙腈中,于30℃搅拌打浆形成悬浮液;然后将2.7g甲磺酸、10ml醋酸和20ml乙腈的混合溶液滴加至悬浮液中;待滴加完毕并且混悬液溶清后加入3%的晶种X,然后将30ml乙腈滴加至悬浮液中,滴加完毕后继续30℃搅拌12h,抽滤干燥得固体粉末8.65g,收率86.5%。HPLC纯度为99.76%,醇解杂质未检出,基毒杂质未增加。其粉末衍射数据与实施例1数据保持基本一致。
实施例5:甲磺酸乐伐替尼晶型X的制备
将10.0g乐伐替尼分散于20ml纯化水和40ml乙腈中,于30℃搅拌打浆形成悬浮液;然后将2.2g甲磺酸、10ml醋酸和30ml乙腈的混合溶液滴加至悬浮液中;待滴加完毕并且混悬液溶清后加入2%的晶种X,然后将60ml乙腈滴加至悬浮液中,滴加完毕后继续15℃搅拌12h,抽滤干燥得固体粉末9.46g,收率94.6%。HPLC纯度为99.78%,醇解杂质未检出,基毒杂质未增加。其粉末衍射数据与实施例1数据保持基本一致。
实施例6:甲磺酸乐伐替尼晶型X的制备
将10.0g乐伐替尼分散于20ml纯化水和60ml乙腈中,于30℃搅拌打浆形成悬浮液;然后将4.5g甲磺酸、20ml醋酸和20ml乙腈的混合溶液滴加至悬浮液中;待滴加完毕并且混悬液溶清后加入5%的晶种X,然后将10ml乙腈滴加至悬浮液中,滴加完毕后继续25℃搅拌12h,抽滤干燥得固体粉末8.56g,收率85.6%。HPLC纯度为99.71%,醇解杂质未检出,基毒杂质未增加。其粉末衍射数据与实施例1数据保持基本一致。
实施例7:甲磺酸乐伐替尼晶型X的制备
将10.0g乐伐替尼分散于60ml纯化水和60ml乙腈中,于20℃搅拌打浆形成悬浮液;然后将2.7g甲磺酸、30ml醋酸和60ml乙腈的混合溶液滴加至悬浮液中;待滴加完毕并且混悬液溶清后加入3%的晶种X,然后将60ml乙腈滴加至悬浮液中,滴加完毕后继续20℃搅拌18h,抽滤干燥得固体粉末9.12g,收率91.2%。HPLC纯度为99.69%,醇解杂质未检出,基毒杂质未增加。其粉末衍射数据与实施例1数据保持基本一致。
实施例8:甲磺酸乐伐替尼晶型X的制备
将10.0g乐伐替尼分散于20ml纯化水和20ml乙腈中,于30℃搅拌打浆形成悬浮液;然后将3.5g甲磺酸、20ml醋酸和30ml乙腈的混合溶液滴加至悬浮液中;待滴加完毕并且混悬液溶清后加入2%的晶种X,然后将30ml乙腈滴加至悬浮液中,滴加完毕后继续25℃搅拌12h,抽滤干燥得固体粉末9.15g,收率91.5%。HPLC纯度为99.72%,醇解杂质未检出,基毒杂质未增加。其粉末衍射数据与实施例1数据保持基本一致。
实施例9:甲磺酸乐伐替尼晶型X的制备
将10.0g乐伐替尼分散于40ml纯化水和40ml乙腈中,于25℃搅拌打浆形成悬浮液;然后将2.7g甲磺酸、10ml醋酸和20ml乙腈的混合溶液滴加至悬浮液中;待滴加完毕并且混悬液溶清后加入1%的晶种X,然后将20ml乙腈滴加至悬浮液中,滴加完毕后继续20℃搅拌24h,抽滤干燥得固体粉末8.93g,收率89.3%。HPLC纯度为99.74%,醇解杂质未检出,基毒杂质未增加。其粉末衍射数据与实施例1数据保持基本一致。
实施例10:甲磺酸乐伐替尼晶型X的制备
