用于癌症侦测的多重化验预测模型
文献发布时间:2023-06-19 09:29:07
背景技术
本发明一般地涉及在一患者中的癌症的辨识,且更特定地涉及对获得自所述患者的一检测样本执行一物理化验,以及所述物理化验的结果的统计分析。
使用次世代定序(NGS)分析循环的游离核苷酸(cell-free nucleotides),比如游离DNA(cfDNA)或游离RNA(cfRNA),被认为是用于癌症的侦测及诊断的一有价值的工具。分析cfDNA相较于传统的肿瘤活检(tumor biopsy)方法可以是有利的;然而,在肿瘤衍生(tumor-derived)的cfDNA中辨识指示癌症的信号面临独特的挑战,特别是对于在其中所述指示癌症的信号尚未被显著表达的目的,比如癌症的早期侦测。作为一示例,要达到对肿瘤衍生断片(tumor-derived fragments)的必要定序深度可能是困难的。作为另一示例,在样本制备及定序时被引入的错误可能使准确辨识指示癌症的信号变得困难。上述种种挑战的结合阻碍了经由使用获得自一对象的cfDNA,以足够的敏感度及特异度精确地预测在所述对象中的癌症特征。
发明内容
本发明的数个实施例提供一种基于在获得自一个体的一检测样本中的cfDNA,产生对所述个体的一癌症预测,比如癌症的一存在或不存在的方法。特定地,来自所述个体的cfDNA被以一个或更多个定序化验(sequencing assays),在本文中也称为物理化验定序以产生数个序列读数(sequence reads)。所述物理化验的数个示例包括一小变异定序化验、全基因组定序化验及甲基化定序化验。所述数个定序化验的所述序列读数是经由对应的计算机分析而被处理。计算机分析在下文中也被称作计算机管道、计算机评估及计算机分析中的任一者。每个计算机分析在考虑干扰信号(例如,噪声)的同时,辨识数个序列读数对产生一癌症预测提供信息的数个特征的数个数值。作为一示例,小变异特征(例如,衍生自通过一小变异定序化验产生的数个序列读数的数个特征)可以包括体细胞变异的一总数。在另一示例中,全基因组特征(例如,衍生自通过一全基因组定序化验产生的数个序列读数的数个特征)可以包括拷贝数目畸变的一总数。作为又一示例,甲基化特征(例如,衍生自通过一甲基化定序化验产生的数个序列读数的数个特征)可以包括过甲基化或低甲基化区域的一总数。非衍生自基于定序的手段的额外特征,比如数个可以意指临床症状及患者信息的基线特征,可以被进一步产生及分析。
在一些实施例中,一个、两个、三个或所有四个类型的特征(例如,数个小变异特征、数个全基因组特征、数个甲基化特征及数个基线特征)可以被提供到一单一预测性癌症模型,所述预测性癌症模型产生一癌症预测。在一些实施例中,不同类型的特征的数值可以被分离地提供到不同的预测性模型。每个分离的预测性模型可以输出一分数,所述分数接着作为进入一整体模型的输入,所述整体模型输出所述癌症预测。
在本文中揭示的数个实施例描述一种用于在一对象中侦测癌症的存在的方法,所述方法包含:获得产生自数个游离核酸的定序资料,所述数个游离核酸在来自所述对象的一检测样本中,其中所述定序资料包含决定自所述数个游离核酸的数个序列读数;使用一适宜的经编程计算机分析所述数个序列读数,以辨识两个或更多个基于定序的特征;及基于对所述两个或更多个特征的分析,侦测癌症的存在。
在本文中揭示的数个实施例进一步描述一种用于在一无症状的对象中侦测癌症的存在的方法,所述方法包含:获得产生自数个游离核酸的定序资料,所述数个游离核酸在来自一无症状对象的一检测样本中;使用一适宜的经编程计算机分析所述定序资料,以辨识两个或更多个基于定序的特征;基于对所述两个或更多个特征的分析,侦测癌症的存在。
在本文中揭示的数个实施例进一步描述一种用于在一无症状的对象中侦测癌症的存在的方法,所述方法包含:获得产生自数个游离核酸的定序资料,所述数个游离核酸在来自所述一无症状对象的一检测样本中;使用一适宜的经编程计算机分析所述定序资料,以辨识两个或更多个基于定序的特征;基于对所述两个或更多个特征的分析,侦测癌症的存在。
在一些实施例中,所述方法侦测三种或更多种不同类型的癌症。在一些实施例中,所述方法侦测五种或更多种不同类型的癌症。在一些实施例中,所述方法侦测十种或更多种不同类型的癌症。在一些实施例中,所述方法侦测二十种或更多种不同类型的癌症。在一些实施例中,所述两种或更多种不同类型的癌症是选自于乳癌、肺癌、前列腺癌、大肠直肠癌、肾癌、子宫癌、胰脏癌、食道癌、淋巴瘤、头颈癌、卵巢癌、肝胆癌、黑色素瘤、子宫颈癌、多发性骨髓瘤、白血病、甲状腺癌、膀胱癌、胃癌、肛门直肠癌及以上的任意组合。
在一些实施例中,所述游离核酸包含游离DNA(cfDNA)。在一些实施例中,所述数个序列读数是产生自一次世代定序(NGS)程序。在一些实施例中,所述数个序列读数是产生自使用合成式定序(sequencing-by-synthesis)的一大量平行定序。在一些实施例中,所述游离核酸包括来自白血球的cf-DNA。
在一些实施例中,所述两个或更多个特征是衍生自:对在所述检测样本中的数个游离核酸的一甲基化定序化验;对在所述检测样本中的数个游离核酸的一全基因组定序化验;及/或对在所述检测样本中的数个游离核酸的一小变异定序化验。
在一些实施例中,所述甲基化定序化验是一全基因组亚硫酸氢盐定序化验(bisulfite sequencing assay)。在一些实施例中,所述甲基化定序化验是一针对性(targeted)亚硫酸氢盐定序化验。在一些实施例中,侦测癌症的存在是基于分析决定自所述甲基化定序化验的两个或更多个特征。在一些实施例中,所述甲基化定序化验特征包含低甲基化计数的一数量、过甲基化计数的一数量、在数个CpG位点处的异常甲基化断片的存在或不存在、每个CpG位点的一低甲基化分数、每个CpG位点的一过甲基化分数、基于数个过甲基化分数的排名,及基于数个低甲基化分数的排名中的一个或更多个。
在一些实施例中,侦测癌症的存在是基于分析决定自所述全基因组定序化验的两个或更多个特征。在一些实施例中,所述全基因组定序化验特征包含遍及一cfDNA样本或一gDNA样本的所述基因组的数个区间段的一个或更多个特征、遍及来自一cfDNA样本或一gDNA样本的所述基因组的数个节段的数个特征、一个或更多个拷贝数目畸变的存在及数个经缩减的维度特征。在一些实施例中,所述方法进一步包含自所述对象的一个或更多个白血球获得基因组DNA的序列资料。
在一些实施例中,所述小变异定序化验是一针对性定序化验,及其中所述序列资料是衍生自数个基因的一针对性检测组合。在一些实施例中,侦测癌症的存在是基于分析决定自所述小变异定序化验的两个或更多个特征。在一些实施例中,所述小变异定序化验包含体细胞变异的一总数、非同义变异的一总数、同义变异的总数、每个基因的一体细胞变异的一存在/不存在、已知与癌症相关联的数个特定基因的体细胞变异的一存在/不存在、每个基因的一体细胞变异的一等位基因频率、根据体细胞变异的AF的顺序统计量,及已知与癌症相关联的数个体细胞变异基于它们的等位基因频率的分类中的一个或更多个。。
在一些实施例中,所述分析进一步包含一个或更多个基线特征,及其中所述基线特征包含一个体的一多基因风险分数或数个临床特征,所述临床特征包含年龄、行为、家族史、症状、解剖学观察及外显生殖细胞系癌症带原中的一个或更多个。
在一些实施例中,所述被侦测的癌症是一乳癌、肺癌、大肠直肠癌、卵巢癌、子宫癌、黑色素瘤、肾癌、胰脏癌、甲状腺癌、胃癌、肝胆癌、食道癌、前列腺癌、淋巴瘤、多发性骨髓瘤、头颈癌、膀胱癌、子宫颈癌或以上的任意组合。
在一些实施例中,所述分析进一步包含侦测在所述检测样本中的一种或更多种病毒衍生核酸的存在,及其中所述癌症侦测是部分基于所述一种或更多种病毒核酸的侦测。在一些实施例中,所述一种或更多种病毒衍生核酸是选自于由:人类乳突瘤病毒、埃-巴二氏病毒、B型肝炎病毒、C型肝炎病毒或以上的任意组合所组成的群组。
在一些实施例中,所述检测样本是一血液样本、一血浆样本、一血清样本、一尿液样本、一脑脊髓液样本、一粪便样本、一唾液样本、一胸膜液样本、一心包液样本、一子宫颈拭子、一唾液样本或一腹膜液样本。
在一些实施例中,所述预测性癌症模型是一回归预测器、一随机森林预测器、一梯度提升机、一单纯贝氏分类器、一类神经网络,或一XGBoost模型中的一个。
附图说明
图1A绘示根据一实施例的,基于数个特征产生一癌症预测的一总体流程,所述数个特征衍生自获得自一个体的一cfDNA样本。
图1B至1D中的每个绘示根据一实施例的,使用获得自一个体的至少一cfDNA样本决定一癌症预测的一总体流程图。
图2绘示根据一实施例,用于执行一定序化验以产生数个序列读数的一方法的一流程104。
图3A是根据一实施例的,用于执行一资料工作流程以分析由一小变异定序化验产生的数个序列读数的一示例性流程300。
图3B是根据一实施例的,用于使用不同类型的过滤器及模型处理数个候选变异的一方法的流程图。
图3C是根据一实施例的一贝氏阶层模型的一应用的一图表。
图3D展示根据一实施例,用于决定真实单一核苷酸变异的一贝氏阶层模型的参数及子模型间的依赖关系。
图3E展示根据一实施例,用于决定真实插入或删除的一贝氏阶层模型的参数及子模型间的依赖关系。
图3F至3G例示根据一实施例的,与一贝氏阶层模型相关联的图表。
图3H是根据一实施例,通过拟合一贝氏阶层模型决定数个参数的一图表。
图3I是根据一实施例使用来自一贝氏阶层模型的数个参数决定一伪阳性的一似然性的一图表。
图3J是根据一实施例的用于训练一贝氏阶层模型的一方法的流程图315。
图3K是根据一实施例的用于评分一给定的核苷酸突变的数个候选变异的一方法的流程图325。
图3L是根据一实施例的用于使用一联合模型处理游离核酸样本及基因组核酸样本的一方法的流程图335。
图3M是根据一实施例的一联合模型的一应用的一图表。
图3N是根据一实施例的,来自数个健康个体的数个样本中的数个观察到的变异的计数的一图表。
图3O是根据一实施例的一联合模型的数个示例性参数的一图表。
图3R至3S是根据一实施例的,由一联合模型决定的数个变异辨认的图表。
图3T是根据一实施例的由一联合模型决定的机率密度的一图表。
图3U是根据一实施例的一联合模型的敏感度及特异度的一图表。
图3V是根据一实施例使用一联合模型自小变异定序化验侦测到的一组基因的一图表。
图3W是根据一实施例使用所述联合模型自小变异定序化验侦测到的展示于图17中的所述一组基因的长度分布的一图表。
图3X是根据一实施例使用一联合模型自小变异定序化验侦测到的另一组基因的一图表。
图3Y是根据一实施例的用于调校一联合模型以处理游离核酸样本及基因组核酸样本的一方法的流程图350。
图3Z是根据一实施例的数个cfDNA样本的候选变异的示例性计数的一表格。
图3AA是根据一实施例的来自数个健康个体的数个cfDNA样本的候选变异的示例性计数的一表格。
图3AB是根据一实施例的基于cfDNA及gDNA的比率而绘制的数个候选变异的一图表。
图4A绘示根据一实施例使用数个训练变异产生一人为分布及一非人为分布的一程序400。
图4B绘示根据一实施例被分类在一人为训练资料类别中的数个序列读数。
图4C绘示根据一实施例被分类在所述非人为训练资料类别中的数个序列读数。
图4D绘示根据一实施例被分类在所述参考等位基因训练资料类别中的数个序列读数。
图4E是根据一实施例,用于提取一距边缘统计距离特征的一过程的一示例性描绘。
图4F是根据一实施例,用于提取一显著性分数特征的一过程的一示例性描绘。
图4G是根据一实施例,用于提取一等位基因部分特征的一过程的一示例性描绘。
图4H及4I绘示根据各种实施例,用于辨识数个边缘变异的示例性分布。
图4J绘示根据一实施例,用于决定一样本特定预测比率的一方块图流程。
图4K绘示根据一实施例用于辨识边缘变异的一边缘变异预测模型的应用。
图4L绘示根据一实施例,辨识并报告侦测自一样本的数个边缘变异的一流程452。
图4M至4O中的每个绘示根据各种实施例被分类在所述人为或非人为类别中的一个当中的示例训练变异的数个特征。
图4P绘示根据一实施例对遍及各个对象样本的数个边缘变异的辨识。
图4Q绘示根据一实施例,在使用不同的边缘过滤器移除边缘变异后,作为在cfDNA中被辨认的变异的一部份而在实性肿瘤及cfDNA两者中被辨认的一致变异。
图4R绘示根据一实施例,在使用不同的边缘过滤器移除边缘变异后,作为在实性肿瘤中被辨认的变异的一部份而在实性肿瘤中及cfDNA两者中被辨认的一致变异。
图4S是根据一实施例的,描述用于一游离基因组研究的一样本集合的数个个体的一表格。
图4T是根据一实施例显示与图4S的游离基因组研究的所述样本集合相关联的癌症的类型的一图表。
图4U是根据一实施例显示与图4S的游离基因组研究的所述样本集合相关联的癌症的类型的另一表格。
图4V展示根据一实施例,使用一个或更多个类型的过滤器及模型决定的经辨认变异的示例性计数的图表。
图4W是根据一实施例的,已知具有乳癌的数个样本的数个示例质量分数的一图表。
图4X是根据一实施例的,已知具有各种类型的癌症且处于不同阶段的样本的经辨认变异的示例性计数的一图表。
图4Y是根据一实施例的,已知具有早期或晚期癌症的数个样本的经辨认变异的示例性计数的一图表。
图4Z是根据一实施例的,已知具有早期或晚期癌症的数个样本的经辨认变异的示例性计数的另一图表。
图5A绘示根据一实施例用于决定数个全基因组特征的两个不同的工作流程的一示例性流程。
图5B绘示根据一实施例的,描述用于辨识衍生自cfDNA及gDNA样本的数个区间段及数个节段的特性的分析的一流程。
图5C是根据一实施例的,与一参考基因组的数个区间段相关联的序列读数的一示例性描绘。
图5E及图5F各自绘示横跨一cfDNA样本及一gDNA样本的一基因组的数个区间段的数个区间段分数,所述样本是获得自一乳癌对象。
图5G及图5H各自绘示横跨决定自一cfDNA样本及一gDNA样本的一基因组的数个区间段的数个区间段分数,所述样本是获得自一非癌症个体。
图5I及5J各自绘示横跨决定自一cfDNA样本及一gDNA样本的一基因组的数个区间段的数个区间段分数,所述样本是获得自一非癌症个体。
图6A例示根据一实施例的,用于通过减少高维度资料的所述维度以决定一分类分数的一流程。
图6B绘示根据一实施例的,用于分析资料以减少资料维度的一取样过程。
图6C绘示根据一实施例的,用于基于自具有缩减的维度的资料习得的信息,分析来自一检测样本的资料的一过程。
图6D绘示根据一实施例的,用于资料分析的一取样过程。
图6E绘示将当前方法(分类分数)与一先前已知的分段方法(z分数)进行比较的一表格。
图6F绘示使用所述分类分数法的改进的预测力,对于所有类型的癌症可被观察到。
图7A是根据一实施例,描述用于辨识来自一对象的数个异常甲基化的断片的一过程的一流程图
图7B是根据一实施例的一示例性p值分数计算的一示图。
图7C是根据一实施例,描述基于数个断片的甲基化状态训练一分类器的一过程的一流程图。
图7D至7F是展示对横跨不同癌症阶段的各种癌症而决定的癌症对数胜算比的数张图表。
图8A绘示根据一实施例的,用于决定可以被用于将一患者分层的数个基线特征的一流程。
图8B绘示基于数个基线特征的不同组合的数个模型的表现。
图9A绘示CCGA研究的数个实验参数。
图9B绘示用以为每个各自的预测性癌症模型决定数个特征的数值的数个实验细节(例如,基因检测组合,定序深度等)。
图9C绘示根据展示于图1B中的实施例的,使用数个小变异特征、数个全基因组特征、及数个甲基化特征以预测癌症的存在的一预测性癌症模型的特异度及敏感度的一接收者操作特征(ROC)曲线。
图10A绘示使用一第一组小变异特征预测癌症的存在的一预测性癌症模型的特异度及敏感度的一接收者操作特征(ROC)曲线。
图10B绘示使用一第二组小变异特征预测癌症的存在的一预测性癌症模型的特异度及敏感度的一接收者操作特征(ROC)曲线。
图10C绘示使用一第三组小变异特征预测癌症的存在的一预测性癌症模型的特异度及敏感度的一接收者操作特征(ROC)曲线。
图10D绘示使用一组全基因组特征预测癌症的存在的一预测性癌症模型的特异度及敏感度的一接收者操作特征(ROC)曲线。
图10E绘示使用一第一组甲基化特征预测癌症的存在的一预测性癌症模型的特异度及敏感度的一接收者操作特征(ROC)曲线。
图10F绘示使用一第二组甲基化特征预测癌症的存在的一预测性癌症模型的特异度及敏感度的一接收者操作特征(ROC)曲线。
图10G绘示使用一第三组甲基化特征预测癌症的存在的一预测性癌症模型的特异度及敏感度的一接收者操作特征(ROC)曲线。
图10H绘示数个单一化验预测性癌症模型中的每一个(例如,应用于衍生自所述小变异定序化验、全基因组定序化验及甲基化定序化验中的每一个的表现的数个预测性癌症模型)的表现。
图10I绘示数个预测性癌症模型对横跨数个不同阶段的不同类型的癌症的表现。如在图10I中所展示的,被分析的数个不同癌症类型被分为两组,具有高5年死亡率(≥25%)的癌症,及具有低5年死亡率(<25%)的癌症。
图10J至10L中的每张图绘示(除了展示于图10I中的所述小变异(在图10J至10L中的每张图中被称为非同义变异)、WG及甲基化预测性癌症模型外)额外的数个预测性癌症模型对不同类型的侵袭性癌症的表现。
图10M绘示数个预测性癌症模型对数个不同阶段的大肠直肠癌的表现。
图10N绘示额外的数个预测性癌症模型对数个不同阶段的大肠直肠癌的表现。
图10O绘示数个预测性癌症模型对数个不同阶段及数个不同类型的乳癌的表现。
图10P绘示数个预测性癌症模型对数个不同阶段及数个不同类型的肺癌的表现。
图10Q至10R绘示一多阶段模型的ROC曲线图形,所述多阶段模型包括使用数个小变异特征产生一癌症预测的一第一模型、使用数个WGS特征产生一癌症预测的一第二模型及使用所述第一模型及所述第二模型的所述数个癌症预测产生一癌症预测的一结合模型。
图10S绘示于图10Q至10R中被绘示的所述第一模型、所述第二模型及所述结合模型的敏感性作为敏感性的一函数的一比较。
图10T至10U绘示一多阶段模型的ROC曲线图形,所述多阶段模型包括使用数个WGS特征产生一癌症预测的一第一模型、使用数个甲基化特征产生一癌症预测的一第二模型及使用所述第一模型及所述第二模型的所述数个癌症预测产生一癌症预测的一结合模型。
图10V至10X绘示一多阶段模型的ROC曲线图形,所述多阶段模型包括使用数个基线特征产生一癌症预测的一第一模型、使用数个甲基化特征产生一癌症预测的一第二模型及使用所述第一模型及所述第二模型的所述数个癌症预测在高信号性癌症、肺癌及HR-癌症各自产生一癌症预测的一结合模型。
图11A绘示预测癌症的存在的一个二阶段预测性癌症模型的特异度及敏感度的一ROC曲线。
图12A绘示一单一阶段预测性癌症模型的特异度、敏感度及曲线下面积(AUC)的一ROC曲线,所述单一阶段预测性癌症模型包括数个小变异特征,所述数个小变异特征包含被包括在一针对性基因检测组合中的数个基因当中的非同义变异的最大等位基因频率(MAF)。
图12B绘示一单一阶段预测性癌症模型的特异度、敏感度及曲线下面积(AUC)的一ROC曲线,所述单一阶段预测性癌症模型包括数个小变异特征,所述数个小变异特征包含被包括在一针对性基因检测组合中的数个基因的顺序统计量。
图12C绘示一单一阶段预测性癌症模型的特异度、敏感度及曲线下面积(AUC)的一ROC曲线,所述单一阶段预测性癌症模型包括数个甲基化特征。
图12D绘示一单一阶段预测性癌症模型的特异度、敏感度及曲线下面积(AUC)的一ROC曲线,所述单一阶段预测性癌症模型包括数个小变异及甲基化特征。
图12E绘示一单一阶段预测性癌症模型的特异度、敏感度及曲线下面积(AUC)的一ROC曲线,所述单一阶段预测性癌症模型包括数个全基因组定序(WGS)特征。
图12F绘示一单一阶段预测性癌症模型的特异度、敏感度及曲线下面积(AUC)的一ROC曲线,所述单一阶段预测性癌症模型包括数个小变异及WGS特征。
图12G绘示一单一阶段预测性癌症模型的特异度、敏感度及曲线下面积(AUC)的一ROC曲线,所述单一阶段预测性癌症模型包括数个甲基化及WGS特征。
图12H绘示一单一阶段预测性癌症模型的特异度、敏感度及曲线下面积(AUC)的一ROC曲线,所述单一阶段预测性癌症模型包括数个小变异、甲基化及WGS特征。
图12I绘示一单一阶段预测性癌症模型的特异度、敏感度及曲线下面积(AUC)的一ROC曲线,所述单一阶段预测性癌症模型包括数个基线特征。
图12J绘示一单一阶段预测性癌症模型的特异度、敏感度及曲线下面积(AUC)的一ROC曲线,所述单一阶段预测性癌症模型包括数个基线及WGS特征。
图12K绘示一单一阶段预测性癌症模型的特异度、敏感度及曲线下面积(AUC)的一ROC曲线,所述单一阶段预测性癌症模型包括数个基线及甲基化特征。
图12L绘示一单一阶段预测性癌症模型的特异度、敏感度及曲线下面积(AUC)的一ROC曲线,所述单一阶段预测性癌症模型包括数个基线、小变异及甲基化特征。
图12M绘示一单一阶段预测性癌症模型的特异度、敏感度及曲线下面积(AUC)的一ROC曲线,所述单一阶段预测性癌症模型包括数个基线及WGS特征。
图12N绘示一单一阶段预测性癌症模型的特异度、敏感度及曲线下面积(AUC)的一ROC曲线,所述单一阶段预测性癌症模型包括数个基线、小变异及WGS特征。
图12O绘示一单一阶段预测性癌症模型的特异度、敏感度及曲线下面积(AUC)的一ROC曲线,所述单一阶段预测性癌症模型包括数个基线、甲基化及WGS特征。
图12P绘示一单一阶段预测性癌症模型的特异度、敏感度及曲线下面积(AUC)的一ROC曲线,所述单一阶段预测性癌症模型包括数个基线、小变异、甲基化及WGS特征。
具体实施方式
附图与下文的描述仅以例示性的方式与优选的实施例相关联。应当注意的是,由下文的讨论,在本文中被揭示的结构及方法的替代实施例将被认为是可以在不背离权利要求所主张之物的原则下被使用的可实施替代方案。
现在将详细参考数个实施例,被参考的实施例的数个示例将被例示在附随的附图中。应注意到的是,在可行处,类似的或相似的参考标号可以被用在附图中,且可以指示类似的或相似的机能。举例而言,在一参考标号后的一字母,比如“预测性癌症模型170A”,指示内文是特定地指涉具有该特定参考标号的元件。在内文中的,没有一后续字母的一参考标号,比如“预测性癌症模型170A”,指涉在附图中的任何或所有带有该参考标号的元件(例如,在内文中的“预测性癌症模型170”意指附图中的“预测性癌症模型170A”及/或“预测性癌症模型170B”)。
“个体”一词意指一人类个体。“健康个体”一词意指被假定为没有一癌症或疾病的一个体。“对象”一词意指已知具有,或潜在具有一癌症或疾病的一个体。
“序列读数”一词意指来自获得自一个体的一样本的核苷酸序列读数。序列读数可以经由本技艺中习知的各种方法被获得。
“读数节段”或“读数”等词汇意指任何核苷酸序列,包括获得自一个体的序列读数及/或衍生自初始序列读数的核苷酸序列,所述初始序列读数来自获得自一个体的一样本。
“单核苷酸变异”或“SNV”等词汇意指在一核苷酸序列,例如,来自一个体的一序列读数,的一位置(例如,位点),一核苷酸替换为一不同的核苷酸。自一第一核碱基X至一第二核碱基Y的一替换可以被记为“X>Y”。举例而言,胞嘧啶至胸腺嘧啶的一SNV可以被记为“C>T”。
“插入或缺少”一词意指在一序列读数中具有一长度及一位置(也被称为一锚点位置)的一个或更多个碱基对的任何插入或删除。一插入对应到一正长度,而一删除对应到一负长度。
“突变”一词意指一个或更多个SNV或插入或缺少。
“真实”或“真实阳性”等词汇意指指示真实生物学情况,举例而言,指示在一个体中的一潜在的癌症、疾病或生殖细胞系突变的存在的一突变。真实突变是衍生自肿瘤的突变,且非由在健康个体中自然发生的突变(例如,频发突变)或其它人为来源比如在核酸样本的化验制备中的处理错误所造成。
“伪阳性”一词意指被不正确地判定为一真实阳性的一突变。
“游离核酸”、“游离DNA”或“cfDNA”等词汇意指在一个体的身体(例如,血流)中循环且源自一个或更多个健康细胞及/或源自一个或更多个癌细胞的核酸断片。此外,cfDNA可能来自其它来源比如病毒、胎儿等。
“基因组核酸”、“基因组DNA”或“gDNA”等词汇意指包括染色体DNA的核酸,所述染色体DNA源自一个或更多个健康(例如,非肿瘤)细胞。在各种实施例中,gDNA可以自一细胞被提取,所述细胞衍生自一血液细胞谱系,比如是一白血球。
“循环肿瘤DNA”或“ctDNA”等词汇意指源自肿瘤细胞或其它类型的癌细胞的核酸断片,所述核酸断片可能作为生物性过程比如濒死细胞的细胞凋亡(apoptosis)或坏死(necrosis)的结果,或被活肿瘤细胞主动地释放而进入一个体的血流。
“替代等位基因”或“ALT”等词汇意指相对于一参考等位基因,例如,对应于一已知基因,具有一个或更多个突变的一等位基因。
“定序深度”或“深度”等词汇意指来自获得自一个体的一样本的数个读数序列的一总数。
“替代深度”或“AD”等词汇意指支持一ALT,例如,包括所述ALT的突变,的一样本中的数个读数节段的一数量
“参考深度”一词意指在一样本中,在一候选变异位置包括一参考等位基因的数个读数节段的一数量。