将10.0g乐伐替尼分散于20ml纯化水和30ml乙腈中,于30℃搅拌打浆形成悬浮液;然后将2.7g甲磺酸、10ml醋酸和20ml乙腈的混合溶液滴加至悬浮液中;待滴加完毕并且混悬液溶清后将20ml乙腈滴加至悬浮液中,滴加完毕后继续25℃搅拌24h,抽滤干燥得固体粉末9.16g,收率91.6%。HPLC纯度为99.73%,醇解杂质未检出,基毒杂质未增加。其粉末衍射数据与实施例1数据保持基本一致。
实施例11:甲磺酸乐伐替尼晶型X的制备
将550.0g乐伐替尼分散于1100ml纯化水和1650ml乙腈中,于30℃搅拌打浆形成悬浮液;然后将148.5g甲磺酸、550ml醋酸和1100ml乙腈的混合溶液滴加至悬浮液中;待滴加完毕并且混悬液溶清后将1100ml乙腈滴加至悬浮液中,滴加完毕后继续25℃搅拌24h,抽滤干燥得固体粉末502g,收率91.2%。HPLC纯度为99.75%,醇解杂质未检出,基毒杂质未增加。其粉末衍射数据与实施例1数据保持基本一致
对比例1:
将20.0g乐伐替尼悬浮于200ml甲醇和20ml水的混合溶剂中,将4.5g甲磺酸(1.0当量)缓慢滴加到该悬浮液中,室温搅拌30分钟,然后加热至60℃保证固体完全溶解。固体完全溶解后趁热过滤,滤液自然降温至室温,室温下搅拌20小时,负压抽滤,所得固体置于25℃恒温干燥过夜,所得固体经检测为乐伐替尼甲磺酸盐晶型X,收率72.0%,HPLC检测纯度99.14%,而醇解杂质为0.13%,基毒杂质为92ppm,超出限度。
对比例2:
将20.0g乐伐替尼悬浮于180ml甲醇和20ml水的混合溶剂中,将4.5g甲磺酸(1.0当量)缓慢滴加到该悬浮液中,室温搅拌30分钟,然后加热至55℃保证固体完全溶解。固体完全溶解后趁热过滤,滤液自然降温至室温,室温下搅拌20小时,负压抽滤,所得固体置于25℃恒温干燥过夜,所得固体经检测为乐伐替尼甲磺酸盐晶型X,收率75.0%,HPLC检测纯度99.21%,醇解杂质为0.15%,基毒杂质为82ppm,超出限度。
上述实施例和对比例的具体数据对比如表2所示。
表2不同工艺所得产品的收率、杂质与纯度数据对比
稳定性试验
将上述实施例制备的晶型样品采用PE袋为内包装、铝箔袋为外包装的方式置于25℃±2℃/60%RH±5%RH条件下进行6个月的加速试验,分别于0天、6个月取样检测晶型、纯度、醇解杂质和基毒杂质,检测结果如下表3所示:
表3本申请实施例样品的加速试验数据
由上表3的数据可知,本申请实施例的样品经过加速实验6个月的考察后,稳定性很好,其晶型和醇解杂质均未发生变化,纯度和基毒杂质也与0天的数据相当或仅少量增加。
因此,相比于现有技术晶型X的制备方法而言,本申请的制备反应条件温和、操作简单,工艺重现性好,所得产品纯度高,降低了能耗,与现有制备工艺需要使用醇类溶剂相比,生成醇解杂质和基毒杂质的风险大大降低。
对于本领域的普通技术人员而言明显的是,在不偏离本申请精神或者范围的情况下,可对本申请化合物、组合物以及其制备方法进行的多种修饰和变化,因此,本申请的保护范围涵盖了对本申请进行的各种修饰和变化,只要所述修饰或变化处于权利要求和其等同实施方式所涵盖的范围内。
- 一种甲磺酸乐伐替尼晶型X的制备方法
- 一种高纯度甲磺酸乐伐替尼晶型C的制备方法