“变异”或“真实变异”等词汇意指位于基因组中的一位置的一突变核苷酸碱基。这样的一变异可能导致在一个体中的癌症发展及/或进展。
“候选变异”、“经辨认变异”或“推定变异”意指一核苷酸序列的,举例而言,在所述基因组中被判定为经突变的一位置的一核苷酸序列的一个或更多个被侦测到的核苷酸变异。一般地,一核苷酸碱基是基于获取自一样本的数个序列读数上的一替代等位基因的存在而被视为一经辨认变异,在所述样本中,所述数个序列读数各自跨越所述基因组中的所述位置。一候选变异的来源可能起初是未知的或未确定的。在处理时,数个候选变异可以被与预期来源比如gDNA(例如,衍生自血液的gDNA)或受癌症影响的细胞(例如,肿瘤衍生的细胞)相关联。此外,数个候选变异可以被辨认为真实阳性。
“拷贝数目畸变”或“CNAs”等词汇意指在体肿瘤细胞(somatic tumor cells)中的拷贝数目的改变。举例而言,CNAs可以意指在一实性肿瘤中的拷贝数目改变。
“拷贝数目变异”或“CNVs”等词汇意指衍生自生殖细胞系细胞的拷贝数目变化或衍生自非肿瘤细胞中的体细胞拷贝数目变化的变化。举例而言,CNVs可以意指可能由于克隆性造血(clonal hematopoiesis)而在白血球中出现的拷贝数目变化。
“拷贝数目事件”一词意指一拷贝数目畸变及一拷贝数目变异中的一个或两者。
1.产生一癌症预测
1.1.整体处理流程
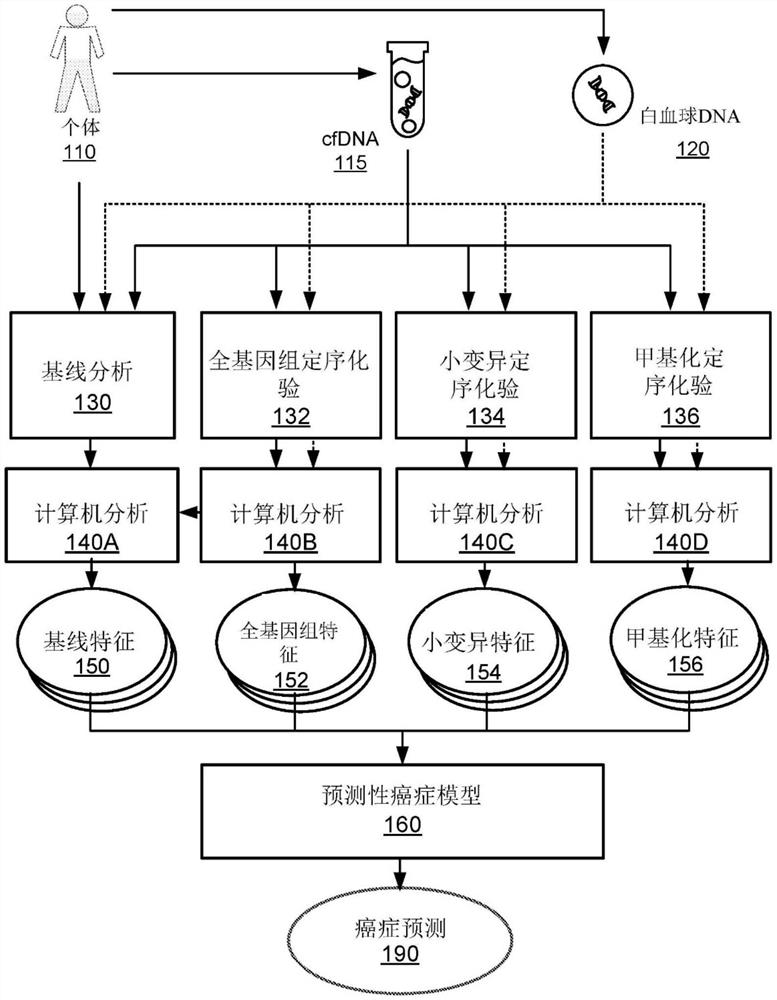
图1A绘示根据一实施例的,基于数个特征产生一癌症预测的一总体流程100,所述数个特征衍生自获得自一个体的一cfDNA样本。进一步参考图1B至1D,其中的每个绘示根据一实施例的,使用获得自一个体的至少一cfDNA样本决定一癌症预测的一总体流程图。
在步骤102,一检测样本自所述个体被获得。一般地,样本可以来自健康对象、已知具有或被怀疑具有癌症的对象,或没有已知先验信息(prior information)的对象。所述检测样本可以是选自于由血液样本、血浆样本、血清样本、尿液样本、粪便样本及唾液样本所组成的一群组的一样本。替代地,所述检测样本可以包含选自于由全血样本、一血液部分样本、一组织活检样本、胸膜液样本、心包液样本、脑脊髓液样本及腹膜液样本所组成的群组的一样本。
如在图1B至1D中的每张图中所展示的,一检测样本可以包括cfDNA115。在各种实施例中,一检测样本可以包括基因组DNA(gDNA)。如在图1B至1D中所展示的,gDNA的一来源的一示例是白血球(WBC)DNA120。
在步骤104中,一个或更多个物理过程分析被执行,至少一个物理过程分析包括对cfDNA115的一基于定序的化验,以产生数个序列读数。参考图1B至1D,物理过程分析的数个示例可以是个体110的一基线分析130或对cfDNA115的一基于定序的化验,比如一全基因组定序化验132、一小变异定序化验134或一甲基化定序化验136的执行。
所述个体110的一基线分析130可以包括所述个体110的一临床分析,及可以由一医师或一医学专业人员执行。在一些实施例中,所述基线分析130可以包括生殖细胞系改变的一分析,所述生殖细胞系改变可以在所述个体110的所述cfDNA115中被侦测到。在一些实施例中,所述基线分析130可以以额外信息,比如被上调或下调的基因的一辨识,执行生殖细胞系改变的所述分析。在其它实施例中,所述基线分析包括数个临床特征(例如,癌症的已知风险因子,比如一对象的年龄、种族、身体质量指数(BMI)、吸烟史、酒类摄取及/或家族癌症史)的分析。这样的额外信息可以由一计算机分析提供,比如如在图1B至1D中绘示的计算机分析140B。所述基线分析130在下文中被进一步描述。
如在下文中所使用地,一小变异定序化验意指产生数个序列读数的一物理化验,所述物理化验典型地经由可以被用于决定数个小变异的针对性基因定序检测组合产生所述数个序列读数。所述数个小变异的数个示例包括单核苷酸变异(SNVs)及/或插入或缺少。替代地,如本领域中的一般技术人员将理解到的,小变异的评估也可以使用一全基因组定序方式或一全外显子组定序方式而被达成。
如在下文中被使用地,一全基因组定序化验意指一物理化验,所述物理化验产生一全基因组或所述全基因组的一大部分的数个序列读数,所述数个序列读数可以被用于判定大型变异,比如拷贝数目变异或拷贝数目畸变。这样的一个物理化验可以应用全基因组定序技术或全外显子组定序技术。
如在下文中被使用地,一甲基化定序化验意指一物理化验,所述物理化验产生数个序列读数,所述数个序列读数可以被用于判定遍及所述基因组的数个CpG位点的所述甲基化状态或甲基化模式。一甲基化定序化验的一示例可以包括用于将未甲基化的胞嘧啶(例如,数个CpG位点)(例如,使用EZ DNA甲基化-黄金套组(EZ DNA Methylation–Goldkit)或一EZ DNA甲基化-闪电套组(EZ DNA Methylation–Lightning kit)(可自ZymoResearch Corp获得))转化为脲嘧啶的亚硫酸氢盐处理。替代地,一酶转化步骤(例如,使用一胞嘧啶脱氨酶(cytosine deaminase)(比如APOBEC-Seq(可自NEBiolabs获得)))可以被用于将未甲基化胞嘧啶转化为脲嘧啶。在转化后,所述经转化的cfDNA分子可以经由一全基因组定序过程或一针对性基因定序检测组合而被定序,且数个序列读数被用于评估在数个CpG位点处的甲基化状态。基于甲基化的定序方式在本领域中是已知的(例如,参见美国专利第2014/0080715号及USSN第16/352,602号,上述申请通过引用被并入本文中)。在另一实施例中,DNA甲基化可以在处于其它上下文中,例如CHG及CHH中的胞嘧啶中发生,其中H是腺嘌呤、胞嘧啶或胸腺嘧啶。处于5-羟甲基胞嘧啶(5-hydroxymethylcytosine)形式的胞嘧啶甲基化及其特征也可以使用本文中揭示的方法及程序而被评估(参见,例如,WO2010/037001号及WO2011/127136号,上述申请通过引用被并入本文中)。在一些实施例中,一甲基化定序化验不需要执行一碱基转化步骤以判定遍及所述基因组的数个CpG位点的甲基化状态。举例而言,这样的甲基化定序化验可以包括PacBio定序或牛津纳米孔定序(OxfordNanopore sequencing)。
所述全基因组定序化验132、小变异定序化验134及甲基化定序化验136中的每个是在所述cfDNA115上被执行以产生所述cfDNA115的数个序列读数。在各种实施例中,所述全基因组定序化验132、小变异定序化验134及甲基化定序化验136中的每个是进一步在所述WBC DNA120上被执行以产生所述WBC DNA120的数个序列读数。在所述全基因组定序化验132、小变异定序化验134及甲基化定序化验136中的每个中被执行的所述数个处理步骤与图2相关联地被进一步详细描述。
在步骤106,作为执行所述基于定序的化验的结果而被产生的所述数个序列读数被处理,以决定数个特征的数值。特征,一般而言,是可获得自物理化验及/或计算机分析的数种类型的信息,可以被用于预测在一个体中的癌症。一般地,用于辨识在一个体中的癌症的任何给定的预测性模型包括一个或更多个特征作为所述模型的组成部分。对于任何给定的患者或样本,一特征会具有一个决定自所述物理及/或计算机分析的数值。这些数值被输入到所述预测性模型中,以产生所述模型的一输出。
数个序列读数通过应用一计算机分析而被处理。一般地,每个计算机分析140代表可以由一处理器或一计算机,下文中称为一处理系统,所执行的一演算法。因此,每个计算机分析分析数个序列读数,及输出基于所述数个序列读数的数值。每个计算机分析是特定用于一给定的基于定序的化验,及因此,每个计算机分析输出一特定类型的,所述基于定序的化验特有的特征。
如图1B至1D中所展示的,产生自应用一全基因定序化验132的应用的数个序列读数被以计算机分析140B处理,所述计算机分析140B又被称为一全基因组计算机分析。所述计算机分析140B输出数个全基因组特征152。产生自一小变异定序化验的应用的数个序列读数被以一计算机分析140C处理,所述计算机分析140C又被称为一小变异计算机分析。所述计算机分析140C输出数个小变异特征154。产生自一甲基化定序化验的应用的数个序列读数被以一计算机分析140D处理,所述计算机分析140D又被称为一甲基化计算机分析。所述计算机分析140C输出数个甲基化特征156。此外,计算机分析140A分析来自所述基线分析130的信息并输出数个基线特征150。
在步骤108,一预测性癌症模型被应用到所述数个特征以产生对所述个体110的一癌症预测。一癌症预测的数个示例包括癌症的一存在或不存在、癌症的一发源组织、癌症的一严重性、阶段、一分级、一癌症亚型、一治疗决定及对一治疗有反应的一似然性。在各种实施例中,由所述预测性癌症模型输出的所述癌症预测是一分数,比如一似然性或机率,所述似然性或机率指示:癌症的一存在或不存在、癌症的一发源组织、癌症的一严重性、阶段、一分级、一癌症亚型、一治疗决定及对一治疗有反应的一似然性中的一项或更多项。
一般地,任何这样的分数可以或者是单一的,比如癌症一般地存在或不存在,一特定类型的癌症的存在/不存在。替代地,这样的分数可以是复数的,而使所述预测性癌症模型的所述输出可以,举例而言,是代表数个类型的癌症中的每个的存在/不存在的一分数、代表数个类型的癌症中的每个的严重性/分级的一分数、代表特定cfDNA源自数种类型的组织中的每个的所述似然性的一分数等。为了描述的清晰性,所述预测性癌症模型的所述输出被一般地称作一组分数,视所述预测性癌症模型是被配置以决定何物,所述组包含一个或更多个分数。
所述预测性癌症模型可以基于所述预测性癌症模型的数个特定特征而被不同地建构。举例而言,所述预测性癌症模型可以包括一个、两个、三个或四个不同类型的特征,比如所述数个基线特征150、数个全基因组特征152、数个小变异特征154及数个甲基化特征156。在一些实施例中,可能有四个分离的预测性癌症模型,每个预测性癌症模型被建构以包括一个类型的特征。在一些实施例中,所述预测性癌症模型是一个两阶段模型,所述两阶段模型包括一第一组子模型及一第二子模型,所述第一组子模型中的每个包括一个类型的特征,所述第二子模型分析所述第一组子模型的输出以决定一癌症预测。每个被特定地架构的预测性癌症模型在下文中与一处理工作流程相关地被描述,所述处理工作流程产生所述预测性癌症模型接收的一个或更多个类型的数个特征的数值。如在下文中所使用地,一工作流流程意指所述物理过程分析、计算机分析的所述表现及一预测性癌症模型的应用。
在各种实施例中,自不同的数个计算机分析输出的数个特征的数值被输入到一单一预测性癌症模型中,以产生一癌症预测。举例而言,参考图1B,数个基线特征150、数个全基因组特征152、数个小变异特征154及数个甲基化特征156中的每个的数值可以被编译(例如,成为一特征向量)并作为输入提供到一预测性癌症模型160。所述预测性癌症模型160输出基于所述被提供的数个特征的所述癌症预测190。
在各种实施例中,一单一工作流流程被执行,以产生一单一癌症预测190,而不需要执行其他工作流流程。因此,自一单一计算机分析输出的数个特征的数值被输入到一单一预测性癌症模型以产生一癌症预测。举例而言,参考图1C,为了产生癌症预测190A,一个体110、cfDNA115及/或WBC DNA120使用一基线分析132,分析所述基线分析130的输出的计算机分析140B而被分析,以获得数个基线特征150的数值,且数个基线特征150的数值作为输入被提供到所述预测性癌症模型170A。作为另一示例,为了产生癌症预测190B,cfDNA115及/或WBC DNA120使用一全基因组定序分析132,分析由所述全基因组定序分析132产生的数个序列读数的计算机分析140B而被分析,以获得数个全基因组特征152的数值,且数个全基因组特征152的所述数值作为输入被提供到所述预测性癌症模型170B。在又一示例中,为了产生癌症预测190C,cfDNA 115及/或WBC DNA120使用一小变异定序分析134,分析由所述小变异定序分析134产生的数个序列读数的计算机分析140C而被分析,以获得数个小变异特征154的数值,且数个小变异特征154的所述数值作为输入被提供到所述预测性癌症模型170C。在又一示例中,为了产生癌症预测190D,cfDNA115及/或WBC DNA120使用一甲基化定序分析136,分析由所述甲基化定序分析136产生的数个序列读数的计算机分析140D而被分析,以获得数个甲基化特征156的数值,且数个甲基化特征156的所述数值作为输入被提供到所述预测性癌症模型170D。
在各种实施例中,一预测性癌症模型(例如,预测性癌症模型170A至170D中的任何一个)可以基于两个类型的特征(例如,选自于数个基线特征150、数个全基因组特征152、数个小变异特征154及数个甲基化特征156的两个特征)产生一癌症预测190A至190D。在一些实施例中,一预测性癌症模型可以基于三个类型的特征(例如,选自于数个基线特征150、数个全基因组特征152、数个小变异特征154及数个甲基化特征156的三个特征)产生一癌症预测190A至190D。
在各种实施例中,一个两阶段预测性癌症模型被应用到所述数个特征以产生一癌症预测,举例而言,在一第一阶段,输出自数个计算机分析中的每个的数个特征的所述数值被分别地输入个别的子模型中。在一第二阶段,每个个别的子模型的输出作为输入被提供到一总体子模型以产生一癌症预测。图1D例示一个两阶段预测性癌症模型195的一示例。在此,数个基线特征150作为输入被提供到预测性模型180A、数个全基因组特征152作为输入被提供到预测性模型180B、数个小变异特征154作为输入被提供到预测性模型180C,及数个甲基化特征156作为输入被提供到预测性模型180D。预测性模型180A、180B、180C及180D中的每个的输出可以作为输入到所述总体预测性模型185中的输入。在各种实施例中,预测性模型180A、180B、180C及180D中的每个的输出是一个或更多个分数。因此,所述总体预测性模型185基于所述一个或更多个分数产生一癌症预测190。
虽然图1D绘示四个分离的预测模型180A、180B、180C及180D的所述输出是作为输入被提供到所述总体预测性模型185,在各种实施例中,额外的或更少的预测模型可以在提供一输入到所述总体预测性模型185中被涉及。举例而言,在一些实施例中,所述四个预测性模型180A、180B、180C及180D中的一个、两个或三个输出作为输入被提供到所述总体预测性模型185的信息。
在各种实施例中,由所述预测性模型180A、180B、180C及180D中的每个输出的数个分数的数量可以相异。举例而言,预测性模型180B可以输出一组分数(在下文中称为一“WGS分数”)、预测性模型150C可以输出一组两个分数(在下文中称为“变异基因分数”及“排序分数”)及预测性模型180C可以输出一组三个分数(在下文中称为“MSUM分数”、“WGBS分数”及“二元分数”)。
在所述预测性癌症模型的所述数个不同实施例的每个中(例如,图1B中的预测性癌症模型160、预测性癌症模型170A至170D,或图1D中的预测性癌症模型195),每个预测性癌症模型可以是一决策树(decision tree)、一系综(例如,套袋(bagging)、提升(boosting)、随机森林(random forest))、梯度提升机、线性回归、单纯贝氏分类器、类神经网络,XGBoost或逻辑回归中的一个。每个预测性癌症模型包括在训练时被调整的,对所述数个特征的数个习得的权重。权重一词在此一般地被用来代表与一模型的任何给定特征相关的习得的量值,无论使用何种特定的机器学习技术。
在训练时,训练资料被处理以产生数个特征的数值,所述特征被用于训练所述预测性癌症模型的所述数个权重。作为一示例,训练资料可以包括获得自数个训练样本的cfDNA及/或WBC DNA,以及一输出标签。举例而言,所述输出标签可以是所述个体是否已知是罹癌的或已知没有癌症(例如,健康),发源癌症组织的一指示,或所述癌症的一严重性的一指示。视展示于图1B至1D中的特定实施例而定,所述预测性癌症模型接收获得自一个或更多个所述物理化验及计算机分析的一个或更多个所述特征的数值,所述一个或更多个所述物理化验及计算机分析与要被训练的所述模型相关。取决于由所述训练中模型输出的所述数个分数及所述训练资料的所述数个输出标签间的差异,所述预测性癌症模型的所述数个权重被最佳化,使所述预测性癌症模型能够做出更加准确的预测。在各种实施例中,一预测性癌症模型可以是一非参数性模型(例如,k-近邻演算法)及因此,所述预测性癌症模型可以不须最佳化参数而被训练以做出更准确的做出预测。
经训练的预测性癌症模型可以被储存及在需要时,举例而言,在图1A的步骤108中的部署时被随后取回。
1.2.物理化验
图2是根据一实施例,用于执行一物理化验(例如,一定序化验)以产生数个序列读数的一方法的一流程图。在此,所述流程图更加详细地绘示图1A的步骤104。所述方法104包括,但不限于下述步骤。举例而言,所述方法104的任何步骤可以包含用于质量控制的一定量次步骤或为本领域的一般技术人员所知的其他实验室化验程序。
一般地,所述数个步骤(例如,步骤205至235)的各种次组合为所述全基因组定序化验、小变异定序化验及甲基化定序化验中的每一个而被执行。特定地,步骤205、215、230及235是为所述全基因组定序化验而被执行。步骤205及215至235是为所述小变异定序化验而被执行。在一些实施例中,步骤205至235中的每一个是为所述甲基化定序化验而被执行。举例而言,应用一针对性基因检测组合亚硫酸氢盐定序的一甲基化定序化验应用步骤205至235中的每个。在一些实施例中,步骤205至215及230至235是为所述甲基化定序化验而被执行。举例而言,应用全基因组亚硫酸氢盐定序的一甲基化定序化验不需要执行步骤220及225。
在步骤205,核酸(DNA或RNA)自一检测样本中被提取。在当前的揭示中,除非另有指示,DNA及RNA可以被可互换地使用。亦即,在变异辨认及质量控制中使用错误来源信息(error source information)的下述数个实施例对于DNA及RNA类型的核酸序列皆适用。然而,为了清晰性及解释的目的,在本文中所描述的数个示例可能聚焦在DNA上。在各种实施例中,DNA(例如,cfDNA)经由一纯化程序被自所述检测样本提取。一般地,任何本领域中已知的方法皆可以被用于纯化DNA。举例而言,核酸可以通过在一管中粒化(pelleting)及/或沉淀所述核酸而被分离。被提取的核酸可以包括cfDNA,或可以包括gDNA比如WBC DNA。
在步骤210中,所述数个cfDNA断片被处理以将未甲基化的胞嘧啶转化为脲嘧啶。在一实施例中,所述方法使用所述DNA的一亚硫酸氢盐处理,所述处理将所述未甲基化的胞嘧啶转化为脲嘧啶而不转化经甲基化的胞嘧啶。举例而言,一商业套组比如一EZ DNA甲基化–黄金(EZ DNA METHYLATION–Gold)、EZ DNA甲基化–直接(EZ DNA METHYLATION–Direct)或EZ DNA甲基化–闪电(EZ DNA METHYLATION–Lightning)套组(可获得自加利福尼亚州尔湾市Zymo Research Corp)被用于所述亚硫酸氢盐转化。在另一实施例中,未甲基化胞嘧啶至脲嘧啶的所述转化是使用一酶反应而被达成。举例而言,所述转化可以使用市面上可获得的一套组,比如APOBEC-Seq(麻萨诸塞州伊普斯维奇,NEBiolabs)进行未甲基化胞嘧啶至脲嘧啶的转化。
在步骤215,一序列文库(sequencing library)被制备。在文库制备时,转接子(adapters),举例而言,包括用于随后的簇生成(cluster generation)及/或定序的一个或更多个定序寡核苷酸(例如,用于合成式定序(sequencing by synthesis(SBS))的已知的P5及P7序列(Illumina,加利福尼亚州圣地亚哥)),经由转接子连接(adapter ligation)被连接到所述数个核酸断片的末端。在一实施例中,数个唯一分子识别码(unique molecularidentifiers,UMI)在转接子连接时被添加到被提取的核酸。所述数个UMI是在转接子连接时被添加到核酸的末端的短核酸序列(例如,4至10个碱基对)。在一些实施例中,UMI是简并的(degenerate)碱基对,作为可以被用以辨识获得自核酸的数个序列读数的一独特标签。如随后所描述的,所述UMI可以在放大过程中与所附着的核酸一同被进一步复制,这提供了在下游分析中辨识源自相同原始核酸断片的数个序列读数的一方式。
在步骤220中,杂合探针被用以富集(enrich)一选定核酸组的一序列文库。数个杂合探针可以被设计以针对目标核酸序列并与所述序列杂合以拉下(pull down)并富集所针对的数个核酸断片,所述数个核酸断片对癌症(或疾病)的存在或不存在、癌症状态、或一癌症分类(例如,癌症类型或发源组织)可能提供信息。依照此步骤,数个杂合拉下探针可以被用于一给定的目标序列或基因。所述数个探针可以于长度上在自约40至约160碱基对(bp)的范围内、自约60至约120碱基对的范围内,或自约70至约100碱基对的范围内。在一实施例中,所述数个探针覆盖所述目标区域或基因的数个相重叠部分。对于针对性基因检测组合定序,所述数个杂合探针是被设计以针对及拉下数个核酸断片,所述数个核酸断片是衍生自被包括在所述针对性基因检测组合中的数个特定基因序列。对于全外显子组定序,所述数个杂合探针是被设计以针对及拉下数个核酸断片,所述数个核酸断片是衍生自在一参考基因组中的数个外显子序列。如本领域中的一般技术人员将轻易理解的,本领域中用于核酸的针对性富集的其它已知方式可以被使用。
在一杂合步骤220后,所述经杂合的数个核酸断片被富集225。举例而言,所述经杂合的数个核酸断片可以使用PCR被捕捉及放大。所述数个目标序列可以被富集以获得数个经富集的序列,所述数个经富集的序列可以随后被定序。这增进了数个序列读数的所述定序深度。
在步骤230中,所述核酸被定序以产生数个序列读数。数个序列读数可以以在本领域中已知的方式被获取。举例而言,数种技术及平台直接平行地从数百万个个别核酸(例如,DNA,比如cfDNA或gDNA)获得数个序列读数。这样的技术可以适宜于执行针对性基因检测组合定序、全表现子组定序、全基因组定序、针对性基因检测组合亚硫酸氢盐定序,及全基因组亚硫酸氢盐定序中的任何程序。
作为一第一示例,数种合成式定序科技依赖当数个萤光核苷酸被并入与被定序的所述模板互补的DNA的一新生股(nascent strand)中时,对所述数个萤光核苷酸的侦测。在一方法中,数个30至50碱基长的寡核苷酸在5'端被共价地锚定到数个玻璃盖玻片。这些被锚定的股执行两个功能。第一,若数个模板股被配置有与所述数个连结于表面的寡核苷酸互补的捕捉尾(capture tail),所述被锚定的股作为对所述数个目标模板股的数个捕捉位点。它们也作为供模板导向引子延伸的引子(primers),所述引子延伸形成所述序列读数的基础。所述数个捕捉引子作为用于序列判定的一固定位置位点而发挥功能,所述序列判定使用合成、侦测及化学切割染剂连结物以移除所述染剂的多个循环。每个循环由添加所述聚合酶/经标记核苷酸混合物、冲洗(rinsing)、成像及染剂的切割所组成。
在一替代方法中,聚合酶被以一萤光供体(donor)分子修饰并在一玻璃玻片上被固定化,而每个核苷酸被以连结到一伽马磷酸盐(gamma-phosphate)的一个受体萤光部分色彩编码(color-coded)。所述系统侦测当一核苷酸被并入到新炼(de novo chain)中时,一经萤光标记的聚合酶及一经萤光修饰的核苷酸间的互动。
任何适宜的合成式定序平台可以被用以辨识突变。数个合成式定序平台包括来自罗氏/454生命科学(Roche/454Life Sciences)的Genome Sequencers、来自Illumina/SOLEXA的GENOME ANALYZER、来自应用生物系统(Applied BioSystems)的SOLID系统及来自螺旋生物科学(Helicos Biosciences)的HELISCOPE系统。数个合成式定序平台也由太平洋生物科学公司(Pacific BioSciences)及VisiGen生物科技(VisiGen Biotechnologies)所描述。在一些实施例中,被定序的数个核酸分子是被连结到一支撑物(例如,固体支撑)。为了在一支撑物上固定化所述核酸,一捕捉序列/通用引子位点可以被添加在所述模板的3'端及/或5'端。所述数个核酸可以通过将所述捕捉序列杂合到被共价地连结到所述支撑物的一互补序列而被连结到所述支撑物。所述捕捉序列(也被称为一通用捕捉序列)是与被连结到一支撑物的一序列互补的一核酸序列,可以双重地作为一通用引子。
作为一捕捉序列的一替代,一耦合对(coupling pair)(比如,例如,抗体/抗原、受体/受质或亲和素-生物素配对)的一成员可以被连结到要被捕捉到一表面上的每个断片,所述表面被涂覆以所述耦合对的一对应的第二成员。在所述捕捉后,所述序列可以被分析,举例而言,通过单分子侦测/定序,包括模板依赖性合成式定序而被分析。在合成式定序中,所述连结于表面的分子在聚合酶存在时被暴露于数个经标记的三磷酸核苷酸。所述模板的所述序列通过数个经标记的核苷酸被并入所述成长炼的所述3'端的顺序而被判定。这可以实时地被完成或可以以步进重复(step-and-repeat)模式被完成。对于实时分析,对每个核酸的不同光学标记可以被并入,及多道雷射可以被应用于数个被并入的核苷酸的激发。
大量平行定序或次世代定序(NGS)技术,包括合成科技(synthesis technology)、焦磷酸测序(pyrosequencing)、离子半导体科技(ion semiconductor technology)、单分子实时定序(single-molecule real-time sequencing)、连接式定序(sequencing byligation)、纳米孔定序(nanopore sequencing)或配对末端定序(paired-endsequencing)。大量平行定序平台的数个示例是Illumina HISEQ或MISEQ、ION PERSONALGENOME MACHINE、PACBIO RSII定序器(PACBIO RSII sequencer)或SEQUEL系统(SEQUELSystem)、Qiagen’s GENEREADER及Oxford MINION。另外的类似的当前大量平行定序科技,以及这些科技的未来世代可以被使用。
在步骤230,所述数个序列读数可以使用本领域中已知的方法被对齐到一参考基因组,以决定对齐位置信息。所述对齐位置信息可以指示在所述参考基因组中的一区域的一起始位置及一末端位置,所述起始位置及所述结束位置对应于一给定序列读数的一起始核苷酸碱基及末端核苷酸碱基。对齐位置信息也可以包括序列读数长度、所述序列读数长度可以自所述起始位置及末端位置被判定。在所述参考基因组中的一区域可以与一基因或一基因的一节段相关联。
在各种实施例中,一序列读数包含被记为R
在步骤235后,所述被对齐的数个序列读数使用一计算机分析,比如如上文中所描述并展示于图1D中的计算机分析140B、140C或140D被处理。所述小变异计算机分析140C、全基因组计算机分析140B、甲基化计算机分析140D及基线计算机分析在下文中被进一步详述。
2.小变异计算机分析
2.1.小变异特征
所述小变异计算机分析140C接收由所述小变异定序化验134产生的数个序列读数,并基于所述数个序列读数决定数个所述小变异特征154的数值。如先前描述的,所述小变异定序化验可以是一基于定序的化验,典型地经由可以被用于决定数个小变异的数个针对性基因定序检测组合产生数个序列读数。所述数个小变异的数个示例包括单核苷酸变异(SNVs)及/或插入或缺少。替代地,如本领域的一般技术人员将理解到的,小变异的评估也可以使用一全基因组定序方式或一全外显子组定序方式被达成。小变异特征154的数个示例包括下述的任何特征:在一对象的cfDNA中的数个体细胞变异的一总数、数个非同义变异的一总数、数个同义变异的一总数、在一基因检测组合中的每个基因的数个体细胞变异的一存在/不存在、已知与癌症相关联的特定数个基因的数个体细胞变异的一存在/不存在、在一基因检测组合中的每个基因的一体细胞变异的一等位基因频率(AF)、在一基因检测组合中的每个基因的一体细胞变异的一最大AF、由一公开可用数据库,比如oncoKB指定的每个类别的一体细胞变异的一AF,及根据数个体细胞变异的AF的数个体细胞变异的一排名顺序。
一般地,所述数个小变异特征154的所述数个特征值是在准确辨识可能指示在所述个体中的癌症的数个体细胞变异上被预测。所述小变异计算机分析140C辨识数个候选变异及从所述数个候选变异中,分辨可能存在所述个体的所述基因组中的数个体细胞变异及不太可能对在所述个体中的癌症有预测性的数个伪阳性变异。更特定地,所述小变异计算机分析140C在考虑到干扰信号比如噪声及/或可以被归因于一基因性来源(例如,来自gDNA或WBC DNA)的变异下,辨识可能衍生自一体细胞来源的存在于cfDNA中的数个候选变异。此外,数个候选变异可以被过滤以移除伪阳性变异,所述伪阳性变异可能由于一人为因素而发生及因此并非指示在所述对象中的癌症。作为一示例,数个伪阳性变异可能是在数个序列读数的边缘处或在接近所述边缘处被侦测到的数个变异,所述变异是由于自发胞嘧啶脱氨及末端修复错误而发生。因此,在滤掉伪阳性变异后存留的数个体细胞变异,及其数个特征,可以被用于决定所述数个小变异特征。
对于数个体细胞变异的所述总数的所述特征,所述小变异计算机分析140C加总在所述基因组或基因检测组合各处被辨识的数个体细胞变异。因此,对于获得自一个体的一cfDNA样本,数个体细胞变异的所述总数的所述特征被表示为在所述样本的所述cfDNA中被辨识的数个体细胞变异的所述总数的一单一,数字性数值。
对于数个非同义变异的所述总数的所述特征,所述小变异计算机分析140C可以进一步过滤所述被辨识的数个体细胞变异以辨识出是非同义变异的数个体细胞变异。如在本领域中周知的,一核酸序列的一非同义变异导致与所述核酸序列相关联的一蛋白质的胺基酸序列的一改变。举例而言,数个非同义变异可能改变一个体的一个或更多个表现型或造成所述个体发展出(或更容易发展出)癌症、癌细胞或其它类型的疾病。因此,所述小变异计算机分析140C通过判定对一个三核苷酸(trinucleotide)的一个或更多个核苷碱基的一修改将造成一不同的氨基酸基于所述经修改的三核苷酸被制造,而判定一候选变异将造成一非同义变异。数个非同义变异的所述总数的一特征值是通过加总在所述基因组各处被辨识的数个非同义变异而被决定。因此,对于获得自一个体的一cfDNA样本,数个非同义变异的所述总数的所述特征是被表示为一单一,数字性数值。
对于数个同义变异的所述总数的所述特征,数个同义变异代表不被分类为非同义变异的其它数个体细胞变异。换言之,所述小变异计算机分析140C可以,如同关于数个非同义变异被描述地,执行对被辨识的数个体细胞变异的过滤,及辨识遍及所述基因组或基因检测组合的所述数个同义变异。因此,对于获得自一个体的一cfDNA样本,数个同义变异的所述总数的所述特征是被表示为一单一数字性数值。
每个基因的数个体细胞变异的一存在/不存在的所述特征可以涉及一cfDNA样本的多个特征数值。举例而言,一针对性基因检测组合可以在所述检测组合中包括500个基因及因此,所述小变异计算机分析140C可以产生500个特征值,每个特征值代表所述基因检测组合中的一基因的数个体细胞变异的一存在或不存在。作为一示例,若一体细胞变异存在所述基因中,则所述特征的所述数值是1。相反地,若一体细胞变异不存在所述基因中,则所述特征的所述数值是0。一般地,任何大小的基因检测组合可以被使用。举例而言,所述基因检测组合可以包含100、200、500、1000、2000、10000或更多个遍及所述基因组的基因。在其它实施例中,所述基因检测组合可以包含自约50至约10000个基因目标、自约100至约2000个基因目标或自约200至约1000个基因目标。
对于已知与癌症相关联的数个特定基因的数个体细胞变异的存在/不存在的所述特征,已知与癌症相关联的数个特定基因可以存取自一公开数据库,比如OncoKB。已知与癌症相关的数个基因的数个示例包括p53、LRP1B、及KRAS。已知与癌症相关联的每个基因可以被与一特征值相关联,比如一1(指示一体细胞变异存在所述基因中)或一0(指示一体细胞变异不存在所述基因中)。
(例如,在一基因检测组合中的)每个基因的一体细胞变异的所述AF意指在所述数个序列读数中的一个或更多个体细胞变异的所述频率。一般地,此特征由遍及所述基因组的每个基因或一基因检测组合的每个基因的一个特征值代表。此特征的所述数值可以是所述基因的数个体细胞变异的数个AF的一统计值。在各种实施例中,此特征意指在所述基因中具有最大AF的一体细胞变异。在一些实施例中,此特征意指所述基因的数个体细胞变异的平均AF。因此,对于有500个基因的一针对性基因检测组合,有500个特征值,所述特征值代表(例如,在一基因检测组合中的)每个基因的一体细胞变异的所述AF。
由公开数据库,例如oncoKB,指定的每个类别的一体细胞变异的所述AF。举例而言,oncoKB将数个基因分类在四个不同的类别中的一个当中。在一实施例中,每个类别的一体细胞变异的所述AF是遍及所述类别中的数个基因的一体细胞变异的一最大AF。在一实施例中,每个类别的一体细胞变异的所述AF是遍及在所述类别中的所述数个基因的数个体细胞变异的一平均AF。
根据数个体细胞变异的AF的数个体细胞变异的排名顺序,意指数个体细胞变异的前N个等位基因频率。一般地,一变异等位基因频率的所述数值可以在0至1间,其中一0变异等位基因频率指示在所述位置没有序列读数拥有所述替代等位基因及其中一1变异等位基因频率指示在所述位置所有序列读数皆拥有所述替代等位基因。在其它实施例中,变异等位基因频率的其它范围及/或数值可以被使用。在各种实施例中,所述排名顺序特征自身是独立于所述数个体细胞变异,而仅由所述前N个变异等位基因频率的数个数值代表。前5个等位基因频率的排名顺序特征的一示例可以被表示为:[0.1,0.08,0.05,0.03,0.02],指示在自0.02至0.1的范围中的,独立于所述数个体细胞变异的5个最高的等位基因频率。
2.2.小变异计算机分析过程总览
一处理系统,比如一处理器或一计算机,执行用于执行所述小变异计算机分析140C的所述代码。图3A是根据一实施例的,用于自数个序列读数决定数个体细胞变异的一方法300。在步骤305A,所述处理系统塌缩(collapses)数个对齐的序列读数。在一实施例中,塌缩数个序列读数包括使用UMIs,及可选地使用来自一输出档案的序列资料的对齐位置信息以将多个序列读数塌缩为一个共识序列(consensus sequence),所述共识序列用于决定一核酸断片或核酸断片的一部份的最有可能的序列。所述独特序列标记可以是自约4至20核酸长。既然所述UMIs经过富集及PCR与所述数个经连接的核酸断片一同被复制,所述序列处理器205可以判定特定数个序列读数源自于一核酸样本中的相同分子。在一些实施例中,具有相同或类似的对齐位置信息(例如,在一阀值偏移量中的起始及末端位置)及包括一共同UMI的数个序列读数被塌缩,及所述处理系统产生一经塌缩读数(在本文中也被称为一共识读数)以代表所述核酸断片。若对应的一对经塌缩读数具有一共同UMI,代表所述发源核酸分子的正向及负向股被捕获,则所述处理系统将所述共识读数被指定为“双股”(duplex);否则,所述经塌缩读数被指定为“非双股”(non-duplex)。在一些实施例中,作为塌缩数个序列读数的一替代或附加,所述处理系统可以对数个序列读数执行其它类型的错误修正。
在步骤305B,所述处理系统基于所述对应的对齐位置信息,缝合(stitches)所述数个经塌缩读数。在一些实施例中,所述处理系统在一第一序列读数及一第二序列读数间比较对齐位置信息,以判定所述第一及第二序列读数的数个核苷酸碱基对是否在所述参考基因组中重叠。在一个使用案例中,回应判定所述第一及第二序列读数间的(例如,一给定数量的核苷酸碱基的)一重叠大于一阀值长度(例如,核苷酸碱基的阀值数量),所述处理系统将所述第一及第二序列读数指定为“缝合的”;否则,所述数个经塌缩读数被指定为“未缝合的”。在一些实施例中,若所述重叠大于所述阀值长度及若所述重叠非一滑动重叠(sliding overlap),则一第一及第二序列读数是缝合的。举例而言,一滑动重叠可以包括一均聚合物延续(homopolymer run)(例如,一单一重复核苷酸碱基)、一双核苷酸延续(例如,二核苷酸碱基序列)或一三核苷酸延续(例如,三核苷酸碱基序列),其中所述均聚合物延续、双核苷酸延续或三核苷酸延续具有至少一阀值长度的数个碱基对。
在步骤305C,所述处理系统将数个读数组合成为数个路径(paths)。在一些实施例中,所述处理系统组合数个读数以产生一目标区域(例如,一基因)的一定向图(directedgraph),举例而言,一德布鲁因图(de Bruijn graph)。所述定向图的数个单向边缘代表在所述目标区域中,k个核苷酸碱基的数个序列(在本文中也被称为“k聚体”(k-mers)),及所述数个边缘由数个顶点(或节点)连接。所述处理系统将数个经塌缩读数对齐到一定向图,而使任何所述经塌缩读数可以由所述数个边缘及对应的数个顶点的一子集依序代表。
在一些实施例中,所述处理系统决定描述数个定向图的数组参数及处理数个定向图。附加地,所述数组参数可以包括自数个经塌缩读数成功对齐的数个k聚体的一计数至由所述定向图中的一节点或边缘代表的一k聚体。所述处理系统储存数个定向图及对应的数组参数,所述数组参数可以被找回以更新数个图或产生新图。举例而言,所述处理系统可以基于所述参数组产生一定向图的一压缩版本(例如,或修改一现存的图)。在一使用案例中,为了滤除具有较低重要性等级的一定向图的资料,所述处理系统移除(例如,“修剪”(trims)或“修整”(prunes))具有少于一阀值的一计数的数个节点或数个边缘,及维持具有多于或等于所述阀值的计数的数个节点或数个边缘。
在步骤305D,所述处理系统自所述数个被组合的路径产生数个候选变异。在一实施例中,所述处理系统通过将一定向图(所述定向图或许已通过在步骤305B中修整数个边缘或数个节点而被压缩)与一基因的一目标区域的一参考序列比较而产生所述数个候选变异。所述处理系统可以将所述定向图的数个边缘对齐到所述参考序列,及将毗邻于所述数个边缘的数个错配的边缘及数个错配的核苷酸碱基的基因组位置纪录为候选变异的位置。在一些实施例中,数个边缘的左方及右方的所述数个错配边缘及数个错配核苷酸碱基的所述基因组位置被纪录为经辨认变异的位置。附加地,所述处理系统可以基于一目标区域的所述定序深度而产生数个候选变异。特别地,所述处理系统可以在辨识具有更大的定序深度的数个目标区域中的数个变异上更具信心。例如,因为一更大数量的序列读数有助于(例如,使用冗余)解决数个序列间的错配或其他碱基对变异。
在一实施例中,所述处理系统使用一模型以为来自一对象的数个序列读数决定数个预期噪声比率,而产生数个候选变异。所述模型可以是一贝氏阶层模型,虽然在一些实施例中,所述处理系统使用一个或更多个不同类型的模型。此外,一贝氏阶层模型可以是许多个可能的模型架构中的一个,所述数个可能的模型架构可以被用于产生数个候选变异,及所述数个可能的架构在它们都为了改进变异辨认的敏感度(sensitivity)/特异度(specificity)而为位置特定性噪声信息建模方面与彼此相关。更特定地,所述处理系统使用来自数个健康个体的样本训练所述模型,以为数个序列读数的每个位置的预期噪声比率建模。
进一步地,多个不同的模型可以被储存在一数据库中或在训练后被取回以供应用。举例而言,一第一模型是被训练以为数个SNV噪声比率建模及一第二模型是被训练以为数个插入删除噪声率建模。进一步地,所述处理系统可以使用所述模型的数个参数以决定在一序列读数中的一个或更多个真实阳性的一似然性。所述处理系统可以(例如,在一对数尺度上)决定基于所述似然性的一质量分数。举例而言,所述质量分数是一Phred质量分数(Phred quality score)Q=10·㏒
在步骤305E,所述处理系统使用一个或更多个类型的模型或过滤器过滤所述数个候选变异。举例而言,所述处理系统使用一联合模型、边缘变异预测模型或真实阳性的相对应的数个似然性或数个质量分数评分所述数个候选变异。
现在参考图3B,图3B是根据一实施例的,用于使用不同类型的过滤器及模型处理数个候选变异的,展示于图3A中的步骤305E的一流程图。在步骤310A,所述处理系统为一核酸样本,例如一cfDNA样本的数个序列读数的噪声建模。所述模型可以如先前描述的是一贝氏阶层模型,所述贝氏阶层模型近似所述数个序列读数的每个位置的预期噪声分布。在步骤310B,所述处理系统使用一联合模型自所述数个序列读数过滤数个候选变异。在一些实施例中,所述处理系统使用所述联合模型决定在所述cfDNA样本中观察到的一给定候选变异是否可能与(例如,来自白血球的)一相对应的gDNA样本的一核苷酸突变相关联。
在步骤310C,所述处理系统使用边缘过滤过滤所述数个候选变异。特别地,所述处理系统可以应用一边缘过滤器,所述边缘过滤器考量在所述样本,例如,cfDNA样本中观察到的所述数个变异,预测一候选变异是一伪阳性,也称为一边缘变异的机率。边缘变异一词意指位在接近一序列读数的一边缘处的一突变,举例而言,距所述序列读数的边缘,在一阀值距离的核苷酸碱基内。特别地,给定一样本的数个候选变异的一集合,所述边缘过滤器可以执行一似然性估计,以决定在所述样本中的边缘变异的一预测比率。给定所述样本的特定条件,所述预测比率可以考虑到两种分布而最佳地解释所述样本的数个候选变异的所述观察到的集合。一个分布描述已知的数个边缘变异的数个特征,而另一个经训练的分布描述已知的数个非边缘变异的数个特征。所述预测比率是一样本特定性的参数,控制所述样本被如何积极地分析以自所述样本辨识及过滤边缘。所述样本的数个边缘变异被过滤及移除,留下数个非边缘变异供随后考虑。非边缘变异一词意指一候选变异,所述候选变异并不被,例如,使用本文中描述的一边缘变异过滤方法判定为缘于一人为过程。在一些情境中,一非边缘变异可能并非一真实变异(例如,在所述基因组中的突变),因为非边缘变异可能由于相对于一个或更多个人为过程的一不同理由而发生。
返回图3A,在步骤305F,所述处理系统输出所述经过滤的数个候选变异(例如,数个体细胞变异)。在此,所述经过滤的数个候选变异可以接着被用以决定在上文中被描述的所述数个小变异特征。
在步骤305G,可选地,衍生自所述数个体细胞变异的所述数个小变异特征可以被用以产生一癌症预测。举例而言,一预测模型,比如展示于图1C中的预测性癌症模型170C,可以被应用到所述数个小变异特征。换言之,所述预测模型(例如,预测性癌症模型170C)可以做为仅使用数个小变异特征154输出一癌症预测190C的一单一化验预测模型。
2.2.1.示例噪声模型
图3C是根据一实施例的一贝氏阶层模型的一应用的一图表。突变A及突变B是为解释的目的而作为示例被展示。在图3C的实施例中,突变A及B被表示为SNVs,虽然在其它实施例中,下文的描述也适用于插入或缺少或其它类型的突变。突变A是位于来自一第一样本的一第一参考等位基因的位置4处的一C>T突变。所述第一样本具有10的一第一替代深度(AD)及1000的一第一总深度。突变B是位于来自一第二样本的一第二参考等位基因的位置3处的一T>G突变。所述第二样本具有1的一第二AD及1200的一第二总深度。仅基于AD(或AF),突变A可能显得是一真实阳性,而突变B可能显得是一伪阳性,因为前者的所述AD(或AF)比后者的所述AD(或AF)大。然而突变A及B可能具有不同的每等位基因及/或等位基因的每个位置的相对噪声水平。事实上,一旦这些不同位置的所述相对噪声水平被计算在内,突变A可能是一伪阳性且突变B可能是一真实阳性。在本文中被描述的数个模型为了根据所述噪声恰当地辨识真实阳性而将此噪声建模。
在图3C中例示的数个机率质量函数(probability mass function,PMF)指示来自一对象的一样本在一位置具有一给定AD计数的机率(或似然性)。使用来自数个健康个体的数个样本的序列资料,所述处理系统训练一模型,数个健康样本的所述数个PDF可以衍生自所述模型。特别地,所述数个PDF是基于m
使用图3C的所述示例进一步例示,来自所述数个健康个体的数个样本代表由y-
图3D展示根据一实施例,用于决定真实单一核苷酸变异的一贝氏阶层模型的数个参数及数个子模型间的依赖关系。在展示于图3D中的所述示例中,
,而被潜在变量z
共同地,所述潜在变量z
共变量x
在位置p处的一SNV的所述预期平均AD计数是由所述参数μ
在其它实施例中,其它函数可以被用于代表μ
在展示于图3D中的所述示例中,所述形状及平均数参数是各自依附于所述协变量x
在其它实施例中,其它函数可以被用于代表
图3E展示根据一实施例,用于决定真实插入或删除的一贝氏阶层模型的数个参数及数个子模型间的依赖关系。相对于展示于图3D中的所述SNV模型,展示于图3E中的用于插入或缺少的所述模型包括不同程度的阶层。所述协变量x
在位置p处的预期平均插入或缺少计数是由所述分布μ
在其它实施例中,其它函数可以被用于代表μ
在(一健康个体的)一人类群体样本i中于位置p处观察到的数个插入或缺少是由所述分布
在其它实施例中,其它函数可以被用以代表
由于数个插入或缺少可能长度各不相同,一额外的长度参数存在所述插入或缺少模型中,所述长度参数不存在用于SNVs的所述模型中。作为结果,展示在图3E中的所述示例性模型具有一额外的阶层(例如,另一子模型),所述阶层同样不存在于上文中讨论的所述数个SNV模型中。在样本i中的位置p处的长度l的数个插入或缺少(例如,多至100或更多个插入或缺少的碱基对)的观察到的计数是由所述随机变量
在其它实施例中,一狄利克雷多项式函数(Dirichlet-Multinomial function)或其它类型的模型可以被用于代表
通过以此方式架构所述模型,所述处理系统可以将插入或缺失强度(亦即,噪声比)的学习与插入或缺失长度分布的学习分离。独立地决定对是否一插入或缺少会在健康的样本中发生的预期及对位于一位置的所述插入或缺少的所述长度的预期的推论可以增进所述模型的所述敏感度。举例而言,所述长度分布可能在所述基因组中的数个位置或区域中,相对于所述插入或缺少强度更加稳定,或反之亦然。
图3F及3G例示根据一实施例的,与一贝氏阶层模型相关联的图表。展示于图3F中的所述图表绘示数个噪声比率,亦即,由一模型特征化的对一给定位置的SNVs或插入或缺少的似然性(或强度)的分布μ
图3H是根据一实施例,通过拟合(fitting)一贝氏阶层模型决定数个参数的一示例性过程的一图表。为了训练一模型,所述处理系统自一组位置的每个位置的数个预期噪声比率的一个事后分布(例如,展示于图3G中的所述图表)叠代地采样。在其它取样演算法当中,所述处理系统可以使用马可夫链蒙地卡罗(Markov chain Monte Carlo,MCMC)法取样,例如,一梅特罗波利斯-黑斯廷斯(MH)演算法(Metropolis–Hastings(MH)algorithm)、定制MH演算法(custom MH algorithms)、吉布斯取样演算法(Gibbs samplingalgorithm)、哈密顿力学基础取样(Hamiltonian mechanics-based sampling)、随机取样。在贝氏推论训练中,数个参数被抽取自所述联合事后分布,以叠代地更新所述模型的数个参数及数个潜在变量的全部(或一些)(例如,
在一实施例中,所述处理系统通过储存μ
图3I是根据一实施例,使用来自一贝氏阶层模型的数个参数决定一伪阳性的一似然性的一图表。所述处理系统可以将展示于图3H中的所述R行乘N列矩阵缩减为例示于图3I中的一R行乘2列矩阵。在一实施例中,所述处理系统横跨所述数个事后样本μ
给定所述数个平均参数,所述处理系统也可以在所述经缩减的矩阵中执行所述数个分布参数的分布重新估计。在一实施例中,在贝氏训练及事后近似后,所述处理系统通过为基于每个位置的一负二项式最大似然性估计器的所述数个分布参数
在应用数个经训练模型时,所述处理系统可以存取所述数个分布(例如,形状)参数
2.3.噪声模型的示例处理流程
图3J是根据一实施例的用于训练一贝氏阶层模型的一方法315的流程图。在步骤320A中,所述处理系统自数个序列读数的一数据库收集数个样本,例如,训练资料。在步骤320B中,所述处理系统使用一马可夫链蒙地卡罗法使用所述数个样本训练所述贝氏阶层模型。在训练时,所述模型可以以所述训练资料为条件保留或拒绝数个序列读数。所述处理系统可以排除具有少于一阀值的深度数值或大于一阀值频率的一AF的数个健康个体的数个序列读数,以便移除对数个序列读数中的目标噪声不具指示性的数个疑似生殖细胞系突变。在其它实施例中,所述处理系统可以决定哪个位置可能含有生殖细胞系变异,并使用类似上述的数个阀值选择性地移除这样的数个位置。在一实施例中,所述处理系统可以将这样的数个位置辨识为具有一小的AF平均值绝对变异数(例如,0、1/2及1),所述AF是来自数个生殖细胞系频率。
所述贝氏阶层模型可以对被包括在所述模型中的多个(或全部)位置同步更新参数。此外,所述模型可以被训练以为每个ALT预期噪声建模。举例而言,SNV的一模型可以对A、T、C及G碱基中的每个突变到其它三个碱基中的每个的突变执行一训练过程四次或更多次以更新数个参数(例如,一对一替换)。在步骤320C中,所述处理系统储存所述贝氏阶层模型的数个参数(例如,由所述马可夫链蒙地卡罗法输出的数个系综参数)。在步骤320D中,所述处理系统基于所述数个参数近似所述噪声分布(例如,由一分布参数及一平均值参数代表)。在步骤320E中,所述处理系统使用来自被用来训练所述贝氏阶层模型的所述数个样本(例如,训练资料)的原始AD计数,执行分布重新估计(例如,最大似然性估计)。
图3K是根据一实施例的用于决定一伪阳性的一似然性的一方法325的流程图。在步骤330A,所述处理系统自一组序列读数辨识一候选变异,例如,位于一序列读数的一位置p的一候选变异,所述序列读数可以是自获得自一个体的一cfDNA样本被定序。在步骤330B,所述处理系统存取特定于所述候选变异的数个参数,例如,分别为分布及平均数比率参数的
在步骤330C,所述处理系统将所述一组序列读数的读数资料(例如,AD或AF)输入(例如,基于一负二项式)的一函数,所述函数由所述数个参数,例如
所述处理系统可以为下游分析使用数个函数,所述数个函数编码相关于一给定训练样本的数个核酸突变的数个噪声水平。在一些实施例中,所述处理系统使用前述的由所述分布及平均数比率参数
2.4.示例联合模型
图3L是根据一实施例的,用于使用一联合模型处理数个游离核酸(例如,cfDNA)样本及数个基因组核酸(例如,gDNA)样本的一方法335的一流程图。在本技艺中寻得的一联合模型的其它示例也可以类似地被应用(参见,美国专利第16/201,912号,其通过引用被并入到本文中)。所述联合模型可以独立于cfDNA及gDNA的核酸的位置。所述方法335可以与展示于图3J及3K中的所述方法315及/或方法325共同被执行。举例而言,所述方法315及325是被执行以决定关于来自数个健康样本的训练资料的数个cfDNA及gDNA样本的数个核苷酸突变的噪声。图3M是根据一实施例的一联合模型的一应用的一图表。方法335的数个步骤在下文中参照图3M而被描述。
在步骤340A中,所述处理系统决定核酸的各个位置的深度及AD,所述核酸来自获得自一对象的一cfDNA样本的数个序列读数。所述cfDNA样本可以是收集自来自所述对象的血浆的一样本。
在步骤340B中,所述处理系统决定核酸的各个位置的深度及AD,所述核酸来自获得自相同对象的一gDNA样本的数个序列读数。所述gDNA可以是收集自来自所述对象的白血球或一肿瘤活检。
在步骤340C中,一联合模型通过为cfDNA的观察到的数个AD建模而决定所述对象的所述cfDNA样本的一“真实”AF的一似然性。在一实施例中,所述联合模型使用一泊松分布函数,以为在所述对象的cfDNA中观察到一给定AD的所述机率建模(也展示于图3M中)。所述泊松分布函数由自cfDNA的所述读数观察到的所述数个深度及所述cfDNA样本的所述真实AF参数化。所述深度及所述真实AF的产出可以是所述泊松分布函数的所述比率参数,所述比率参数代表cfDNA的平均预期AF。
P(AD
所述噪声成分噪声
在步骤340D中,所述联合模型通过为gDNA的观察到的数个AD建模而决定所述对象的所述gDNA样本的一“真实”AF的一似然性。在一实施例中,所述联合模型使用一泊松分布函数,以为在所述对象的gDNA中观察到一给定AD的所述机率建模(也展示于图3M中)。所述泊松分布函数由自gDNA的所述数个序列读数观察到的所述数个深度及所述gDNA样本的所述真实AF参数化。虽然所述数个参数值基于自所述对象的所述对应的样本观察到的所述数个数值而异,所述联合模型可以使用一相同函数为gDNA及cfDNA的真实AF的所述数个似然性建模。
P(AD
既然cfDNA的所述真实AF,以及gDNA的所述真实AF,是一特定对象的生理的固有性质,判定来自上述两个来源的所述真实AF的一确切数值可能不必然是实用的。此外,各种噪声来源也将不确定性导入所述真实AF的所述估计值中。因此,所述联合模型使用数值近似以决定真实AF的所述事后分布,所述事后分布以来自一对象的所述观察到的资料(例如,深度及AD)及数个相对应的噪声参数为条件:
所述联合模型使用具有一先验,举例而言,一均匀分布,的贝叶斯定理(Bayes’theorem)决定所述事后分布。用于cfDNA及gDNA的数个先验可以是相同的(例如,范围自0至1的一均匀分布)及是独立于彼此的。
在一实施例中,给定来自cfDNA的所述样本的观察到的资料的一固定集合,所述联合模型通过变化cfDNA的所述参数、真实AF,使用一似然性函数决定cfDNA的真实AF的所述事后分布。此外,给定来自gDNA的所述样本的观察到的资料的一固定集合,所述联合模型通过变化所述gDNA的所述参数、真实AF,使用另一似然性函数决定gDNA的真实AF的所述事后分布。对于cfDNA及gDNA两者,所述联合模型数值近似所述输出事后分布,所述数值近似是通过拟合一负二项式(NB):
在一实施例中,所述联合模型使用所述负二项式的下述数个参数执行数值近似,所述数值近似可以提供计算速度上的一改进:
P(AF|深度,AD)∝NB(AD,尺寸=
其中:
既然在cfDNA及gDNA间观察到的资料不同,为cfDNA的所述负二项式决定的所述数个参数会与为gDNA的所述负二项式决定的所述数个参数不同。
在步骤340E中,所述处理系统使用所述数个似然性,决定所述cfDNA样本的所述真实AF大于所述gDNA样本的所述真实AF的一函数的一机率。所述函数可以包括一个或更多个参数,举例而言,据经验决定的(empirically-determined)k值及p值,并参考图3N至3O被更加详细地描述。所述机率代表来自cfDNA的所述数个序列读数的至少一些核苷酸突变未在参考组织的数个序列读数中被找到的一信心水平。所述处理系统可以将此信息提供至其它程序供下游分析。举例而言,一高机率指示来自一对象的cfDNA的数个序列读数及未在gDNA的数个序列读数中找到的数个核苷酸突变可能源自所述对象中的一肿瘤或其它癌症来源。相对地,低机率指示在cfDNA中观察到的数个核苷酸突变可能不是源自于所述对象的潜在癌细胞或其他患病细胞。而是,所述数个核苷酸突变可以被归因于在健康个体中由于比如生殖细胞系突变、克隆性造血(形成白血球DNA的次族群的独特突变)、嵌合现象(mosaicism)、化疗或诱变性治疗、技术性人为因素等而自然发生的突变。
在一实施例中,所述处理系统基于所述一个或更多个参数(例如,下述的k及p)判定所述事后机率满足一选定的标准(criteria)。给定所述cfDNA及gDNA的所述数个序列,数个变异的所述数个分布是条件独立的。亦即,所述处理系统假定在所述cfDNA或gDNA样本中的一个当中存在的数个ALT及噪声不被另一样本的数个ALT及噪声影响,反之亦然。因此,给定来自cfDNA及gDNA两个来源的所述观察到的信息及数个噪声参数,所述处理系统在判定观察到cfDNA的一特定真实AF及gDNA的一特定真实AF中,将AD的所述数个预期分布的所述数个机率视为独立事件:
在绘于图3M中的所述示例性3D绘图中,所述机率P(AF
在一实施例中,所述处理系统通过判定所述联合似然性满足一给定条件的所述部分,判定所述给定条件是被所述事后机率满足。所述给定条件可以基于所述k及p参数,其中p代表供比较的一阀值机率。举例而言,所述处理系统决定cfDNA的真实AF是大于或等于gDNA的所述真实AF乘以k的所述事后机率,及所述事后机率是否大于p:
P(AF
如展示在上述的等式中地,所述处理系统决定cfDNA的所述真实AF的所述似然性的一累积和F
为了解释由所述数个cfDNA及gDNA样本中的噪声导入的,在所述真实AF的所述数个估计值中的噪声,所述联合模型可以使用先前参照图3C至3I被描述的,所述处理系统的其它模型(例如,贝氏阶层模型)。在一实施例中,展示于上述数个等式P(AD
在一示例中,所述联合模型使用由数个cfDNA特定参数参数化的一函数,决定cfDNA的所述真实AF的一噪声水平。所述数个cfDNA特定参数可以使用一贝氏阶层模型而被衍生。所述贝氏阶层模型是以一组cfDNA样本,例如,来自数个健康个体的一组cfDNA样本被训练。此外,所述联合模型使用由数个gDNA特定参数参数化的另一函数,决定gDNA的所述真实AF的一噪声水平。所述数个gDNA特定参数可以使用另一贝氏阶层模型而被衍生。所述贝氏阶层模型是以一组gDNA样本,例如,来自相同的数个健康个体的一组gDNA样本被训练。在一实施例中,所述数个函数是具有一平均参数m及分布参数
在其它实施例中,所述处理系统可以使用一不同类型的函数与cfDNA及/或gDNA的数个类型的参数。既然所述数个cfDNA特定参数及数个gDNA特定参数是使用不同组的训练资料而被衍生,所述数个参数可以不同于彼此,及特定于各自类型的核酸样本。举例而言,数个cfDNA样本可以较数个gDNA样本具有更大的AF变异,及因此
2.5.联合模型的示例
展示在下述数张附图中的数个示例性结果是由所述处理系统使用一个或更多个经训练的模型决定,所述一个或数个经训练的模型可以包括数个联合模型及数个贝氏阶层模型。为了比较的目的,一些示例性结果是使用一经验性阀值或一简单模型被决定,及是被称为“经验性阀值”示例及在数个附图中被表示为“阀值”;这些示例性结果并非使用所述数个经训练的模型中的一个获得。在各种实施例中,所述数个结果是使用数个示例针对性定序化验中的一个产生。所述数个示例针对性定序化验包括“针对性定序化验A”及“针对性定序化验B”,在本文中及在附图中也分别称为“化验A”及“化验B”。
在对一针对性定序化验执行的一示例性过程中,两管全血自健康个体(自陈无癌症诊断)被抽入Streck采血管(Streck BCT)中。在血浆被自所述全血分离后,血浆被储存于-80℃。在化验处理时,cfDNA自两管血浆被提取并池化。科里尔(Corielle)基因组DNA被断片化至180碱基对(bp)的一平均尺寸,接着使用磁珠被尺寸选择至一较紧密的分布。所述文库制备方案为低输入游离DNA(cfDNA)与经剪切的gDNA而被最佳化。数个唯一分子识别码(UMI)在转接子连接时被整合到所述数个DNA分子中。接着流通槽簇集转接子序列(Flowcell clustering adapter sequences)及双样本指标(dual sample indices)于以PCR进行的文库制备放大被整合。数个文库使用一针对性捕捉板被富集。
目标DNA分子首先使用数个生物素化(biotinlyated)的单股DNA杂合探针被捕捉,及接着使用链霉亲和素(streptavidin)磁珠被富集。非目标分子使用数个随后的清洗步骤被移除。所述HiSeq X试剂套组v2.5(HiSeq X Reagent Kit v2.5)(Illumina,加利福尼亚州圣地亚哥)被用于流通槽簇集及定序。每流通槽四个文库被多重扩增(multipled)。双指标启动子混合被包括,以使双样本指标读取能够进行。对读取1、读取2、指标读取1及指标读取2,读数长度被分别设定为150、150、8及8。在读取1及读取2中的头六个碱基读数是UMI序列。
图3N是根据一实施例的,来自数个健康个体的数个样本中的数个观察到的变异的计数的一图表。每个数据点对应到所述数个个体中给定的一个(横跨一系列核酸位置的)一位置。被所述联合模型用于联合似然性计算的所述参数k及p可以通过以来自数个健康个体的数组cfDNA及gDNA样本及/或已知有癌症的数个样本进行交叉验证而被经验性地选择(例如,以调节敏感度阀值)。展示于图3N中的所述数个示例性结果是以化验B获得,及使用数个血浆样本于所述cfDNA及使用数个白血球样本于所述gDNA。对于给定的参数值k(如展示于图3N中的“k0”)及p,所述图表描绘数个变异的一平均数量,所述平均数量代表所述对应样本的伪阳性的一计算出的置信区间上界(upper confidence bound,UCB)。所述图表指示伪阳性的数量随着p的数值增加而减少。此外,描绘出的所述曲线对较低的k值,例如,离1.0较近,有较大的伪阳性数量。虚线指示一个变异的一目标,尽管经验性结果显示,对于在1.0及5.0间的k值,及在0.5及1.0间的p值,伪阳性的所述平均数量通常落在1至5个变异的范围内。
数个参数的所述选择可能涉及一目标敏感度(使用k及p调整)及目标错误(例如,所述置信区间上界)间的一取舍。对于给定的k及p值对,相对应的伪阳性平均数量可能在数值上相近,尽管所述敏感性数值可能展现较大的变化。在一些实施例中,相较于可能被用做特异度的一测量值的cfDNA的阳性一致率(percent positive agreement,PPA),所述敏感度是使用肿瘤的阳性一致率而被测量。
在上述的等式中“肿瘤”代表使用一组参数自一ctDNA样本辨认出的平均变异的数量,及“cfDNA”代表使用相同的一组参数自所述相应的cfDNA样本辨认出的平均变异的数量。
在一实施例中,交叉检验被执行,以估计所述联合模型对(一给定类型的组织的)数个序列读数的预期拟合度,所述序列读数不同于用于训练所述联合模型的所述数个序列读数。举例而言,所述数个序列读数可以获得自具有肺癌、前列腺癌及乳癌等的组织。为了避免或减少对于任何给定类型的癌症组织过度拟合所述联合模型的程度,使用一组数个类型的癌症组织的数个样本衍生出的数个参数值被用于评估已知具有一不同类型的癌症组织的其它样本的统计结果。举例而言,肺癌及前列腺癌组织的数个参数值被应用到具有乳癌组织的一样本。在一些实施例中,来自最大化所述敏感度的所述肺癌及前列腺癌组织资料的一个或更多个最低k值被选择以被应用到所述乳癌样本。参数值也可以使用其它限制而被选择,比如自伪阳性的一目标平均数量的一阀值偏差,或每个样本的至多3的95%UCB。所述处理系统可以循环多种类型的组织,以交叉验证数组癌症特定性参数。
图3O是根据一实施例的一联合模型的数个示例性参数的一图表。k的所述数个参数值可以被决定为在数个gDNA样本中观察到的AF的一函数,及可以基于一特定类型的癌症组织,例如,如例示的乳癌、肺癌或前列腺癌而不同。曲线345A代表乳癌及前列腺癌组织的数个参数值,及曲线345B代表肺癌组织的数个参数值。虽然目前为止的数个示例一般性地,及参考在其中这些参数为固定的实作来描述k及p,实际上k及p可以作为在所述gDNA样本中观察到的AF的任何函数而变化。在展示于图3O中的所述示例中,所述函数是具有一枢纽值(hinge value)(或下阀值),例如三分之一,的一枢纽损失函数(hinge loss function)。特定地,对大于或等于所述枢纽值的AF
当k的一固定值不会准确地捕捉并分类非与肿瘤或疾病相关的事件时,所述联合模型可以根据一枢纽损失函数或另一函数改变k,以防备这些效应。所述枢纽损失函数示例是特定地针对处理杂合性丢失(LOH)事件。LOH事件是当一基因的拷贝一自一个体的父母中的一个丢失时所发生的生殖细胞系突变。LOH事件可能促成一gDNA样本的观察到的AF的显著比例。通过将所述k值限制到所述枢纽损失函数的所述预先决定的上阀值,所述联合模型可以对在多数序列读数中侦测真实阳性达成更大的敏感度,同时也控制若非如此则会由于LOH的存在而被标示为真实阳性的伪阳性的数量。在其它实施例中,k及p可以基于训练资料而被选择,所述训练资料是特定于一给定的感兴趣的应用,例如,具有一目标群体或定序化验。
在一些实施例中,所述联合模型将一gDNA样本的所述AF及所述gDNA样本的一质量分数皆纳入考量,以防备过低加权低AF候选变异。一噪声模型的所述质量分数可以被用于在一Phred标尺上估计错误的机率。此外,所述联合模型可以对所述枢纽函数使用一经修改的分段函数(piecewise function)。举例而言,所述分段函数包括两个或更多个附加部分。一个部分是基于所述gDNA样本的所述AF的一线性函数,及另一个部分是基于所述gDNA样本的所述质量分数的一指数函数。给定一质量分数阀值及一最大AF比例系数k
在上述的计算中,P(非错误)是所述gDNA样本的一等位基因并非一错误的所述机率,P(错误)是所述gDNA样本的所述等位基因是一错误的所述机率,及P(错误)
2.5.1.联合模型的示例变异辨识
图3R至3S是根据一实施例的,由一联合模型决定的数个变异辨认的图表。展示于图3R中的所述数个示例性结果是使用针对性定序化验A及已知受早期癌症影响的数个样本获得。展示于图3S中的所述数个示例性结果是使用针对性定序化验B及已知受晚期癌症影响的数个样本获得。在图3R至3S中的所述数个图形分享一个共同的x轴,所述x轴代表gDNA的观察到的AF。进一步地,所述数个图形指示cfDNA及gDNA的数个样本的观察到的AF的所述比例的一变异度在晚期癌症比在早期癌症更大。所述变异辨认器240判定数对AF
图3T是根据一实施例的一联合模型决定的机率密度的一图表。展示在图3T中的所述数个示例性结果是使用来自乳房、肺部及前列腺组织样本的数个序列读数,所述数个组织样本具有等于0的一观察到的gDNA的AF。图3T例示所述联合模型的一些一般要点,而不论所述特定实作。在于gDNA中没有ALT被观察到(AF
图3U是根据一实施例的一联合模型的敏感度及特异度的一图表。所述处理系统使用化验A及B,及使用数个健康样本以及已知具有乳癌、肺癌及前列腺癌的数个样本,决定所述数个敏感度(例如,PPA
图3V是根据一实施例使用一联合模型自针对性定序化验侦测到的一组基因的一图表。所述组包括在克隆性造血当中普遍突变的数个基因。所述处理系统使用化验A及B与已知具有乳癌、肺癌及前列腺癌的数个样本判定所述结果。所述数个测试“阀值X”及“联合模型X”并不包括数个非同义突变,而所述数个测试“阀值Y”及“联合模型Y”包括数个非同义突变。使用所述联合模型225获得的所述数个示例结果相较于使用一经验性阀值侦测到的所述计数,减少了自各种类型组织的数个样本侦测到的数个生殖细胞系突变。举例而言,如由化验B的所述图表以肺癌所例示的,“阀值X”及“阀值Y”分别导致计数到5及6个侦测到的TET2基因。所述“联合模型X”及“联合模型Y”分别导致计数到2及3个侦测到的TET2基因,这显示所述联合模型225提供增进的敏感度。
图3W是根据一实施例使用一联合模型自针对性定序化验侦测到的,展示于图3V中的所述一组基因的长度分布的一图表。一般地,源自于肿瘤或患病细胞的核酸断片比源自数个参考等位基因的核酸断片具有更短的(例如,核苷酸)长度。如于化验B的乳癌样本的所述盒状图结果中展示的,对所述TET2基因的侦测到的数个ALT及数个参考等位基因的长度中位数间的差异对于“阀值X”及“阀值Y”两者皆是近似零。相对地,对所述TET2基因的侦测到的数个ALT及数个参考等位基因的长度中位数间的差异对于“联合模型X”及“联合模型Y”两者皆是近于-5。因此,所述处理系统可以以更大的信心判定所述侦测到的数个ALT潜在源自一肿瘤或一患病细胞,而非一参考等位基因。此外,所述数个示例结果指示所述联合模型可以在具有不同噪声水平的数个样本中执行数个序列读数的数个短断片的变异辨认。
图3X是根据一实施例使用一联合模型自针对性定序化验侦测到的另一组基因的一图表。所述数个示例性结果显示所述联合模型侦测数个驱动基因(driver genes)的敏感度可比得上并未使用一模型的数个过滤器的敏感度。亦即,相对于使用一经验性阀值获得的所述结果,所述联合模型并未显著地过度过滤被侦测到的数个驱动基因。
2.6.联合模型的示例调校
图3Y是根据一实施例的用于调校一联合模型以处理游离核酸(例如,cfDNA)样本及基因组核酸(例如,gDNA)样本的一方法350的流程图。所述方法350可以与展示于图3J至3L中的所述方法315、325及/或335,或另一类似方法共同被执行。举例而言,所述方法350是使用一联合模型被执行,以决定所述方法350的步骤355A的机率。相关于图3Y、3Z及3AA而被描述的数个示例参考一对象的血液(例如,白血球)作为所述gDNA样本的来源,然而应注意的是,在其它实施例中,所述gDNA可以是来自一不同类型的生物样本。所述处理系统可以作为一决策树实施所述方法350的至少一部份,以过滤或处理在所述cfDNA样本中的数个候选变异。举例而言,所述处理系统决定一候选变异是否可能与所述gDNA样本相关联,或一关联是否是不确定的。一关联可能指示所述变异可以被归因于由于在所述gDNA样本中的一突变(例如,由于数个因素比如生殖细胞系突变、克隆性造血、人为因素、边缘变异、人类白血球抗原(human leukocyte antigens),比如HLA-A等)及因此可能不是肿瘤衍生的及不指示癌症或疾病。所述方法350可以包括与在一些实施例中连同图3Y而被描述的那些步骤不同的或额外的步骤,或以与连同图3Y而被描述的顺序不同的顺序执行数个步骤。
在步骤355A中,所述处理系统决定一cfDNA样本的所述真实替代频率大于一gDNA样本的一真实替代频率的一函数的一机率。步骤355A可以对应于展示于图3L中的所述方法335的先前描述的步骤340E。
在步骤355B中,所述处理系统判定所述机率是否少于一阀值机率。作为一示例,所述阀值机率可以是0.8,然而在实践中,所述阀值机率可以是在0.5及0.999间的任何数值(例如,基于一所欲的过滤策略而被决定),可以是静止的或动态的,随基因而异及/或由位置,或其他巨观因素等所设定。对判定所述机率是大于或等于所述阀值机率作出回应,所述处理系统判定所述候选变异可能并非与所述gDNA样本,比如包括一对象的白血球的一抽血相关联,亦即,并非血液衍生的。举例而言,所述数个候选变异典型地并不存在于一健康个体的所述gDNA样本的数个序列读数中。因此,所述处理系统可以将所述候选变异辨认为潜在地与癌症或疾病相关联,例如,潜在的是肿瘤衍生的,一真实阳性。
在步骤355C中,所述处理系统判定所述gDNA样本的所述替代深度是否显著的相同于或不同于零。举例而言,所述处理系统使用一噪声模型使用所述候选变异的一质量分数执行一评估。所述处理系统也可以将所述替代深度与一阀值深度相比较,例如,判定所述替代深度是否少于或等于一阀值深度。作为一示例,所述阀值深度可以是0或1读数。对判定所述gDNA样本的所述替代深度是显著不同于零做出回应,所述处理系统判定存在所述候选变异是与非由癌症或疾病造成的核苷酸突变相关的正向证据。举例而言,所述候选变异是血液衍生的,基于在健康的白血球的数个序列读数中可能典型地发生的突变。
对判定所述gDNA样本的所述替代深度非显著非零作出回应,所述处理系统判定所述候选变异可能与所述gDNA样本相关联,但并不作出对所述候选变异的一来源的一判定。换言之,所述处理系统可能对所述候选变异是血液衍生的或肿瘤衍生的是不确定的。在一些实施例中,所述处理系统可以选择多个阀值深度中的一个,用于与所述替代深度比较。所述选择可以基于经处理样本、噪声水平、信心水平或其它因素的一个类型。
在步骤355D中,所述处理系统判定所述gDNA样本的数个序列读数的一gDNA深度质量分数。在一实施例中,所述处理系统使用所述gDNA样本的所述一替代深度计算所述gDNA深度质量分数,其中C是一预先决定的常数(例如,2),以使用一弱先验(weak prior)平滑化所述gDNA深度质量分数,这避免了除零计算(divide-by-zero computations)
在步骤355E中,所述处理系统判定所述gDNA样本的数个序列读数的一比例。所述比例可以代表在所述经处理样本中观察到的cfDNA频率及观察到的gDNA频率。在一实施例中,所述处理系统使用所述cfDNA样本及所述gDNA样本的所述深度及替代深度计算所述比例:
所述处理系统可以使用数个所述预先决定的常数C
在步骤355F中,所述处理系统判定所述gDNA深度质量分数是否大于或等于一阀值分数(例如,1)及所述比例是否少于一阀值比例(例如,6)。对判定所述gDNA深度质量分数少于所述阀值分数或所述比例大于或等于所述阀值比例做出回应,所述处理系统判定关于所述候选变异与所述gDNA样本的关联有不确定证据。换句话说,所述处理系统可能对所述候选变异是否是血液衍生的或肿瘤衍生的是不确定的,因为所述候选变异显得“可能为血液”,但并无决定性证据显示对应的突变是在健康血液细胞中寻得。
在步骤355G中,对判定所述gDNA深度质量分数大于或等于所述阀值分数及所述比例是少于所述阀值比例做出回应,所述处理系统判定所述候选变异是可能与所述gDNA样本的一核苷酸突变相关联。换言之,所述处理系统判定虽然并无一对应突变是在健康血液细胞中寻得的决定性证据,所述候选变异显得较正常而言更像血液。
因此,所述处理系统可能使用所述比例及gDNA深度质量分数调校所述联合模型,以在决定特定候选变异是否应作为伪阳性(例如,初始被预测为肿瘤衍生,但实为血液衍生)、真实阳性或由于不足以分入两个类别中的任一个的证据或信心而作为不确定被滤出上,提供更大的颗粒性。举例而言,基于所述方法350的所述结果,所述处理系统可以修改所述联合模型的一枢纽损失函数的一个或更多个参数(例如,k参数)。在一些实施例中,所述处理系统使用所述方法350的一个或更多个步骤以将数个候选变异指派到不同的类别,举例而言,“绝对”、“可能”或“不确定”与gDNA相关(例如,如在图3Z及3AA中展示的)。
在各种实施例中,所述处理系统在参考展示于图3Y中的方法350的所述流程图而被描述的数个步骤外,使用一个或更多个过滤器处理数个候选变异。所述处理系统可以作为一决策树的一部分以一顺序执行所述数个过滤器,在所述决策树中,所述处理系统持续检查所述数个过滤器的标准,直到一个给定的候选变异“离开”所决策树,例如,因为所述给定候选变异在满足所述标准的至少一个时被过滤。一被过滤的候选变异可能指示所述候选变异可以被归因于在健康个体中自然发生的突变的一来源或原因(例如,与白血球gDNA相关联)或由于流程错误。
在一些实施例中,所述处理系统回应判定一cfDNA样本的数个序列读数没有质量分数,而过滤所述数个序列读数的数个候选变异。所述处理系统可以使用一噪声模型判定数个候选变异的数个质量分数。所述处理系统可以无碱基对齐而判定所述数个质量分数。在一些实施例中,所述质量分数对一些样本或候选变异而言可能由于缺乏给所述联合模型的训练资料或由于在对一给定的候选变异产生有用的参数上失败的不良训练资料而是缺失的。举例而言,在数个序列读数中的高噪声水平可能导致有用训练资料的不可获得。所述处理系统可以基于一单一变异是否被处理或所述处理系统是否控制一针对性检测组合而调校所述联合模型的特异度及选择性。如其它数个示例,所述处理系统回应判定所述候选变异是一边缘变异人为因素、具有少于一阀值的cfDNA深度(例如,200序列读数)、具有少于一阀值的cfDNA质量分数(例如,60)或对应于人类白血球抗原(HLA),例如,HLA-A而过滤一候选变异。既然与HLA-A相关联的序列可能是难以对齐的,所述处理系统可以对这些区域中的数个序列执行一定制过滤或变异辨认过程。
在一些实施例中,所述处理系统过滤被判定与生殖细胞系突变相关联的数个候选变异。所述处理系统可以通过判定一候选变异以对应于一给定的生殖细胞系突变事件的一适当频率发生,及是存在于已知与生殖细胞系事件相关联(例如,在一核苷酸序列中)的特定一个或更多个位置而判定所述候选突变是生殖细胞系的。此外,所述处理系统可以判定gDNA频率的一点估计,其中C是一常数(例如,0.5):
所述处理系统可以回应判定点
图3Z是根据一实施例的数个cfDNA样本的候选变异的示例性计数的一表格。在图3Z、3AA及3AB中的示例资料是使用数个序列变异产生,所述数个序列变异获得自数个个体的一样本集合。所述数个cfDNA样本包括来自已知具有癌症或另一类型的疾病的数个个体。在展示于图3Z中的示例中,所述处理系统使用图Y的所述方法350以判定所述数个候选变异中的23805个是“绝对”与gDNA相关联(例如,由生殖细胞系突变或血液中的克隆性造血而被解释)及所述数个候选变异中的1360个是可能与gDNA相关联(例如,“较可能为血液”或大于一阀值信心水平)。因此,所述处理系统可以自所述联合模型或另一管道滤出这些候选变异,例如,而使这些候选变异被分类为血液衍生。所述处理系统可以决定既不将2607个不确定(例如,像是血液(bloodish))候选变异的所述计数分类为肿瘤衍生,也不分类为血液衍生。因此,通过调校所述联合模型,举例而言,使用来自所述方法350的所述gDNA比例及gDNA深度质量分数调校所述联合模型,所述处理系统增进分类候选变异来源的颗粒性(例如,不同水平的信心)。图3AA是根据一实施例的来自数个健康个体的数个cfDNA样本的候选变异的示例性计数的一表格。在图3Z及3AA中展示的所述示例计数是由所述处理系统判定的。所述处理系统使用200个读数的一阀值深度、(例如,在一Phred标尺上的)60的阀值质量分数、在具有0.005的生殖细胞系突变频率均方差阀值的对应位置处的质量分数、0.3的阀值gDNA频率点估计、0.05的阀值人为因素再现率、7的阀值局部序列重复计数、0.8的阀值机率(例如,一cfDNA样本的所述真实替代频率是大于一gDNA样本的一真实替代频率的一函数的机率)、0的阀值gDNA深度、1的阀值gDNA深度质量分数及6的阀值gDNA样本比例。此外,所述处理系统200滤出不具有质量分数的候选变异、体细胞变异及HLA-A区域。
图3AB是根据一实施例的基于cfDNA及gDNA的比率而绘制的数个候选变异的一图表。对于一对象的数个被绘制的候选变异中的每个,所述x轴数值代表在数个gDNA样本中观察到的所述AF,及y轴代表在所述对象的一相对应的cfDNA样本中观察到的所述AF。展示在图3AB中的所述示例包括由一联合模型使用一枢纽函数,比如在图3O中例示的曲线345A或曲线345B,所传递的数个候选变异。对于此示例资料及上文中列举的数个参数,所述处理系统200判定具有一相对高的AF
为了增进抓取这些候选变异的准确度,所述处理系统可以使用如在上文中参考图3Y而被描述的所述数个过滤器。进一步地,所述处理系统可以通过在特定条件下使用一枢纽函数的更积极的数个参数来调校所述联合模型。举例而言,回应判定所述gDNA样本的所述AD是大于一阀值深度(例如,0),所述处理系统(例如,对于展示于图3Y中的所述方法350的步骤355B)使用一更大的机率阀值,所述gDNA样本的所述AD大于一阀值深度是健康样本中的血液中的核苷酸突变的支持性证据。在一些实施例中,所述处理系统使用所述更大的机率阀值决定一经修改的枢纽函数(或用于分类真实及伪阳性的另一类型的函数)。举例而言,所述经修改的函数可以具有一更急遽的截止(例如,相较于图3O的曲线345A及345B),所述截止会沿着图3AB中的对角虚线滤除所述集簇的至少一些候选变异。所述处理系统也可以使用如分别在所述方法350的步骤355D及355E中决定的所述gDNA样本质量分数或比例来调校所述经修改的函数。
2.7.示例边缘过滤
边缘过滤被执行(例如,展示图3B中的步骤310C),以滤出由于它们与序列读数的所述边缘的邻近而可能是伪阳性的数个候选变异。
2.7.1.来自人为因素及非边缘变异的特征的示例训练分布
图4A绘示根据一实施例使用训练变异产生一人为分布及一非人为分布的一程序。所述边缘过滤器在一训练程序400中使用来自数个先前样本(例如,训练样本)的训练资料405产生所述人为分布440及非人为分布445。一旦被产生,所述人为分布440及非人为分布445可以个别被储存(例如,在所述模型数据库215中),用于随后在一被需要的时间取回。
训练资料405包括各种序列读数。在所述训练资料405中的数个序列读数可以对应于在所述基因组上的各个位置。在各种实施例中,在所述训练资料405中的数个序列读数是获得自多于一个训练样本。
所述边缘过滤器将在所述训练资料405中的数个序列读数分类到一人为训练资料410A类别、参考等位基因训练资料430类别,或非人为训练资料410B类别中的一个。在各种实施例中,回应判定数个序列读数并不满足被放置在所述人为训练资料410A类别、参考等位基因训练资料430类别,或非人为训练资料410B类别中的任何一个的标准,在所述训练资料405中的所述数个序列读数可以被分类到一无结果或一无分类类别中。
如在图4A中所展示的,可能有多组人为训练资料410A、多组参考等位基因训练资料430,及多组非人为训练资料410B。一般地,在一组别中的数个序列读数跨越(重叠)在所述基因组中的一共同位置。在各种实施例中,在一组中的数个序列读数衍生自一单一训练样本(例如,获得自一单一个体的一训练样本)及跨越在所述基因组中的所述共同位置。举例而言,给定获得自M个不同个体的M个不同训练样本的数个序列读数,可以有M个不同组,每个组包括来自M个不同训练样本中的一个的数个序列读数。虽然随后的描述涉及数个组的跨越在所述基因组上的一共同位置的数个序列读数,所述描述可以被进一步扩张到跨越在所述基因组上的其它位置的其它组的数个序列读数。
对应于在所述基因组上的一共同位置的数个序列读数包括:1)在不同于所述参考等位基因(例如,一ALT)的所述位置包括一核苷酸碱基的数个序列读数及2)在与所述参考等位基因相符合的位置包括一核苷酸碱基的数个序列读数。所述边缘过滤器将包括一ALT的数个序列读数分类到所述人为训练资料410A或非人为训练资料410B中的一个。特定地,满足一个或更多个标准的数个序列读数被分类为人为训练资料410A。所述标准可以是所述ALT的突变的一个类型及所述ALT在所述序列读数上的一位置的一结合。关于突变的一类型的一示例,被分类为人为训练资料的数个序列读数包括一个替代等位基因,所述替代等位基因是一胞嘧啶至胸腺嘧啶(C>T)核苷酸碱基替换或一鸟嘌呤至腺嘌呤(G>A)核苷酸碱基替换。关于所述替代等位基因的所述位置的一示例,所述替代等位基因是距一序列读数的一边缘少于一阀值数量的碱基对。在一实施中,碱基对的所述阀值数量是25个核苷酸碱基对,然而,所述阀值数量可以依实施而变化。
图4B绘示根据一实施例被分类在一人为训练资料410A类别中的数个序列读数。此外,所述数个序列读数中的每个满足一个或更多个标准。举例而言,每个序列读数包括一替代等位基因475A,所述替代等位基因475A是一C>T核苷酸碱基替换。此外,在每个序列读数上的所述替代等位基因2375A是位在一边缘距离450A处,所述边缘距离450A少于一阀值边缘距离460。
具有被分类进入所述非人为训练资料410B中的一替代等位基因的数个序列读数全是具有不满足被分类为人为训练资料410A的标准的一替代等位基因的其它序列读数。举例而言,包括非C>T或G>A核苷酸碱基替换中的一个的一替代等位基因的任何序列读数是被分类为一非边缘训练变异。作为另一个示例,不论核苷酸突变的类型,包括距一序列读数的一边缘大于一阀值数量的碱基对的一替代等位基因的任何序列读数,被分类为非人为训练资料410B。在一实施中,所述碱基对的所述阀值数量是25个核苷酸碱基对,然而,所述阀值数量可以依实施而不同。
图4C绘示根据一实施例被分类在一非人为训练资料410B类别中的数个序列读数。在此,所述数个序列读数中的每个包括不满足两个标准的一替代等位基因475B。举例而言,无论所述替代等位基因475B的所述位置,每个替代等位基因475B可以是一非C>T或非G>A核苷酸碱基替换。作为另一实施例,每个替代等位基因475B是一C>T或G>A苷酸碱基替换,但定位得具有大于所述阀值边缘距离460的一边缘距离450B。
参考所述参考等位基因训练资料430类别,包括所述参考等位基因的数个序列读数被分类在所述参考等位基因训练资料430类别中。图4D绘示与根据一实施例被分类在所述参考等位基因训练资料430类别中的,基因组中的相同位置相对应的数个序列读数。作为一示例,展示于图4D中的所述数个序列读数各包括(与展示于图1B中的所述胞嘧啶核苷酸碱基162符合的)所述参考等位基因442。此外,这些包括所述参考等位基因442的序列读数是被分类在所述参考等位基因训练资料430中,无论在所述参考等位基因及所述序列读数的所述边缘间的所述边缘距离450C。
回到图4A,所述边缘过滤器自被分类在所述人为训练资料410A、非人为训练资料410B及参考等位基因训练资料430中的每个当中的数个序列读数的数个组提取数个特征。数个序列读数的每个组对应于在所述基因组中的相同位置。特定地,数个人为特征420及数个非人为特征425是被提取自在所述人为训练资料410A、非人为训练资料410B及参考等位基因训练资料430中的一个、两个或所有三个中的数个序列读数。人为特征420及非人为特征425的数个示例包括距边缘特征的一统计距离、一显著性分数特征,及一等位基因部分特征。这些特征中的每个在下文中与图4E至4G相关联地被进一步详细描述。
图4E是根据一实施例,用于提取一距边缘统计距离特征的一过程的一示例性描绘。在此,所述边缘过滤器分别自在所述人为训练资料410A中的数个序列读数的一群组及在所述非人为训练资料410B中的数个序列读数的一群组提取人为及非人为距边缘统计距离422A及422B特征。每个距边缘统计距离422A及422B特征可以代表在数个序列读数上的数个替代等位基因475及数个序列读数的所述对应边缘间的距离(例如,核苷酸碱基对的数量)的一平均、中位数或众数。更特定的,距边缘422A的人为统计距离代表横跨在所述人为训练资料410A的一群组中的数个序列读数的边缘距离450A(参见图4B)的结合。类似地,距边缘422B的非人为统计距离代表横跨在所述人为训练资料410B的一群组中的数个序列读数的边缘距离450B(参见图4C)的结合。
图4F是根据一实施例,用于提取一显著性分数特征的一过程的一示例性描绘。所述边缘过滤器自在所述人为训练资料410A中的数个序列读数的一群组及在所述参考等位基因训练资料430中的数个序列读数的一群组的结合提取所述人为显著性分数423A特征。类似地,所述边缘过滤器自在所述非人为训练资料410B中的数个序列读数的一群组及在所述参考等位基因训练资料430中的数个序列读数的一群组的一结合提取所述非人为显著性分数423B特征。一般地,来自所述人为训练资料410A、非人为训练资料410B及参考等位基因训练资料430的数个序列读数的所述数个群组对应到在所述基因组上的一共同位置。因此,对于每个位置,可以有对那个位置的一人为显著性分数423A及一非人为显著性分数423B。虽然随后的描述是关于提取一人为显著性分数423A的所述过程,相同的描述适用于提取一非人为显著性分数423B的所述过程。
所述人为显著性分数423A特征是在所述人为训练资料410A中的一组数个序列读数上的数个替代等位基因475A(例如,在距离一序列读数的边缘的距离或其他测量值方面),与在所述参考等位基因训练资料430中的一组数个序列读数上的数个参考等位基因442的所述位置,是否显著不同到一统计上显著的程度。特定地,人为显著性分数423A是在所述人为训练资料410A中的数个替代等位基因475A的数个边缘距离450A(参见图4B)及在所述参考等位基因训练资料430中的数个参考等位基因442的数个边缘距离450C(参见图4D)间的比较。
在各种实施例中,所述边缘过滤器对数个边缘距离间的所述比较执行一统计显著性检测。作为一示例,所述统计显著性检测是一威尔克森排序和检定(Wilcoxon rank-sumtest)。在此,所述边缘过滤器依每个边缘距离450A及450C分别为在所述人工训练资料410A中的每个序列读数及在所述参考等位基因训练资料430中的所述等位基因训练资料指定一排序。举例而言,具有最大的边缘距离450A或450C的一序列读数可以被指定最高的排序(例如,排序=1),具有次大的边缘距离450A或450C的所述序列读数可以被指定次高的排序(例如,排序=2),依此类推。所述边缘过滤器将在所述人造训练资料410A中的数个序列读数的所述中位数排序与在所述参考等位基因训练资料430中的数个序列读数的所述中位数排序相比较,以决定在所述人为训练资料410A中的数个替代等位基因475的所述位置是否与在所述参考等位基因训练资料430A中的数个参考等位基因442的位置相异。作为一示例,所述数个中位数排序间的比较可以收获一p值,所述p值代表所述数个中位数排序是否显著不同的一统计显著性分数。在各种实施例中,所述人为显著性分数423A是由一Phred分数代表,所述Phred分数可以被表达为:
Phred分数=-10log
其中P是所述p值分数。总合而言,一低人为显著性分数423A表明在数个中位数排序间的所述差异并非统计显著的,而一高人为显著性分数423A表明在数个中位数排序间的所述差异是统计显著的。
图4G是根据一实施例,用于提取一等位基因部分特征的一过程的一示例性描绘。所述等位基因部分特征意指替代等位基因475A或475B的所述等位基因部分。特定地,人为等位基因部分424A意指替代等位基因475A的所述等位基因部分(参见图4B),而非人为等位基因部分424B意指替代等位基因475B的所述等位基因部分(参见图4C)。所述等位基因部分代表数个序列读数的所述部分,所述数个序列读数对应于在所述基因组中包括所述替代等基因的位置。举例而言,在所述人为训练资料410A中可能有共X个包括所述替代等位基因475A的序列读数。在所述非人为训练资料410B中也可能有共Y个包括所述替代等位基因475B的序列读数。此外,在所述参考等位基因训练资料430中也可能有共Z个具有所述参考等位基因的序列读数。因此,所述替代等位基因475A的所述人为等位基因部分424A可以被记为等位基因部分
回到图4A,所述边缘过滤器编码自遍及所述基因组的各个位置的数个群组的数个序列读数的提取的数个人为特征420,以产生一人为分布440。此外,所述边缘过滤器编码自遍及所述基因组的各个位置的数个群组的数个序列读数提取的数个非人为特征425,以产生一非人为分布445。图4绘示一特定实施例,在所述特定实施例中,三个不同的特征420A被用于产生一人为分布440及三个不同的特征420B被用于产生一非人为分布445。在另一实施例中,更少或更多个每个类型的特征420A或420B被用于产生一人为分布440或非人为分布445。
图4H及4I绘示根据各种实施例,用于辨识数个边缘变异的数个示例分布。特定地,图4H绘示产生自人为特征420或非人为特征425的一个类型的一分布440或445。虽然图4G为了例示起见而绘示一常态分布,在实施中,分布440及445可以依所述特征420或425的所述数个数值而变化。
在另一实施例中,所述边缘过滤器可以使用多个人为特征420或非人为特征425以产生一单一分布440或445。举例而言,图4I绘示产生自两个类型的人为特征420或两个类型的非人为特征425的一分布440或445。在此,所述分布440或445描述在一第一特征及一第二特征间的一关系。在进一步的实施例中,一分布440或445可以代表三个或更多个类型的人为特征420或非人为特征425间的关系。
2.7.2.对辨识边缘变异的一样本特定比率的示例判定
图4J绘示根据一实施例,用于决定一样本特定预测比率的一方块图流程462。一般地,所述边缘过滤器进行对所述样本480中的数个经辨认变异的一全样本分析,以决定特定于所述样本480的所述预测比率486。换言之,展示于图4J中的所述过程462可以对每个样本480执行一次。
一经辨认变异482的数个序列读数被获得自一样本480。一般地,一经辨认变异482的所述数个序列读数意指跨越在所述基因组中所述经辨识的变异对应的位置的一组序列读数。
对于每个经辨认变异,所述边缘过滤器自所述经辨认变异482的所述数个序列读数提取数个特征484。自一经辨认变异482的所述数个序列读数提取的每个特征484可以是在所述数个序列读数中的数个替代等位基因距边缘的一统计距离、一替代等位基因的一等位基因部分、一显著性分数、另一类型的特征或上述的一些组合。所述边缘过滤器将横跨所述样本480的数个经辨认变异而被提取的数个特征484的值作为输入施加到一样本特定比率预测模型485,所述模型决定所述样本480的一预测比率486。所述样本480的所述预测比率486意指为边缘变异的经辨认变异的一预测比率。在各种实施例中,所述预测比率是在0及1间的一数值,例如,是含括性的(inclusive)。
如在图4J中所展示的,所述样本特定比率预测模型485使用所述先前产生的人为分布440及非人为分布445两者。所述样本特定比率预测模型485通过鉴于所述人为分布440及非人为分布445分析提取自所述样本480中的数个经辨认变异的数个序列读数的所述数个特征484而判定所述预测比率486。作为一示例,给定所述人为分布440及非人为分布445,所述样本特定比率预测模型485执行一拟合优度(goodness of fit)以判定一预测比率486,所述预测比率486解释所述数个观察到的特征。在一实施中,所述样本特定比率预测模型485执行一最大似然性估计以估计鉴于所述人为分布440及非人为分布445,最大化观察到所述数个特征484的似然性的所述预测比率。然而,其它实施可以使用其它程序。
在一实施例中,所述估计的所述似然性等式可以被表示为:
L(w|x)=w*(L(x)|d
其中w是预测比率486、x代表所述数个特征484、d
如展示于图4J中的,所述边缘过滤器可以自一经辨认变异482的数个序列读数提取多个特征484,并将所述数个特征484提供至所述比率预测模型485。举例而言,可能有三个类型的特征(例如,在所述数个序列读数中的数个替代等位基因距边缘的统计距离、一替代等位基因的一等位基因部分或一显著性分数)。进一步推广,假设n个不同类型的特征484(例如,x
总合而言,回应判定提取自所述样本480中的所述数个经辨认变异的数个序列读数的数个特征484的分布较类似所述人为分布440而非所述非人为分布445,所述比率预测模型485决定一高预测比率486,所述高预测比率486指示一高预测比率的经辨认变异可能是边缘变异。替代地,回应判定提取自所述样本480中的所述数个变异的数个序列读数的数个特征484的分布较类似所述非人为分布445而非所述人为分布440,所述比率预测模型485决定一低预测比率486,所述低预测比率486指示一低预测比率的经辨认变异可能是边缘变异。如下文中讨论的,所述预测比率486可以被用于控制在一样本中数个边缘变异被辨识的“积极性”水平。因此,被指派一高预测比率486的一样本可以被积极地过滤(例如,使用较广的标准滤除大量的可能边缘变异)而被指派一低预测比率486的一样本可以被较不积极地过滤。
2.7.3.用于辨识一边缘变异的示例变异特定分析
图4K绘示根据一实施例用于辨识边缘变异的一边缘变异预测模型492的应用。在一变异特定分析464中,所述边缘过滤器分析一经辨认变异482的数个序列读数以决定所述经辨认变异是否是一边缘变异。绘示于图4K中的所述程序可以对每个经辨认变异或侦测自一单一样本480的一组经辨认变异被执行。
在一实施例中,所述边缘过滤器基于所述经辨认变异的突变的一个类型过滤数个经辨认变异。在此,并非所述C>T或G>A突变类型的一经辨认变异可以被自动分类为一非边缘变异。替代地,属于所述C>T或G>A的任何经辨认变异在下文中描述的随后步骤中被进一步地分析。
如展示于图4K中的,所述边缘过滤器自一经辨认变异482的所述数个序列读数提取数个特征484。一经辨认变异482的所述数个序列读数的数个被提取的特征484可以是如在图4J中展示的提取自一经辨认变异482的所述数个序列读数的相同特征484。亦即,所述数个特征484在其它类型的特征当中,可以是下述的一个或更多个:距所述数个序列读数中的数个替代等位基因的边缘的一统计距离、一替代等位基因的一等位基因部分或一显著性分数。
所述边缘过滤器提供所述数个经提取的特征484的数值作为对所述边缘变异预测模型492的输入。如展示于图4K中的,所述边缘变异预测模型492使用先前产生的所述人为分布440及所述非人为分布445两者。所述边缘变异预测模型492产生多个分数,比如代表所述经辨认变异是一边缘变异的一似然性的一人为分数494以及代表所述经辨认变异是一非边缘变异的一似然性的一非人为分数496。
特定地,所述边缘变异预测模型492鉴于所述人为分布440及所述非人为分布445而决定观察到一经辨认变异482的所述数个序列读数的所述数个特征484的所述机率。在一实施例中,所述边缘变异预测模型492通过鉴于所述人为分布440分析所述数个特征484而决定所述人为分数494及通过鉴于所述非人为分布445分析所述数个特征484而决定所述非人为分数496。
作为一视觉化示例,回头参考展示于图4H中的所述示例分布,所述边缘变异预测模型492基于一特征484坐落于所述x轴的何处而辨识一机率。在此示例中,所述经辨识机率可以是由所述边缘变异预测模型492输出的一分数,比如所述人为分数494或非人为分数496。
如展示于图4K中的,所述边缘过滤器将所述人为分数494及非人为分数496与(如在图4J中描述的)所述样本特定预测比率486结合。所述结合收获所述边缘变异机率498,所述边缘变异机率498代表所述经辨认变异是一过程人为因素的一结果的似然性。
在一实施例中,边缘变异机率498可以被表示为鉴于提取自所述经辨认变异482的数个序列读数的所述数个特征484,所述经辨认变异是一边缘变异的所述事后机率。所述人为分数494、所述非人为分数496及所述样本特定预测比率486的所述结合可以被表示为:
所述边缘过滤器可以将所述边缘变异机率498与一阀值比较。回应判定所述边缘变异机率498大于所述阀值,所述边缘过滤器判定所述经辨认变异是一边缘变异。回应判定所述边缘变异机率498少于所述阀值,所述边缘过滤器判定所述经辨认变异是一非边缘变异。
2.7.4.用于辨识一边缘变异的示例变异特定分析
图4L绘示根据一实施例,辨识并报告侦测来自一样本的数个边缘变异的一流程452。来自各种序列读数的数个经辨认变异自一样本被接收454A。一样本特定预测比率基于来自所述样本的所述数个经辨认变异的数个序列读数而对所述样本被决定454B。作为一示例,一预测比率是通过执行一最大似然性估计而被决定。在此,所述预测比率是一参数值,所述参数值鉴于先前产生的分布(例如,给定特定条件)最大化观察到所述数个经辨认变异的数个序列读数的数个特征484的所述似然性。
对于每个经辨认变异,一个或更多个特征484自所述变异的所述数个序列读数被提取454C。所述数个被提取的特征484的数个数值作为输入被应用454D到一经训练的模型,以获得一人为分数494。所述人为分数494代表所述经辨认变异是一边缘变异(例如,一程序人为因素的结果)的一似然性。所述经训练模型进一步输出一非人为分数496,所述非人为分数496代表所述经辨认变异是一非边缘变异(例如,非一程序人为因素的结果)的一似然性。
对于每个经辨认变异,一边缘变异机率498通过结合所述经辨认变异的所述人为分数494、所述经辨认变异的所述非人为分数496及所述样本特定预测比率486而被产生498。基于所述边缘变异机率498,所述经辨认变异可以被报告454E为一边缘变异(例如,作为一程序人为因素的一结果而被辨识的变异)。
2.7.5.边缘过滤器的数个示例
下述数个示例被提出以提供本领域的一般技术人员如何制造及使用所揭示的实施例的一完整揭示及描述,及并非意在限制被认作本发明的范围。已努力确保所用数字(例如,量、温度、浓度等)的准确性,但一些实验误差及偏差应被允许。将被本领域一般技术人员所理解的是,鉴于本揭示,许多修改及改变可以在不脱离本发明所意欲包括的范围的情况下在被例示的特定实施例中被作出。
2.7.5.1.分类人为因素及清理训练样本
图4M、4N及4O中的每个绘示根据各种实施例被分类在所述人为或非人为类别中的一个当中的示例训练变异的数个特征。展示在图4M、4N及4O中的数个示例包括使用展示于图4A中的所述程序400决定的人为分布及非人为分布。数个cfDNA样本通过一抽血自具有乳癌、肺癌或前列腺癌中的一个的数个对象被获得。所述样本集合对每个类型的癌症(乳癌、肺癌及前列腺癌)包括至少50个对象。对于所有参与对象,血液在活检(之前或之后)六周内被同步抽取。
所述边缘过滤器将包括所述基因组上的一特定位点的一替代等位基因的数个序列读数分入人为及非人为组,如同在下文中被描述的。此外,包括所述基因组上的所述特定位点的所述替代等位基因的所述数个序列读数被包括为稍后被使用的参考等位基因资料,以决定所述数个序列读数的数个特征。
所述边缘过滤器基于两个标准将包括所述替代等位基因的数个序列读数分类到所述人为或非人为类别中。一第一标准包括25个核苷酸碱基对的一阀值距离。因此,被分类在所述人为类别中的数个序列变异包括一替代等位基因,所述替代等位基因在距所述序列读数的所述边缘25个核苷酸对内。一第二标准是一个类型的核苷酸碱基突变。特定地,被分类入所述人为类别的数个序列读数包括一替代等位基因,所述替代等位基因是C>T或G>A突变中的一个。所述边缘过滤器将包括不满足这两个标准的一替代等位基因的数个序列读数分入所述非人为类别。
所述边缘过滤器自一经辨认变异的数个序列读数提取数个特征,所述数个序列读数包括包括一替代等位基因的序列读数以及包括所述参考等位基因的序列读数。在此,被提取的所述三个类型的特征包括:1)数个替代等位基因距所述序列读数的所述边缘的中位数距离、2)所述替代等位基因的等位基因部分、及3)显著性分数。所述三个类型的被提取特征被编译及用以产生展示于图4M至4O中的所述人为分布及非人为分布。
图4M至4O中的每个展示一人为分布(左)及非人为分布(右)。每个分布绘示提取自被分类为人为训练资料或非人为训练资料的数个序列读数的两个特征间的一关系。特定地,图4M绘示所述显著性分数及距边缘的中位数距离间的一关系。图4N绘示所述等位基因部分的分布及距边缘的中位数距离间的一关系。图4O绘示所述等位基因部分及显著性分数间的一关系。
数个趋势在展示于图4M至4O中的所述人为分布及非人为分布间被观察到。值得注意的是,在所述人为类别中的边缘变异倾向于具有高显著性分数(例如,如展示于图4M及4O中,边缘变异在显著性分数100处的高集中度),而在所述非人为类别中的非边缘变异倾向具有远远较低的显著性分数。此外,距所述边缘的一较低的中位数距离与边缘变异的一较高的集中度相关联。举例而言,图4M及图26N绘示具有一替代等位基因的边缘变异相对于距一边缘25核苷酸碱基的一中位数距离,在位于或接近距一边缘零核苷酸碱基的一中位数距离有一较高浓度。值得注意的,一较大数量的非边缘变异也包括在距一序列读数的所述边缘25核苷酸碱基内的一替代等位基因(参见图4M及4N)。这显示有一群体的非C>T及非G>A核苷酸碱基替换被辨识为经辨认变异。
图4P绘示根据一实施例对遍及各个对象样本的数个边缘变异及非边缘变异的辨识。图4P包括来自各种对象样本的资料。特别地,图4P将数个对象样本的经辨识的边缘变异及非边缘变异(y轴)作为距数个序列读数的所述边缘的所述中位数距离(x轴)的一函数。经辨识的数个边缘变异带有斜纹影线图样(diagonal hatching patterns)地被展示,而数个非边缘变异被不带有图样地被展示(例如,单色)
图4P显示对于每个对象样本,所述边缘过滤器的所述过滤方法可以不同地辨识边缘变异及非边缘变异。举例而言,MSK-VP-0082(例如,自上方数来第五个样本)包括一大数量的边缘变异,所述边缘变异展现距所述边缘10及25核苷酸碱基对间的一中位数距离。此外,MSK-VP-VL-0081(例如,自上方数来第六个样本)包括一大量的非边缘变异,所述非边缘变异展现距所述边缘10及25核苷酸碱基对间的一中位数距离。此样本特定过滤相较于在所有样本运用相同过滤方法的过滤器,允许边缘变异的更准确的辨识及移除。非样本特定过滤器的数个示例可以运用一固定截止,所述固定截止是基于一特征,比如一等位基因频率,而使若一替代等位基因的所述等位基因频率比一固定的阀值量更大,则对应于所述替代等位基因的所述经辨认变异被分类为一边缘变异。
图4Q绘示根据一实施例,在使用不同的边缘过滤器移除边缘变异后,作为在cfDNA中被辨认的变异的一部份而在实性肿瘤及cfDNA两者中被辨认的一致变异。图4R绘示根据一实施例,在使用不同的边缘过滤器移除边缘变异后,作为在实性肿瘤中被辨认的变异的一部份而在实性肿瘤及cfDNA两者中被辨认的一致变异。特定地,图4Q及图4R绘示依所应用的所述边缘变异过滤器(例如,无边缘变异过滤器、简单边缘变异过滤器或样本特定边缘变异过滤器)而变化的一致的数量。
对于展示于图4Q及图4R中的资料集,样本被获得自数个对象及使用上文描述的所述化验程序被处理。被包括在一初始集合中的数个候选变异尚未经历进一步过滤以移除边缘变异。
在两个分离的情况中,在所述初始集合中的这些候选变异被所述边缘过滤器进一步过滤以辨识及移除数个边缘变异。一第一情况包括一第一过滤器的应用,所述第一过滤器在下文中称作一简单边缘变异过滤器。所述简单边缘变异过滤器移除展现出落在一阀值距离下的,距数个序列读数的边缘的一中位数距离的数个经辨认变异。在此,所述阀值距离是基于被分类在所述人为训练资料类别中的数个训练序列读数中的数个边缘变异的位置而被决定。特定地,所述阀值距离是被表示为所述数个边缘变异距序列读数的边缘的所述中位数距离及所述数个边缘变异距序列读数的边缘的所述中位数距离的所述中位数绝对离差(median absolute deviation)的总和。所述简单边缘变异过滤器是移除满足此阀值距离标准的所有变异的一简单无差别过滤器。所述第二过滤器意指所述边缘过滤程序。在此,所述样本特定边缘变异过滤器在考虑对所述样本观察到的数个经辨认变异的分布的同时,辨识数个边缘变异。
在使用所述简单边缘变异过滤器或所述样本特定边缘变异过滤器移除边缘变异后留存的所述非边缘变异被保留,为与一常规方法相比较而被分析。如在下文中所称的,所述常规方法意指使用一常规过程,特别是可行癌症目标的纪念斯隆凯特林综合突变分析管道(Memorial Sloan Kettering Integrated Mutation Profiling of ActionableCancer Targets(MSK-IMPACT)Pipeline)自实性肿瘤样本辨识基因组变异(Cheng,D.等人,可行癌症目标的纪念斯隆凯特林综合突变分析管道(MSK-IMPACT),用于实性肿瘤分子肿瘤学的一基于杂合捕捉的次世代定序临床化验,分子诊断学杂志,17(3),p.251-264)
在此,为非边缘变异及由所述常规方法侦测到的经辨认变异被称为一致变异。
图4Q绘示在应用一边缘过滤器(或非应用一边缘过滤器)后在一cfDNA样本中被侦测到的一致变异及在实性肿瘤组织中被侦测到的数个经辨认变异除以在cfDNA中被侦测到的所述非边缘变异。此比例可以被表示为:
图4R绘示在应用一边缘过滤器(或非应用一边缘过滤器)后在一cfDNA样本中被侦测到的一致变异及在实性肿瘤组织中被侦测到的数个经辨认变异除以在实性肿瘤组织中被侦测到的数个经辨认变异。此比例可以被表示为:
展示于图4Q及图4R中的一致变异的百分比描述数个有趣的趋势。相较于绘示于图4Q中的一致变异的所述百分比,一显著较大百分比的一致变异被展示在图4R中。作为一示例,在乳癌中被侦测到的一致变异除以单在cfDNA中侦测到的经辨认变异的百分比是9.8%。这显著低于在乳癌中被侦测到的一致变异除以在实性肿瘤组织中被侦测到的经辨认变异的73%。这显示在cfDNA样本中的非边缘变异的辨识比在实性肿瘤组织中辨识变异的常规方法(不论癌症类型)达到较高的敏感度。
关于图4Q中的所述简单边缘变异过滤器,所述简单边缘变异过滤器的应用增加所述数个经辨认变异的特异度。举例而言,相较于所述无边缘变异过滤器,所述简单边缘变异过滤器的应用增加在乳癌、肺癌及前列腺癌中侦测到的经辨认变异的特异度(例如,分别为9.5%至11%、45%至49%及22%至27%)。然而,这在特异度上的增加以敏感度为代价,如展示在图4R中的。与所述无边缘变异过滤器相较,所述简单边缘变异过滤器的应用减少在乳癌、肺癌及前列腺癌侦测到的经辨认变异的所述敏感度(例如分别为73%至69%、73%至70%及76%至71%)。
相对地,一样本特定边缘变异过滤器的应用在不牺牲敏感度下增进特异度。如展示于图4Q中的,相较于所述无边缘变异过滤器,所述样本特定边缘变异过滤器的应用增加在乳癌、肺癌及前列腺癌中侦测到的经辨认变异的特异度(例如,分别为9.5%至9.8%、45%至47%及22%至27%)。此外,如展示在图4R中的,与所述无边缘变异过滤器相较,所述样本特定边缘变异过滤器的应用维持在乳癌、肺癌及前列腺癌侦测到的经辨认变异的所述敏感度(例如分别保持在73%、73%及76%)。
2.8.结合的过滤及评分的示例
在下述的图4S至4Z中的所述示例资料是使用数个序列读数产生的。所述数个序列读数获得自一游离基因组研究的数个个体的一样本集合及使用在本文中描述的一个或更多个方法(例如,噪声建模、联合建模、边缘过滤、非同义过滤等)被处理。所述样本集合包括健康个体,血液样本(例如,cfDNA)被获得自所述健康个体。此外,所述样本集合包括已知具有至少一个类型的癌症的数个个体,血液样本及组织样本(例如,肿瘤或gDNA)被获得自所述个体。所述资料是收集自遍及美国及加拿大的约140间中心的个体。
图4S是根据一实施例的,描述用于一游离基因组研究的一样本集合的数个个体的一表格。所述样本集合包括已知至少具有乳癌、肺癌、前列腺癌、大肠直肠癌及其它类型的癌症的个体。所述数个个体的人口数据(demographic data)(例如,年龄、性别及族群)也被展示在图4S中。图4T是根据一实施例显示与图4S的游离基因组研究的所述样本集合相关联的癌症的类型的一图表。如在图4T中所展示的,所述循环游离基因组图谱(circulatingcell-free genome atlas,CCGA)样本集合包括下列癌症类型:乳癌、肺癌、前列腺癌、大肠直肠癌、肾癌、子宫癌、胰脏癌、食道癌、淋巴瘤、头颈癌、卵巢癌、肝胆癌、黑色素瘤、子宫颈癌、多发性骨髓瘤、白血病、甲状腺癌、膀胱癌、胃癌及肛门直肠癌。图4U是根据一实施例的描述图4S的游离基因组研究的所述样本集合的另一表格。特定地,所述表格展示基于癌症的临床阶段被组织起来的,已知具有癌症的所述数个样本的计数。
图4V展示根据一实施例,使用一个或更多个类型的过滤器及模型决定的经辨认变异的示例性计数的数个图表。所述数个图表中的每个包括所述样本集合的数个数据点,所述数个数据点被描绘在代表所述对应个体的年龄的一x轴上及代表由所述处理系统处理后的经辨认变异的一数量的一y轴上。图表466A包括来自使用噪声建模处理所述样本集合的数个序列读数的结果。图表466B包括来自在所述噪声建模外使用联合建模及边缘过滤处理所述样本集合的数个序列读数的结果。图466C包括来自在所述联合建模、边缘过滤及噪声建模外,使用非同义过滤处理所述样本集合的数个序列读数的结果。
如由图表的进展所例示地,经辨识变异的所述数量随着过滤的程度增加而逐渐减少。因此,所述数个示例显示由所述处理系统进行的非同义过滤、联合建模、边缘过滤及噪声建模可以成功地辨识及移除一显著量的伪阳性。
因此,所述处理系统提供一更准确的变异辨认器,所述变异辨认器减轻来自各种噪声或人为因素的影响。使用揭示的方法分析来自血液样本的cfDNA的针对性化验可能能够捕捉肿瘤相关的生物学特征。一略为成比例的关联性可以在所述图表中,在所述经辨认变异的计数及所述个体的年龄间被观察到(例如,在图表466A中更加明显)此外,如预期地,比起非癌症样本,癌症样本有更大的经辨认变异计数。
图4W是根据一实施例的,已知具有各种类型的癌症及处于癌症的不同阶段的数个样本的经辨认变异的示例计数的一表格。图4X是根据一实施例的,已知具有各种类型的癌症且处于癌症的不同阶段的样本的经辨认变异的示例性计数的一图表。如由已知具有乳癌、大肠直肠癌、肺癌或前列腺癌的样本的盒状图所展示的,经辨认变异的所述中位数倾向于随癌症的分期自I至IV而增加,及与所述癌症样本的所述数量相较,所述非癌症样本的所述数量相对较低。
图4Y是根据一实施例的,已知具有早期或晚期癌症的样本的经辨认变异的示例性计数的一图表。图4Z是根据一实施例的,已知具有早期或晚期癌症的数个样本的经辨认变异的示例性计数的另一图表。特定地,图4Y及图4Z各自展示来自与乳癌(例如,HER2+、HR+|HER2-、TNBC)及肺癌(例如,肺腺癌、小细胞肺癌及鳞状细胞癌)相关联的cdstg1lh_编组基因的数个序列读数的数个经辨认变异。图4Y至4Z展示经辨认变异的所述数量倾向于随着癌症自早期至晚期进展而增加的一趋势。所述示例资料指示所述处理系统可以侦测基因中的数个序列的不同亚型或变异。此外,相较于癌症样本的所述数量,非癌症样本的所述数量相对较低。
3.全基因组计算机分析
3.1.全基因组特征
再度简要地参照图1B至1D,所述全基因组计算机分析140C接收由所述全基因组定序化验132产生的数个序列读数,及基于所述数个序列读数决定数个全基因组特征152的数值。全基因组特征152的数个示例包括下述的任何特征:决定自遍及所述基因组的数个区间段中的每个的一区间段的数个特征(例如,遍及来自一cfDNA样本的所述基因组的数个区间段的区间段分数及区间段变异数及/或遍及来自一gDNA样本的所述基因组的数个区间段的区间段分数及区间段变异数)、遍及所述基因组的数个节段的数个特性(例如,遍及来自一cfDNA样本的所述基因组的数个节段的节段分数及节段变异数及/或遍及来自一gDNA样本的所述基因组的数个节段的节段分数及节段变异数)、拷贝数目畸变的总数、每条染色体(或一染色体的部分,比如一染色体臂)的拷贝数目畸变的存在,及经缩减的维度特征。
一般地,区间段分数及节段分数代表一经标准化的序列读数计数。特定地,区间段分数及节段分数代表代表被分类在一区间段或节段中的数个序列读数的一总数,所述总数基于在所述区间段或节段中的预期序列读数(例如,基于训练资料被预期)的一总数而被标准化。
在一些实施例中,拷贝数目畸变的所述总数是一样本的一单一,数值性的特征值。举例而言,每条染色体的拷贝数目畸变的存在可以是0或1的一数值,所述数值指示一拷贝数目畸变是否位于一特定染色体上。在此,拷贝数目畸变在一工作流程上被断定,所述工作流程可以自其它拷贝数目事件,例如在非体细胞来源中发生(例如,在血液中发生)的一拷贝数目变异中准确地区辨拷贝数目畸变。所述拷贝数目畸变侦测工作流的一示例在下文中被描述。
经维度缩减的特征意指(相较于原始序列读数的资料)已被缩减到一较低维度空间,同时依然代表所述原始序列读数的主要特性的特征。作为一示例,遍及所述基因组的数个区间段的区间段分数可以被缩减到一较低维度空间及由经缩减的维度特征代表。经缩减的维度特征可以经由一维度缩减过程,比如主成分分析(PCA)而被产生。
一般地,前述的数个全基因组特征152的特征值是经由一个或两个工作流程而被决定。图5A绘示根据一实施例,用于决定数个全基因组特征的两个不同的工作流程的一示例性流程。步骤505及506A至C在下文中将被称为拷贝数目畸变侦测工作流程及步骤505及508A至C在下文中将被称为维度缩减工作流程。
3.2.拷贝数目畸变侦测工作流程
首先参考所述拷贝数目畸变侦测工作流程,遍及所述基因组的数个区间段的所述特性的特征、遍及所述基因组的数个节段的所述特性及每条染色体的拷贝数目畸变的存在可以经由所述拷贝数目畸变侦测工作流程被决定。在本领域中已知的一拷贝数目畸变工作流程,而非所描述的工作流程也可以被使用(参见,例如,美国专利申请第16/352,214号,上述专利申请通过引用被并入本文中)。
举例而言,如展示于图5A中的,在步骤505,衍生自一cfDNA样本的数个序列读数被获得,及可选地,在步骤506A,衍生自一gDNA样本的数个序列读数被获得。如上文中与图1B至1D关联地被描述的,衍生自cfDNA115及/或自gDNA(例如,WBC DNA120)的所述数个序列读数可以被接收自一全基因组定序化验132。一般而言,在本领域中已知的任何全基因组定序化验可以被使用(参见,美国专利第2013/0040824号及美国专利第2013/0325360号,上述申请通过引用被并入到本文中)。在另一实施例中,如本领域中已知的,衍生自一全基因组亚硫酸氢盐定序化验或衍生自一针对性检测组合的数个序列读数可以被用于判定数个全基因组特征的数值。
在步骤506B,衍生自cfDNA及gDNA中的每个的数个序列读数被分析,以辨识遍及一基因组的数个区间段及数个节段的数个特性。一般地,一区间段包括一基因组的一系列核苷酸碱基。一节段意指一个或更多个区间段。因此,每个序列读数被分类到区间段及/或节段中,所述区间段及/或节段包括对应到所述序列读数的一系列的核苷酸碱基。所述基因组的每个统计显著区间段或节段包括被分类到指示一拷贝数目事件的所述区间段或节段中的数个序列读数(或经标准化的数个序列读数)的一总数。一般地,一统计显著区间段或节段包括一序列读数计数,即使计入可能的混淆因素,所述序列读数计数也显著不同于对所述区间段或节段的一预期序列读数计数。可能的混淆因素的数个示例包括处理偏误(processing biases)、在所述区间段或节段中的变异性,或在所述样本(例如,cfDNA样本或gDNA样本)中的一总体噪声水平。因此,一统计显著区间段及/或一统计显著节段的所述序列读数计数很可能指示一生物性异常,比如在所述样本中的一拷贝数目事件的存在。
一般地,数个cfDNA序列读数的所述分析及数个gDNA序列读数的所述分析独立于彼此地被执行。在各种实施例中,数个cfDNA序列读数的所述分析及数个gDNA序列读数的所述分析是平行地被执行。在一些实施例中,数个cfDNA序列读数的所述分析及数个gDNA序列读数的所述分析是在不同的时间被执行,所述时间依所述数个序列读数何时被获得而定。
现在参考图5B,图5B是根据一实施例的,描述用于辨识衍生自cfDNA及gDNA样本的数个区间段及数个节段的特性的分析的一示例流程。特定地,图5B绘示包括在展示于图5A中的步骤506B当中的额外步骤。因此,步骤510A至510I可以对一cfDNA样本执行,及类似地,步骤510A至510I可以分离地对一gDNA样本执行。
在步骤510A,一区间段序列读数计数对一参考基因组的每个区间段被决定。一般地,每个区间段代表所述基因组的若干个相邻的核苷酸碱基。一基因组可以由许多(例如,数百甚至数千个)区间段组成。在一些实施例中,在每个区间段中的核苷酸碱基的数量于在所述基因组中的所有区间段间皆恒定。在一些实施例中,在每个区间段中的核苷酸碱基的所述数量对所述基因组中的每个区间段各不相同。在一实施例中,在每个区间段中的核苷酸碱基的所述数量是介于10千碱基(kb)及1百万碱基(mb)间。在一实施例中,在每个区间段中的核苷酸碱基的所述数量是介于25千碱基(kb)及200kb间。在一实施例中,在每个区间段中的核苷酸碱基的所述数量是介于40kb及100kb间。在一实施例中,在每个区间段中的核苷酸碱基的所述数量是介于45kb及75kb间。在一实施例中,在每个区间段中的核苷酸碱基的所述数量是50kb。在实践中,其它区间段尺寸也可以被使用。
一区间段的所述序列读数计数代表被分类到所述区间段中的数个序列读数的一总数。若所述序列读数跨越一阀值数量的被包括在所述区间段中(亦即,被对齐或绘制到一区间段)的核苷酸碱基,则所述序列读数被分类到所述区间段中。在一实施例中,被分类到一区间段中的每个序列读数跨越至少一个被包括在所述区间段中的核苷酸碱基。现在参考图5C,图5C是根据一实施例的,与一参考基因组512的数个区间段514相关联的序列读数516的一示例性描绘。序列读数516A、序列读数516B及序列读数516C可以个包括一不同数量的核苷酸碱基,及可以跨越所述数个区间段514的一个或更多个。
如展示于图5C中的,相较于在一区间段(例如,区间段514B)中的数个核苷酸碱基的所述数量,序列读数516A包括较少的核苷酸碱基。在此,序列读数516A是被分类在区间段514B中。序列读数516B跨越被包括在区间段514C及514D两者中的数个核苷酸碱基。因此,序列读数516B被分类到区间段514C及区间段514D两者中。序列读数516C跨越被包括在区间段514B、区间段514C及514D中的数个核苷酸碱基。因此,序列读数516C被分类到区间段514B、区间段514C及区间段514D中的每个当中。
为了决定每个区间段的所述区间段序列读数计数,被分类到每个区间段中的所述数个序列读数被量化。因此,展示于图5C中的区间段514A具有为零的一区间段序列读数计数、区间段514B具有二的一区间段序列读数计数(例如,序列读数516A及序列读数516B)、区间段514C具有二的一区间段序列读数计数(例如,序列读数516B及序列读数516C)、区间段514D具有二的一区间段序列读数计数(例如,序列读数516B及序列读数516C),及区间段514E具有一的一区间段序列读数计数(例如,序列读数516C)。
返回图5B,在步骤510B,每个区间段的区间段序列读数计数被标准化以移除一个或更多个不同的处理偏误。一般地,一区间段的所述区间段序列读数计数基于先前为所述相同区间段决定的处理偏误而被标准化。在一实施例中,标准化所述区间段序列读数计数涉及以代表所述处理偏误的一数值除所述区间段序列读数计数。在一实施例中,标准化所述区间段序列读数计数涉及自所述区间段序列读数计数减去代表所述处理偏误的一数值。对一区间段的一处理偏误的数个示例可以包括鸟嘌呤-胞嘧啶(GC)含量偏误、可作图性偏误或如本领域中所知的,经由一主成分分析捕捉到的其它形式的偏误。对一区间段的处理偏误可以使用先前训练资料,例如,获得自健康个体的数个序列读数而被判定。
举例而言,在一实施例中,在步骤510C,每个区间段的一区间段分数是通过以所述区间段的所述预期区间段序列读数计数来修改所述区间段的所述区间段序列读数计数而被决定。步骤510C用于标准化所述观察到的区间段序列读数计数,而使若特定区间段横跨许多样本一致地具有一高序列读数计数(例如,高预期区间段序列读数计数),则所述被观察到的区间段序列读数计数的所述标准化解释该趋势。所述区间段的所述预期序列读数计数可以自训练资料(例如,来自数个健康个体的序列读数)被决定。对每个区间段的所述预期序列读数计数的产生在下文中被更详细地描述。
在一实施例中,一区间段的一区间段分数可以被表示为所述区间段的所述观察到的序列读数计数与所述区间段的所述预期序列读数计数的比率的对数。举例而言,区间段i的区间段分数b
在其它实施例中,所述区间段的所述区间段分数可以被表示为所述区间段的所述观察到的序列读数计数与所述区间段的所述预期序列读数计数间的比率(例如,
在此,在所述第一组518A中的数个区间段的所述观察到的序列读数计数及预期序列读数计数可能并不显著地不同。然而,在所述第二组518B中的数个区间段的所述观察到的序列读数计数可能显著高于所述数个区间段的对应预期读数计数。因此,在所述第二组518B中的数个区间段中的每个的所述区间段分数高于在所述第一组518A中的数个区间段中的每个的所述区间段分数。在所述第二组518B中的数个区间段的较高的区间段分数指示在区间段M、区间段M+1及区间段M+2中的所述观察到的序列读数计数是一拷贝数目事件的一结果的一较高的似然性。
所述第一组518A及第二组518B的区间段的不同区间段分数例示了以所述区间段的所述对应的预期序列读数计数标准化每个区间段的所述观察到的序列读数计数的益处。特定地,在展示于图5D中的所述示例中,在所述第一组518A中的数个区间段的所述观察到的序列读数计数及在所述第二组518B中的数个区间段的所述观察到的序列读数计数可能不显著地不同于彼此。
在此,横跨数个区间段的数个区间段分数对辨识一拷贝数目畸变可以提供信息。此外,如上文所描述的,不同区间段的所述区间段分数可以作为一全基因组特征。
返回图5B,在步骤510D,一区间段变异度估计对每个区间段被决定。在此,所述区间段变异性估计代表所述区间段的一预期变异性,所述预期变异性由代表所述样本中的变异性的一水平的一膨胀系数进一步调整。换句话说,所述区间段变异性估计代表决定自先前训练样本的所述区间段的所述预期变异性的结合,以及当前样本(例如,cfDNA或gDNA样本)的一膨胀系数,所述膨胀系数并未被计入所述区间段的所述预期变异性中。所述区间段变异性可以影响一区间段分数是否指示一拷贝数量畸变。如在上文中讨论过的,所述区间段变异性可以作为一全基因组特征。
为了提供一示例,一区间段i的一区间段变异性估计(var
var
其中var
为了决定所述样本的所述膨胀系数I
更特定地,为了对每个训练样本决定数个系数值,来自所述训练样本的数个序列读数可以被用于决定所述参考基因组的每个区间段的z分数。区间段i的一z分数可以被表示为:
其中b
一第一曲线拟合是在每个训练样本的所述区间段z分数及z分数的理论性分布间被执行。在此,z分数的一示例理论性分布是一常态分布。在一实施例中,所述第一曲线拟合是一线性稳健回归拟合,所述线性稳健回归拟合收获一斜率值。因此,在一训练样本的区间段z分数及z分数的所述理论性分布间执行所述第一曲线拟合收获一斜率数值。所述第一曲线拟合对多个训练样本被执行多次,以计算多个斜率数值。
一第二曲线拟合是在数个训练样本的斜率数值及离差间被执行。作为一示例,一训练样本的所述离差可以是一中位数绝对成对离差(median absolute pairwisedeviation,MAPD),所述中位数绝对成对离差代表遍及所述讯练样本的相邻区间段的区间段分数间的绝对值差异的中位数。在一实施例中,所述第二曲线拟合是一线性稳健回归拟合。在另一实施例中,所述第二曲线拟合可以是一高次方多项式拟合。所述第二曲线拟合收获数个系数值,在所述第二曲线拟合是一线性稳健回归拟合的所述实施例中,所述数个系数值包括一斜率系数及一截距系数。由所述第二曲线拟合收获的所述数个系数值被储存为数个样本变异系数。
所述样本的所述离差代表遍及所述样本的数个区间段中的序列读数计数的变化性的一度量。在一实施例中,所述样本的所述离差是一中位数绝对成对离差(MAPD)及可以通过分析相邻区间段的序列读数计数而被计算。特定地,所述MAPD代表遍及所述样本的相邻区间段的区间段分数间的绝对值差异的中位数。数学上,所述MAPD可以被表示为:
其中b
所述膨胀系数I
I
在此,所述“斜率”及“截距”系数中的每个是样本变异系数,而σ
在步骤510E,每个区间段被分析以基于所述区间段的所述区间段分数及区间段变异性估计,决定所述区间段是否是统计显著的。对于每个区间段i,所述区间段的所述区间段分数(b
为了判定一区间段是否是一统计显著区间段,所述区间段的所述z分数被与一阀值比较。若所述区间段的所述z分数是大于所述阀值,所述区间段被认为是一统计显著区间段。相反地,若所述区间段的所述z分数是少于所述阀值,所述区间段不被认为是一统计显著区间段。在一实施例中,若一区间段的z分数是大于2,则所述区间段被认为是统计显著的。在其它实施例中,若一区间段的z分数是大于2.5、3、3.5或4,则所述区间段被认为是统计显著的。在一实施例中,若一区间段的z分数是少于-2,则所述区间段被认为是统计显著的。在其它实施例中,若一区间段的z分数是少于-2.5、-3、-3.5或-4,则所述区间段被认为是统计显著的。所述统计显著区间段可以指示存在一样本(例如,cfDNA或gDNA样本)中的一个或更多个拷贝数目事件。
在步骤510F,所述参考基因处的数个节段被产生。每个节段是由所述参考基因组的一个或更多个区间段组成,及具有一统计序列读数计数。一统计序列读数计数的数个示例可以是一平均区间段序列读数计数、一中位数区间段序列读数计数等。一般地,所述参考基因组的每个被产生的节段拥有一统计序列读数计数,所述统计序列读数计数不同于一相邻节段的一统计序列读数计数。因此,一第一节段可以具有显著不同于一第二的,相邻的节段的一平均区间段序列读数计数的一平均区间段序列读数计数。
在各种实施例中,所述参考基因组的数个节段的所述产生可以包括两个分离的阶段。一第一阶段可以包括基于每个节段中的所述数个区间段的区间段序列读数计数的差异,将所述参考基因组分段为数个初始节段的一初始分段。所述第二阶段可以包括一重分段过程,所述重分段过程涉及将一个或更多个所述初始节段重新结合为更大的节段。在此,所述第二阶段考量经由所述初始分段过程被创造的所述数个节段的长度,以结合数个伪阳性节段,所述伪阳性节段是在所述初始分段过程中发生的过度分段的一结果。
更具体地参考所述初始分段过程,所述初始分段过程的一示例包括执行一循环二元分段演算法,以基于在数个节段中的数个区间段的所述区间段序列读数计数,将所述参考基因组的数个部分递回地分解为数个节段。在其它实施例中,其它演算法可以被用以执行所述参考基因组的一初始分段。作为所述循环二元分段过程的一示例,所述演算法在所述参考基因组中辨识一断点,而使由所述断点形成的一第一节段包括在所述第一节段中的数个区间段的一统计区间段序列读数计数,所述在所述第一节段中的数个区间段的一统计区间段序列读数计数显著不同于在由所述断点形成的第二节段中的数个区间段的一统计区间段序列读数计数。因此,所述循环二元分段过程收获许多节段,其中在一第一节段中的数个区间段的所述统计区间段序列读数计数显著不同于在一个第二的,相邻的节段中的数个区间段的所述统计区间段序列读数计数。
所述初始分段过程在产生数个初始节段时,可以进一步考虑每个区间段的所述区间段变异估计。举例而言,当计算在一节段中的数个区间段的一统计区间段序列读数计数时,每个区间段i可以被指定一权重,所述权重取决于所述区间段的所述区间段变异性估计(例如,var
现在参考所述重新分段过程,所述过程分析由所述初始分段过程创造的所述数个节段,及辨识数对应被重新结合的,被错误分离的节段。所述重新分段过程可以解释在所述初始分段过程中未被考虑的,数个节段的一特征。举例而言,一节段的一特征可以是所述节段的所述长度。因此,一对被错误分离的节段可以意指相邻节段,所述相邻节段当鉴于所述成对节段的长度进行考虑时,并不具有显著相异的统计区间段序列读数计数。较长的节段一般地与所述统计区间段序列读数计数的一较高变异相关联。因此,初始被判定为各具有相异于彼此的统计区间段序列读数计数的相邻节段,可以通过考虑每个节段的所述长度而被视为一对被错误分离的节段。
在所述配对中的被错误分离的节段被结合。因此,执行所述初始分段及重新分段程序导致一参考基因组的被产生的数个节段,所述数个节段考虑到由于每个节段的不同长度而发生的变异性。
在步骤510G,一节段分数基于节段的一观察到的节段序列读数计数及所述节段的一预期节段序列读数计数而对每个节段被决定。所述节段的一观察到的节段序列读数计数代表被分类在所述节段中的数个观察到的序列读数的所述总数。因此,所述节段的一观察到的节段读数计数可以通过加总被包括在所述节段中的数个区间段的所述观察到的区间段读数计数而被决定。类似地,所述预期节段序列读数计数代表在被包括在所述节段中的数个区间段间的预期序列读数计数。因此,一节段的所述预期节段序列读数计数可以通过量化被包括在所述节段中的数个区间段的所述预期区间段读数计数而被计算。被包括在所述节段中的数个区间段的所述预期读数计数可以被决定自先前训练资料,比如来自健康个体的数个序列读数。如上文所描述的,所述节段分数可以指示一拷贝数目畸变。因此,所述节段分数可以作为一全基因组特征。
一节段的所述节段分数可以被表示为所述节段序列读数计数及所述节段的所述预期节段序列读数计数的比率。在一实施例中,一节段的所述节段分数可以被表示为所述节段的观察到的序列读数计数及所述节段的所述预期序列读数计数的比率的对数。节段k的节段分数s
在其它实施例中,所述节段的所述节段分数可以表示为所述比率的平方根中的一个(例如,
在步骤510H,一节段变异性估计对每个节段被决定。一般地,所述节段变异性估计代表所述节段的所述序列读数计数是如何偏差。所述节段变异性可以影响一节段分数是否指示一拷贝数目畸变。如上文中所讨论的,所述节段变异性可以做为一全基因组特征
在一实施例中,所述节段变异性估计可以通过使用被包括在所述节段中的数个区间段的所述区间段变异性估计,及通过一节段膨胀系数(I
var
其中平均(var
所述节段膨胀系数解释在所述节段层次的所述增加的离差,所述离差相较于处于所述区间段层次的离差典型地较高。在各种实施例中,所述节段膨胀系数可以根据所述节段的尺寸调整。举例而言,由大量区间段组成的一较大的节段将被指定一节段膨胀系数,所述节段膨胀系数大于被指定给由较少区间段组成的一较小的节段的一节段膨胀系数。因此,所述节段膨胀系数说明在较长的节段中发生的较高水平的离差。在各种实施例中,被指定给一第一样本的一节段的所述节段膨胀系数不同于被指定给一第二样本的相同节段的所述节段膨胀系数。在各种实施例中,具有一特定长度的一节段的所述节段膨胀系数I
在各种实施例中,每个节段的所述节段变异性估计可以通过分析数个训练样本而被决定。举例而言,一旦所述数个节段在步骤510F中被产生,来自数个训练样本的数个序列读数被分析,以决定每个被产生的节段的一预期节段序列读数计数及每个节段的一预期节段变异性估计。
每个节段的所述节段变异性估计可以被表示为使用由所述样本膨胀系数调整的所述数个训练样本而被决定的,每个节段的所述预期节段变异性估计。举例而言,一节段k的所述节段变异性估计(var
其中
在步骤510I,每个节段被分析,以基于所述节段的所述节段分数及所述节段变异性估计,判定所述节段是否统计上显著。对于每个节段k,所述节段的所述节段分数(s
为了判定一节段是否是一统计显著节段,所述节段的所述z分数被与一阀值比较。若所述节段的所述z分数是大于所述阀值,所述节段被认为是一统计显著节段。相反地,若所述节段的所述z分数是少于所述阀值,所述节段不被认为是一统计显著节段。在一实施例中,若一节段的z分数是大于2,则所述节段被认为是统计显著的。在其它实施例中,若一节段的z分数是大于2.5、3、3.5或4,则所述节段被认为是统计显著的。在一些实施例中,若一节段的z分数是少于-2,则所述节段被认为是统计显著的。在其它实施例中,若一节段的z分数是少于-2.5、-3、-3.5或-4,则所述节段被认为是统计显著的。所述统计显著节段可以指示存在一样本(例如,cfDNA或gDNA样本)中的一个或更多个拷贝数目事件。
返回图5A,在步骤506C,在所述cfDNA中的数个拷贝数目畸变根据(例如,在步骤510E决定的)数个统计显著区间段及/或(例如,在步骤510I决定的)数个统计显著节段而被辨识。特定地,所述cfDNA样本的数个统计显著区间段/节段被与所述gDNA样本的对应区间段比较。所述cfDNA样本的数个统计显著节段及区间段与所述gDNA样本的对应的数个节段及区间段间的所述比较收获一判定,所述判定是关于所述cfDNA样本的数个统计显著节段及区间段是否与所述gDNA样本的对应的数个节段及区间段对齐。如在下文中所使用的,经对齐的节段或区间段意指所述节段或区间段在所述cfDNA样本及所述gDNA样本中皆是统计显著的。相反地,未对齐的或非对齐的数个节段或区间段意指所述数个节段或区间段在一样本(例如,cfDNA样本)中是统计显著的,但在另一样本(例如,gDNA样本)中不是统计显著的。
一般地,若所述cfDNA样本的数个统计显著区间段及统计显著节段是与所述gDNA样本的同样是统计显著的对应的区间段及节段对齐,这显示相同的拷贝数目事件存在于所述cfDNA样本及所述gDNA样本中。因此,所述拷贝数目事件的来源可能是由于一非肿瘤事件(例如,一生殖细胞系或体细胞非肿瘤事件)及所述拷贝数目事件很可能是一拷贝数目变异。
相反地,若所述cfDNA样本的数个统计显著区间段及统计显著节段是与所述gDNA样本的非统计显著的区间段及节段对齐,这显示所述拷贝数目事件存在于所述cfDNA样本中但在所述gDNA样本中缺少。在此情况中,在所述cfDNA样本中的所述拷贝数目事件是由于一体细胞肿瘤事件及所述拷贝数目事件是一拷贝数目畸变。
辨识在所述cfDNA样本中被侦测到的一拷贝数目事件的来源,在滤出缘于一生殖细胞系或体细胞非肿瘤变异的拷贝数目事件上是有益的。这改进了正确地辨识出缘于一实性肿瘤的存在的数个拷贝数目畸变的能力。
在此,遍及一cfDNA样本的所述基因组的经辨认的拷贝数目畸变的存在可以作为一全基因组特征。在各种实施例中,所述拷贝数目畸变的位置被进一步考虑。举例而言,一全基因组特征可以是在一染色体的一特定区域中的拷贝数目畸变的数量。
3.2.1.示例1:在一癌症样本中源自体细胞肿瘤来源的拷贝数目畸变
图5E及5F各自绘示横跨一cfDNA样本及一gDNA样本的一基因组的数个区间段的数个区间段分数,所述样本是获得自一癌症对象。在此,所述癌症患者被临床诊断出第一期乳癌。一血液检测样本经由一抽血被获得自所述癌症患者,及被收集在一采血管中。所述血液样本管于1600g被离心,血浆及血沉棕黄层(buffy coat)成分被分别提取,并储存于负20℃。cfDNA使用QIAAMP循环核酸套组(Circulating Nucleic Acid kit)(马里兰州日耳曼城,Qiagen)自血浆被提取及被池化。在所述血沉棕黄层中的白血球被裂解,且gDNA使用一DNEASY血液及组织套组(DNEASY Blood and Tissue kit)(马里兰州日耳曼城,Qiagen)被提取。定序文库自所述被提取的cfDNA样本及所述gDNA样本使用TRUSEQ纳米DNA试剂(TRUSEQ Nano DNA reagents)(加利福尼亚州圣地亚哥,Illumina)被制备。在文库制备后,所述cfDNA定序文库及gDNA定序文库使用一HiSeqX定序仪(HiSeqX sequencer)(加利福尼亚州圣地亚哥,Illumina)被定序以自所述cfDNA及gDNA样本两者获得数个序列读数。特定地,数个cfDNA序列读数及数个gDNA序列读数通过在35x的一覆盖深度执行全基因组定序而被获得。
特定地参照展示于图5E及5F中的资料,在图5E及5F中的每个当中的每个指标代表所述参考基因组的一区间段的一区间段分数。被展示于x轴上的被选择的数个区间段代表来自所述癌症患者的染色体1至22的数个核苷酸序列。每个区间段的所述区间段分数相对于对所述区间段的预期序列读数计数而被标准化,及因此,缺少一拷贝数目事件的一cfDNA样本或一gDNA样本将绘出最小地偏离于零的区间段分数。
数个未对齐指标(例如,在图5E及5F中被标示为“+”)意指所述cfDNA样本的数个区间段及/或节段,所述数个区间段/节段不同于所述gDNA样本的数个对应的区间段及/或节段。举例而言,若所述gDNA样本的所述对应的区间段并非统计上显著的,则所述cfDNA样本的一统计显著区间段在图5E中被绘示为一未对齐指标。类似地,若所述gDNA样本的所述对应的区间段是统计上显著的,则所述cfDNA样本的一非统计显著区间段在图5E中被绘示为一未对齐指标。此外,若一cfDNA样本的一节段不同于所述gDNA样本的所述对应节段(例如,统计上显著的对非统计上显著的),则在所述cfDNA样本的所述节段中的所有区间段使用未对齐指标被绘示。
对齐区间段指标(例如,在图5E及5F中被标示为“x”)意指在所述cfDNA样本及所述gDNA样本中的对齐的数个区间段。举例而言,若所述gDNA样本的所述对应区间段也是统计上显著的,所述cfDNA样本的一统计显著区间段被绘示为一对齐区间段指标。类似地,若所述gDNA样本的所述对应区间段也是非统计上显著的,所述cfDNA样本的一非统计显著区间段被绘示为一对齐区间段指标。
数个对齐节段指标(例如,在图5E及5F中被标示为
参考图5E,所述cfDNA样本包括一统计显著节段520A,所述节段520A包括具有高于零的区间段分数的数个区间段。此外,所述cfDNA样本包括一统计显著节段522A,所述节段522A包括具有低于零的区间段分数的数个区间段。再者,所述cfDNA样本包括统计显著区间段524A及526A,因为所述区间段524A及526A中的每个具有高于零的一区间段分数。每个统计显著节段(例如,520A及522A)及统计显著区间段(例如,524A及526A)指示一拷贝数目事件。
参考图5F,所述gDNA样本包括节段520B及节段522B,所述节段520B及节段522B各包括数个区间段,所述数个区间段具有不显著地不同于零的数个区间段分数。在此,所述gDNA样本的节段520B是所述cfDNA样本的节段的520A的所述对应节段。此外,所述gDNA样本的节段522B是所述cfDNA样本的节段的522A的所述对应节段。所述gDNA样本也包括统计显著区间段526B,所述区间段526B是所述cfDNA样本的区间段526A的对应区间段。
在此,在所述cfDNA样本中的数个所述统计显著节段(例如,节段520A及520B)未与所述gDNA样本中的所述对应的节段(例如,节段520B及522B)对齐。特定地,所述cfDNA样本的统计显著节段520A是未与所述gDNA样本的节段520B对齐的。此外,所述cfDNA样本的节段522A是未与所述gDNA样本的节段522B对齐的。这指示由所述统计显著节段520A及522A中的每个所代表的所述拷贝数目事件很可能是由于一体细胞肿瘤事件。
此外,所述cfDNA样本的区间段526A与所述gDNA样本的区间段526B对齐。因此,由所述cfDNA样本的区间段526A所代表的所述拷贝数目事件很可能是由于一生殖细胞系或体细胞非肿瘤事件。
3.2.2.示例2:在一非癌症样本中源自体细胞肿瘤来源的潜在拷贝数目畸变
图5G及5I各自绘示横跨决定自一cfDNA样本及一gDNA样本的一基因组的数个区间段的数个区间段分数,所述样本是获得自一非癌症个体。在此,由于所述个体未被诊断出癌症,所述个体可以是癌症的早期侦测的一候选者。一血液检测样本经由一抽血被获得自所述非癌症个体,及cfDNA与gDNA被提取。提取及定序cfDNA及gDNA样本以产生供分析的数个序列读数是根据上文中于示例1中描述的过程被执行。
如在图5G中所展示地,所述cfDNA样本包括一统计显著节段532A,所述节段532A包括数个区间段,所述数个区间段具有高于零的区间段分数。此外,所述cfDNA样本包括一统计显著区间段530A,所述区间段530A包括高于零的一区间段分数。所述统计显著节段532A及统计显著区间段530A指示拷贝数目事件。如展示于图5H中地,所述gDNA样本包括节段532B,所述节段532B包括数个区间段,所述数个区间段具有不显著不同于零的区间段分数。所述gDNA样本的节段532B是所述cfDNA样本的节段532A的所述对应节段。此外,所述gDNA样本也包括统计显著区间段530B,所述区间段530B是所述cfDNA样本的区间段530A的所述对应区间段。
所述cfDNA样本的区间段530A与gDNA样本的区间段530B对齐。因此,由所述cfDNA样本的区间段530A所代表的所述拷贝数目事件可能是缘于一生殖细胞系或体细胞非肿瘤事件。在所述cfDNA样本中的所述统计显著节段532A未与在所述gDNA样本中的所述对应节段532B对齐。这指示由所述统计显著节段532A代表的所述拷贝数目事件可能是缘于一体细胞肿瘤事件。这展示了一健康个体(例如,未被诊断出癌症)可以潜在的通过使用获得自所述个体的cfDNA及gDNA样本辨认可能的拷贝数目畸变,为癌症的早期侦测而被筛检。
3.2.3.示例3:在一非癌症样本中源自一生殖细胞系或体细胞非肿瘤来源的拷贝数目变异
图5I及5J各自绘示决定自横跨一cfDNA样本及一gDNA样本的一基因组的数个区间段的数个区间段分数,所述样本是获得自一非癌症个体。在此,由于所述个体未被诊断出癌症,所述个体可以是癌症的早期侦测的一候选者。一血液检测样本经由一抽血被获得自所述非癌症个体,及cfDNA与gDNA被提取。提取与定序cfDNA及gDNA样本以产生供分析的数个序列读数是根据上文中于示例1中被描述的过程被执行。
如在图5I中所展示地,所述cfDNA样本包括一统计显著节段534A,所述节段534A包括数个区间段,所述数个区间段具有高于零的区间段分数。此外,所述cfDNA样本包括一统计显著区间段536A,所述区间段536A包括高于零的一区间段分数。所述统计显著节段534A及统计显著区间段536A指示拷贝数目事件。如展示于图5J中地,所述gDNA样本包括节段534B。所述gDNA样本的节段534B是所述cfDNA样本的节段534A的所述对应节段。在此,所述统计显著节段534B包括数个区间段的至少一子集,所述数个区间段具有不显著偏离零的数个区间段分数。换言之,所述节段层次分析允许一统计显著节段534B的一辨识,所述节段534B包括一子集的数个区间段,所述数个区间段个别地将不会被辨识为一统计显著区间段。这展现了除执行一区间段层次的分析外执行一节段层次分析,以辨识拷贝数目事件的益处。所述gDNA样本额外地包括统计显著区间段536B,所述区间段536B是所述cfDNA样本的区间段536A的所述对应区间段。
在此,在所述cfDNA样本中的所述统计显著节段534A与在所述gDNA样本中的所述对应统计显著节段534B对齐。这指示由所述统计显著节段534A代表的所述拷贝数目事件很可能是缘于一生殖细胞系或体细胞非肿瘤事件。此外,所述cfDNA样本的区间段536A与所述gDNA样本的区间段536B对齐。因此,由所述cfDNA样本的区间段536A代表的所述拷贝数目事件也可能是缘于一生殖细胞系或体细胞非肿瘤事件。
3.3.维度缩减工作流程
再次返回图5A,所述维度缩减工作流程(例如,步骤505,508A至C)一般地分析高维度资料及执行一维度缩减以获得一较低维度资料,所述较低维度资料代表所述数个原始序列读数的所述数个主要特性。这样的较低维度资料可以是作为数个全基因组特征的数个维度缩减特征。作为一示例,高维度资料可以是遍及所述基因组的数个区间段的区间段分数,所述区间段分数在一些实施例中,可能包括数千个数值。因此,较低维度资料(例如,数个维度缩减特征)可以是一较小数目的数个数值,所述数个数值是缩减自遍及所述基因组的数个区间段的数千个区间段分数。
所述维度缩减工作流程在步骤505处开始。在所述步骤505,衍生自一cfDNA样本的数个序列读数被获得。衍生自cfDNA115的所述数个序列读数可以自一全基因组定序化验132被接收。
在步骤508A,在序列读数计数展现低变异性的,遍及所述基因组的数个基因组区域经由一个或更多个标准的使用而被辨认。在各种实施例中,数个基因组区域可以意指所述基因组的数个区间段,所述数个区间段在上文中与所述拷贝数目畸变侦测工作流程关联地被讨论。
所述标准被制定以通过在资料收集时辨认并去除系统性误差或其它类型的非疾病相关噪声而增进高维度的资料的品质。换言之,通过应用所述条件,不适合后续分析的,典型地充满噪声的数个区间段可以被去除。在一些实施例中,在步骤508A受到分析的数个序列读数已被预处理以修正偏误或错误。所述预处理是使用一个或更多个方法,比如标准化、GC偏误的修正、缘于PCR过度放大的偏误的修正等。
在一些实施例中,一个或更多个标准被建立以在资料收集时排除可能含有系统性误差或其它类型的非疾病相关噪声的核酸定序资料。如本文中揭示的,序列资料可以包括任何生物性样本的数个序列读数,所述生物性样本包括但不限于游离核酸样本。
在一些实施例中,仅来自数个健康对象的资料被用于建立所述一个或更多个标准,以避免来自与一或更多疾病情况相关的资料的干扰。在一些实施例中,如在本文中所揭示的一标准可以关于数个基因组或染色体区域而被建立。举例而言,数个核酸序列读数可以被对齐到一参考基因组的数个区域,及所述数个序列读数的一个或更多个特性可以被用于判定与特定基因组区域相关联的资料是否较少地与混淆来自所述基因组区域的信息的一基线噪声相关联。因此,所述特定的基因组区域可以被自后续分析去除。数个示例性特性包括但不限于,举例而言,读数的数量、所述数个读数的可作图性等。
在一些实施例中,所述数个基因组区域具有相同尺寸。在一些实施例中,所述数个基因组区域可以具有不同的尺寸。在一些实施例中,一基因组区域可通过在所述区域中的数个核酸残基(nucleic acid residues)的数量而被辨识。在一些实施例中,一基因组区域可以通过其位置及在所述区域中的数个核酸残基的数量而被辨识。任何适宜的尺寸可以被用于界定基因组区域。举例而言,一基因组区域可以包括10kb或更少、20kb或更少、30kb或更少、40kb或更少、50kb或更少、60kb或更少、70kb或更少、80kb或更少、90kb或更少、100kb或更少、110kb或更少、120kb或更少、130kb或更少、140kb或更少、150kb或更少、160kb或更少、170kb或更少、180kb或更少、190kb或更少、200kb或更少、或250kb或更少。
可能与系统性误差相关联的数个区域被辨认。在一些实施例中,一个或更多个标准可以被界定以减少或消除对应于这些较多噪声的区域的资料。举例而言,一高变异性过滤器可以被创造,以允许使用者丢弃对应于所有具有高于一阀值的资料变异性的区域的资料。在其它实施例中,一低变异性过滤器可以被创造,以将后续分析集中于具有低于一阀值的资料变异性的资料。
作为一例示,一人类单倍参考基因组包括超过三十亿个碱基,所述三十亿个碱基可以被分为约30000个区域(或区间段)。若一实验数值,举例而言,与所述特定区域或区间段对齐的数个序列读数的一总数,对每个区间段被观察,每个对象可以对应到超过30000个测量值。在一低或高变异性过滤器被施加后,对应于一对象的测量值的所述数量可以被减少一显著比例;举例而言,包含但不限于约50%或更少、约45%或更少、约40%或更少、约35%或更少、约30%或更少、约25%或更少、20%或更少、15%或更少、10%或更少、或5%或更少。在一些实施例中,对应于一对象的测量值的所述数量可以被减少50%或更多,比如约55%、60%、65%、或70%或更多。举例而言,一对象,原本具有超过30000个对应的测量值,在一高或低变异度过滤器被施用后,可以具有减少30%以上的测量值(例如,约20000个)。
在步骤508B,建立自所述先前步骤的所述一个或更多个标准可以被应用到一训练群组的一生物性资料集(也被称为“训练资料”)。如在本文中所揭示的,所述训练群组包括数个健康对象及已知具有一个或更多个病况(medical conditions)的数个对象(也被称为“患病对象”)。举例而言,对于定序资料,先前在步骤508A中被决定的一个或更多个标准(例如,一低或高变异性过滤器)被应用到所述训练群组的资料,以完全移除与在所述过滤器中被界定的染色体区域相关联的资料部分。在一些实施例中,所述被假定为充满噪声的资料仅被部分移除。在一些实施例中,未被移除的所述被假定为充满噪声的资料可以被指定一加权系数,以减少它们在整体资料集中的重要性。
一旦资料筛选已对所述训练资料的所述生物性资料集执行,所述剩余的训练资料,也被称为“经筛选训练资料”或“经过滤训练资料”经受进一步分析,以提取反映数个健康对象及数个已知具有一个或更多个病况的对象间的差异的数个特征。如前所述,所述原始训练资料包括来自数个健康对象及数个患病对象的资料。所述经过滤的训练资料构成所述原始训练资料的一部分,及因此也包括来自数个健康对象及数个已知具有一病况的对象两者的资料。被假定的是,在所述经过滤训练资料中的最大的变异来自来自所述数个健康对象的资料及来自所述数个患病对象的资料间的差异。本质上,被假定的是,与一健康对象相关联的资料比起与来自任何患病对象的资料,应与另一健康对象的资料更加类似;及反之亦然。
如同所述原始训练资料,所述经过滤的训练资料也是高维度的。在一些实施例中,所述经过滤的训练资料经受进一步分析以减少资料维度,及所述数个健康与患病对象间的差异基于所述被减少的维度而被界定。对于一给定对象,约20000个经过滤测量值可以被进一步减少到少数数据点。举例而言,所述约20000个经过滤的测量值可以基于少数被提取的特征(例如,数个主成分)而被变换,以渲染一些数据点。在一些实施例中,在维度的减少后,有5个或更少个特征;6个或更少个特征;7个或更少个特征;8个或更少个特征;9个或更少个特征;10个或更少个特征;12个或更少个特征;15个或更少个特征;或20个或更少个特征。在一些实施例中,所述经过滤测量值可以具有多于20个特征。所述经过滤的测量值可以接着基于所述经选择的特征而被转换。举例而言,具有二个20000个经过滤的测量值的一样本可以被转换及减少至五个或更少的资料点。在一些实施例中,具有二个20000个经过滤的测量值的一样本可以被转换及减少到多于五个资料点,比如10、15、20个等。
如在本文中揭示的,来自在所述经过滤训练资料集中的所有对象的所述经转换资料点经受进一步分析,以提取数个关联或模式,所述数个关联或模式反映在所述经过滤资料集中的数个子集间的差异。在一些实施例中,进一步的分析包括一个二项式逻辑回归程序;举例而言,用于决定一对象具有癌症对不具有癌症的所述似然性。在一些实施例中,进一步的分析包括一多项式逻辑回归程序;举例而言,用于在一对象具有癌症的所述似然性外,判定癌症的所述类型。
在步骤508C,可选地,数个分数对每个对象被计算。在一些实施例中,一分数是由一预测性癌症模型170B输出的一癌症预测(例如,展示于图1C中的癌症预测190B)。
现在参考图6A,图6A例示根据一实施例的,用于通过减少高维度资料的所述维度以决定一分类分数的一流程600。
在所述资料筛选部分,高维度的资料初始被处理以增进质量。在一些实施例中,对齐到一参考基因组的一特定区域的数个序列读数的所述数量被标准化。举例而言,健康对象资料605A可以包括来自数个健康对象(也被称为基线对象)的一群组的数个序列读数,及来自所述数个基线对象的资料可以被用于建立所述标准化标准(normalizationstandards)。在一些实施例中,来自所述数个基线对象的数个序列读数被对齐到已被分为数个区域的一参考基因组。假定在所述定序过程中没有显著的偏误,在所述基因组中的不同区域应以约略相同的水平被覆盖。因此,被对齐到一特定区域的数个序列读数的所述数量应与对齐到尺寸相同的另一区域的那些序列读数相同。
在一示例中,遍及不同基因组区域的来自一基线对象的数个序列读数的所述数量可以被写为
在一些实施例中,一对象的数个序列读数可以被标准化至遍及所述对象的所有染色体区域的所述平均读数计数。当i保持恒定,来自基因组区域1至m的数个序列读数及所述数个区域的对应尺寸可以被用于计算对象i的数个序列读数的一平均预测数量,举例而言,基于所述等式:
其中
如在本文中所揭示的,任何对象的横跨不同基因组区域的资料可以被用作一控制组,以标准化一基因组区域的所述数个序列读数。在此,被用作资料标准化的基准的一平均读数,可以对一健康控制组对象、一组控制组对象,或一检测对象本身而被计算。
在一些实施例中,一对象的数个序列读数可以对来自一组对象(例如,一组n个健康对象)的一总体平均计数而被标准化。
在一些实施例中,对应于一特定区域的一对象的数个序列读数可以使用多个方式,应用来自所述对象本身的不同区域及横跨不同控制组对象的资料两者而被标准化。
在一面向中,在本文中揭示的是用于基于数个模式,建立用于筛选供进一步分析的资料的一模板的数个方法。所述数个模式是收集自来自数个健康对象(例如,数个基线健康对象605A及数个参考健康对象605B)的资料。在数个优选实施例中,数个参考健康对象605B与数个基线健康对象605A并不重叠或仅有少数重叠。数个游离核酸的定序资料被用于例示所述数个概念。然而,本领域的一般技术人员将了解到,当前的方法也可以被应用到其它材料的定序资料或非定序资料。
在一些实施例中,在一基线或参考健康对象群组中的健康对象的所述数量可以变化。在一些实施例中,对在所述基线及参考健康对象群组中的数个健康对象的选择标准是相同的。在一些实施例中,对在所述基线及参考健康对象群组中的数个健康对象的选择标准是不同的。
在一些实施例中,一高或低变异度过滤器是使用来自数个健康参考对象605B的资料被建立。如在本文中揭示的,来自所述健康参考对象605B的所述资料可以被预处理(例如,经历各种标准化步骤);举例而言,基于来自数个健康对象605A的基线控制组资料而被预处理。举例而言,来自数个健康及癌症对象两者的训练资料可以被预处理。在一些实施例中,原始序列读数资料(raw sequence read data)可以被直接用于设立一高或低变异性过滤器。
在一些实施例中,(例如,来自健康对象资料605A的)每个健康对象的数个序列读数可以被对齐到参考基因组的数个染色体区域。每个染色体区域的所述变异性可以被评量;举例而言,通过横跨在所述控制组中的所有健康对象,比较一特定基因组区域的序列读数的数量。作为一例示,不被预期具有癌症的数个健康对象可以被包括作为参考控制组。所述数个健康对象包括但不限于不具有癌症家族史的对象或健康且年轻(例如,低于35或30岁)的对象。在一些实施例中,在所述参考控制组中的数个健康对象可以满足其它条件:举例而言,仅有健康女性会被包括在乳癌分析的一控制组中。仅有男性会被包括在前列腺癌分析的一控制组中。在一些实施例中,对于主要或仅在一特定族群中发现的疾病,只有来自所述相同族群的人被用于建立所述参考控制组。
举例而言,对于数个控制组健康对象(n)的一群组,若我们计算对齐到一基因组区域的数个序列读数的数量,对于每个基因组区域将有n个数值。数个参数,比如一平均数或中位数计数、标准差(SD)、中位数绝对离差(MAD)或四分差(interquartile range,IQR)可以基于所述n个计数数值被计算及被用于决定一基因组区域是否被视为低或高变异性。任何用于计算这些参数的方法皆可以被使用。
举例而言,在对象1至n中的区域j的数个序列读数数量可以被表示为
在一些实施例中,数个高或低变异性过滤器可以只对所述基因组的一部分被创造;举例而言,只对一特定染色体或所述染色体的一部分被创造。
在一些实施例中,训练资料650C包括来自数个健康对象及数个已知有一病况的对象(也称为患病对象)的生物性资料(例如,定序资料)。在一些实施例中,先前已被包括在所述基线控制组或参考控制组中的数个资料关联健康对象将会被自训练资料650C排除,以可能地避免特定偏误。
在一些实施例中,使用健康对象资料650A及所述低或高变异性过滤器605D获得的数个标准化参数可以被应用到训练资料605C,以渲染新的及经过滤的训练资料610A供后续分析。
在一些实施例中,经过滤训练资料610A包含数个健康及患病对象的经平衡资料;举例而言,数个健康及患病对象的所述数量彼此间约在5%或10%内。在一些实施例中,经过滤训练资料610A包含数个健康及患病对象的未平衡资料;举例而言,数个健康及患病对象的所述数量与彼此差异大于10%。在后一个情况中,数个方法可以被应用以减少未平衡资料的冲击。
在一些实施例中,经过滤训练资料610A经受进一步的分析,以创造预测模型610B。预测模型610B被用于预测一对象是否具有一特定病况。
在一些实施例中,预测模型610B反映数个健康及患病对象间的数个差异。在一些实施例中,在预测模型610B中被使用的所述数个差异可以通过对经过滤训练资料610A应用,举例而言,逻辑回归而被获得。在一些实施例中,经过滤训练资料610A(例如,对齐到一参考基因组的数个特定区域的序列读数的数量)可以直接被用在一逻辑回归分析中。在一些实施例中,经过滤训练资料610A经历一维度缩减,以将所述资料集减少及可能转换到小许多的一尺寸。举例而言,主成分分析(PCA)可以被用于减少一资料集的所述尺寸约100000倍或更少、约90000倍或更少、约80000倍或更少、约70000倍或更少、约60000倍或更少、约50000倍或更少、约40000倍或更少、约30000倍或更少、约20000倍或更少、约10000倍或更少、约9000倍或更少、约8000倍或更少、约7000倍或更少、约6000倍或更少、约5000倍或更少、约4000倍或更少、约3000倍或更少、约2000倍或更少、约1000倍或更少或约500倍或更少。在一些实施例中,一资料集的所述尺寸可以被减少多于100000倍。在一些实施例中,一资料集的所述尺寸可以被减少数百倍或更少。如在本文中所揭示的,虽然一资料集的所述尺寸被减少,样本的所述数量可以被保持。举例而言,在PCA后,1000个样本的一资料集可以依然保持1000个样本,但每个样本的复杂度被减少(例如,自对应于25000个特征至对应于5个或更少个特征)。因此,在本文中揭示的数个方法可以在大幅减少需要的计算机储存空间的同时,增进资料处理的效率及准确性。
一旦一预测模型被建立,它可以被应用到检测资料615A。检测资料615A可以被取自于一检测对象,所述检测对象关于一病况的状态是未知的。在一些实施例中,来自数个已知状态的检测对象的资料也可以为了验证目的被使用。在一些实施例中,检测资料615A会被预处理,比如经历标准化、GC含量修正等。
在一些实施例中,一高或低变异性过滤器605D被应用到检测资料615A,以移除可能对应于系统性误差的数个染色体区域中的资料。在一些实施例中,预处理及一低或高过滤器皆可以被应用到检测资料615A,以渲染经过滤测试资料以供进一步处理。
在一些实施例中,当预测模型610B被应用到经过滤测试资料610A时,一分类分数可以作为一机率分数被计算,以代表特定病况存在被分析的所述检测对象中的所述似然性。在这样的实施例中,展示于图6A中的所述预测模型610B可以是展示于图1C中的所述预测性癌症模型170B。因此,所述分类分数可以是一癌症预测190B。所述分类分数可以是一个二项式分类分数;举例而言,非癌症对癌症。在一些实施例中,所述分类分数可以是一多项式分类分数;举例而言,非癌症、肝癌、肺癌、乳癌、前列腺癌等。如上文中所讨论的,所述分类分数可以代表一全基因组特征。
图6B绘示根据一实施例的,用于分析资料以减少资料维度的一取样过程630。特定地,所述取样过程630更详细地意指(展示于图6A中)的步骤610A及610B。所述样本资料分析过程630以过滤训练资料开始步骤635A。举例而言,对于定序资料,仅有对应于假定地是低变异性的基因组区域的资料在步骤635A被接收。如在本文中所揭示的,训练资料包括来自数个健康及患病个体的资料。在一高或低变异性过滤器已被应用后,所述经过滤训练资料应依然包括来自数个健康及患病个体两者的生物性资料。在一些实施例中,所述数个患病对象是已被诊断出至少一个类型的癌症的数个患者。
在步骤635B,所述经过滤训练资料使用数个交叉验证方法被分离。在一些实施例中,所述数个交叉验证方法包括但不限于穷举性方法(exhaustive methods)如留p交叉验证(leave-p-out cross-validation,LpO CV),其中p可以具有将创造一有效划分(validpartition)的任何数值,或留一交叉验证(LOOCV),其中p=1。在一些实施例中,所述数个交叉验证方法包括但不限于非穷举性方法,比如保持除外法(holdout method)、重复随机次取样验证法(repeated random sub-sampling validation method)或一分层或非分层k折交叉验证法,其中k具有将创造一有效划分的任何数值。如在本文中揭示的,一交叉验证程序将所述经过滤训练资料划分为处于一预先决定的比例分割的一训练子集及一验证子集的不同配对。举例而言,在步骤635B处被绘示的所述第一训练子集及第一验证子集代表在一k折交叉验证试验的一折时的一80:20划分。在所述相同的k折交叉验证试验的另一折数中,所述经过滤训练资料将被划分为处于相同百分率比例的一不同配对的训练及验证子集。在一些实施例中,多个交叉验证试验被应用,其中一对训练及验证子集的所述划分比率在每个试验中可以不同。如在本文中所揭示的,所述数个子集可以随机地被创造。在一些实施例中,所述数个子集是被创造而使每个子集包括来自数个健康及患病对象两者的资料。在一些实施例中,仅有所述数个子集中的一个包括来自数个健康及患病对象两者的资料。举例而言,所述训练子集必须包括数个健康及患病对象两者。
在一些实施例中,一训练子集构成所述经过滤训练资料的一大部分;举例而言,高至所述经过滤训练资料的60%、高至65%、高至70%、高至75%、高至80%、高至85%、高至90%或高至95%。在一些实施例中,经过滤训练资料的一非常大的集组的多于95%可以被用作为所述训练资料。为了避免训练偏误,保存至少5%的未经触及资料作为一测试子集通常是良好的做法。亦即,此子集将永不会被用作训练资料及将只被用以验证作为结果的模型。
在步骤635C,来自所述第一训练子集的资料(例如,序列读数的计数或自所述序列读数的计数衍生的量值)可以被用以衍生数个维度缩减成分,所述数个维度缩减成分捕捉了数个健康及患病对象间的一个或更多个差异。举例而言,被辨认为具有约10000至约20000个低变异性区域的样本可以具有10000至20000个相对应的计数数值(或衍生数值,比如一相对计数数值,一对数计数数值等。)。通过使用一维度缩减方法,比如主成分分析(PCA),辨认及选择数个维度缩减成分是可能的,所述维度缩减成分的一示例是主成分(PCs),所述主成分代表在所述第一训练子集中的资料当中数个最大的变异。这些维度缩减成分可以被用于将所述10000至20000个数值缩减到一较低维度空间,其中在所述缩减维度空间中的每个数值对应到所述数个被选择的PC中的一个。作为一示例,所述较高维度数值可能是遍及所述基因组的N个区间段的N个区间段分数。因此,所述维度缩减过程将所述N个区间段分数缩减到一经缩减数量的数值,所述数值中的每个对应到一维度缩减的成分。在一些实施例中,5个或更少的维度缩减成分被选择。在一些实施例中,10个或更少的维度缩减成分被选择。在一些实施例中,15个或更少的维度缩减成分被选择。在一些实施例中,20个或更少的维度缩减成分被选择。在一些实施例中,25个或更少的维度缩减成分被选择。在一些实施例中,30个或更少的维度缩减成分被选择。在一些实施例中,35个或更少的维度缩减成分被选择。在一些实施例中,40个或更少的维度缩减成分被选择。在一些实施例中,45个或更少的维度缩减成分被选择。在一些实施例中,50个或更少的维度缩减成分被选择。在一些实施例中,60个或更少的维度缩减成分被选择。在一些实施例中,70个或更少的维度缩减成分被选择。在一些实施例中,80个或更少的维度缩减成分被选择。在一些实施例中,90个或更少的维度缩减成分被选择。在一些实施例中,100个或更少的维度缩减成分被选择。在一些实施例中,多于100个维度缩减成分被选择。
这些经由所述维度缩减方法(例如,经由PCA)被辨认的维度缩减成分可以在部署时(例如,在展示于图6D中的步骤中)被使用,以将高维度的资料缩减到较低维度的资料(例如,数个经缩减的维度特征)。
在步骤635D,一个或更多个成分权重可以被衍生,所述权重反映所述数个维度缩减成分的每个的相对贡献。举例而言,对于每个维度缩减成分,一成分比重被指派到来自每个低变异性区域的每个数值,以反映所述资料的所述各自的重要性。对所述观察到的差异贡献较多的一区域将会被指派一较大的成分权重;反之亦然。在一些实施例中,所述成分比重是特定于一成分的及也是区域特定的,但对于所有对象皆是相同的,举例而言,
在步骤635E,在所述第一训练子集中的资料基于所述经提取的维度缩减成分(例如,几个经筛选的PC)而被变换。在一些实施例中,所述经变换资料的维度比已自所述原始未过滤资料缩减的所述经过滤训练资料的维度小很多。所述数个概念被例示如下。
例示在一低变异性过滤器被应用前的对象1的资料,其中m是区域的总数。
例示在一低变异性过滤器被应用前的对象1的资料,其中m′是区域的总数。在一低变异性过滤器被应用后,基因组区域的总数被缩减到m′,m′可显著小于m。举例而言,一对象的未过滤资料可以包括30000个或更多个成分,每个与一基因组区域相关联。在一低变异性过滤器被应用后,所述数个基因组区域的一大部分可以因具有高变异性而被去除;举例而言,所述相同对象的经过滤资料可以包括20000个成分或更少,每个成分与一低变异性基因组区域相关联。
在步骤635E,所述经过滤资料的资料维度可以基于经提取的维度缩减成分的所述数量而被进一步缩减。举例而言,若k个主成分被选择,所述经过滤资料的所述维度可以被缩减至k。经选择的PC的所述数量可以比所述经过滤资料的维度小得多。举例而言,当仅有5个PC被选择时,对象1的经过滤读数资料(F读数)的资料维度可以被进一步缩减到5,比如下方的表示:
因此,与一大量低变异性区域相关联的质量资料(例如,数个读数数量)可以被缩减并变换为少数数字性数值。在一些实施例中,一成分比重(例如,
在步骤635F,一分类方法被应用到每个对象的所述经变换资料,以提供一分类分数。在一些实施例中,所述分类分数可以是一个二项式或多项式机率分数。举例而言,在癌症的一个二项式分类中,逻辑回归可以被应用以计算一机率分数,其中0代表无癌症的似然性,而1代表具有癌症的最高度的确定性。超过0.5的一分数指示所述对象比起不具癌症,更可能具有癌症。逻辑回归产生一方程式的数个系数(及其标准差及显著性水平),以预测所述感兴趣的特性的存在的机率的一分对数变换(logit transformation)。使用相同的示例来例示由逻辑回归决定的机率,一对象具有癌症的所述机率(p)可以被写成下述:
分对数(p)=b
其中衍生自PC1的每个经变换及缩减的资料被指派一权重。所述分对数变换被界定为对数胜算:
及机率p
p的数值可以通过插入所述分对数(p)值而被计算。在一些实施例中,可能在一分对数表中查询数值。
在一些实施例中,一多项式分类方式可以被采用,以将数个对象分入不同的癌症类型。举例而言,现存的多项式分类技术可以被分类为(i)变换至二项式(ii)自二项式延伸及(iii)阶层式分类。在一变换至二项式方式中,一多分类问题可以基于一一对剩余(one-vs-rest)或一对一方式被变换为多个二项式问题。自二项式演算法的示例性延伸包括但不限于类神经网络、决策树、k最近邻法、简单贝氏、支持向量机及极限学习机等。阶层式分类通过划分所述输出空间,亦即划分为一树状,而处理所述多项式分类问题。每个亲节点被划分为多个子节点,及所述过程被持续直到每个子节点仅代表一类别。数个方法基于阶层式分类被提出。在一些实施例中,多项式逻辑回归可以被应用。给定一组独立变数(可能是实值的、二项式值的、分类值的等),所述多项式逻辑回归被用以预测一类别分布依赖的变数的所述不同可能结果的机率。
在步骤635G,所述经过滤训练资料被分隔为一第二训练子集及一第二测试/验证子集,及635B至635F的步骤在一个或更多个精炼循环(也被称为“一个或更多个交叉验证循环”)中被重复。如在本文中揭示的,在所述交叉验证程序中,所述数个验证子集本身在不同折数上少有(例如,在重复随机取样中)或完全没有(LOOCV、LpO CV、k折)重叠。
在一精炼循环中,一预先决定的条件(例如,一成本函数)可以被应用以最佳化所述分类结果。在一些实施例中,在一分类函数中的一个或更多个参数使用训练资料子集被精炼及在所述交叉验证过程的每折中由验证或保持除外子集验证。在一些实施例中,数个PC特定的权重及/或区域特定权重可以被精炼,以最佳化分类结果。
在一些实施例中,所述经过滤训练资料的一小部分可以被保持在一旁,在所述交叉验证程序的任何折中不作为一训练子集的一部分,以更佳地估计过拟合。
在步骤635H,数个所述经精炼参数被用于计算数个分类分数。如在本文中揭示的,数个所述经精炼参数可以作为对癌症以及癌症类型的一预测模型而发挥功能。使用多个类型的生物性资料建构一预测模型是可能的;所述生物性资料包括但不限于,举例而言,核酸定序资料(游离对非游离、全基因组定序资料、全基因组甲基化定序资料、RNA定序资料、针对性检测组合定序资料)、蛋白质定序资料、组织病理学资料、家族史资料、族群学资料等。
在一面向中,在本文中被揭示的是用于分类一对象为具有一特定病况的方法,所述分类是基于使用训练资料建立的所述数个维度缩减成分及所述数个成分权重(或经精炼成分权重)。图6C绘示根据一实施例的,用于基于自具有缩减的维度的资料习得的信息,分析来自一检测样本的资料的一过程。过程640例示来自一对象(所述对象相关于一病况的状态是未知的)的检测资料,如何可以被用于计算一分类分数及作为诊断所述对象是否可能具有所述病况的一基础。
在步骤645A,检测资料被接收自一检测样本,所述检测样本来自其状态为未知的所述对象。在一些实施例中,所述检测资料与来自数个所述基线健康对象的资料所包括的属于相同的类型。在一些实施例中,所述检测资料与来自数个所述参考健康对象的资料属于相同的类型。样本资料类型包括但不限于用于侦测目标突变的定序资料、全基因组定序资料、RNA定序资料及用于侦测甲基化的全基因组定序资料。在一些实施例中,所述检测资料可以被校准及调整以增进质量(例如,标准化、GC含量修正等。)。
在步骤645B,所述检测资料的数个高维度数值使用在训练时被决定的数个成分权重被缩减到一较低维度空间。举例而言,所述数个高维度数值可以是读数资料,比如一区间段分数(例如,被分类在遍及所述基因组的数个区间段中的数个序列读数的所述经标准化数量)。首先,一资料筛选可以使用一先前界定的低变异性过滤器被执行。有利地,所述基于过滤器的方式是直截了当的,及可以通过改变所述参考量值的阀值而被容易地调整。所述参考量值的阀值是为在一参考基因组中的数个基因组区域而被计算的。因此,(例如,由于在特定区间段中的噪声的存在)高度可变的高维度数值可以被过滤并移除。接着,在所述资料选择步骤后留存的所述数个高维度数值可以使用一维度缩减过程,比如PCA,而在维度上被缩减。缘自所述维度缩减过程的数个维度缩减成分的所述数量可以基于在训练时被决定的维度缩减成分的数量。举例而言,若所述维度缩减过程产生5个主成分,则对所述检测样本的数个数值的所述维度缩减过程也产生5个主成分。每个维度缩减成分通过在训练时被决定的一对应的成分权重而被加权。因此,所述较低维度资料可以被计算。
作为一示例,有5个维度缩减成分,则5个较低维度资料可以被产生。特定地,经过滤读数资料(F读数)的所述资料维度可以被减少至5个(较低维度资料)(F读数
其中
在步骤645C,一分类分数可以使用所述缩减维度资料,对所述检测对象被计算。在各种实施例中,所述分类分数可以是由一预测模型,比如展示于图1C中的预测性癌症模型输出的一癌症预测。换句话说,所述预测模型可以作为一单一化验预测模型,所述单一化验预测模型仅基于数个全基因组特征152输出一癌症预测190B。
图6D绘示根据一实施例的,用于资料分析的一取样过程。如关联于图6B而被详述的,维度的缩减可以在所述资料分析的多个位点进行。
在一些实施例中,一特定水平的资料筛选可以在初始的资料处理时发生:例如,在标准化、GC含量修正及其它初始资料校正步骤。拒绝明显缺陷的数个序列读数及因此减少资料的数量是可能的。如例示在图6D中的,资料维度缩减可以以一低或高变异性过滤器的应用而进行。举例而言,一参考基因组可以被分为数个区域,所述数个区域的数个示例在元件610中被编号为1、2、3...、m-1、及m。所述数个区域可以在大小上相等或不相等。
如在本文中所揭示的,一低变异性过滤器指定将被选择供进一步处理的所述数个基因组区域的一子集。举例而言,以一铺面阴影指示的所述数个区域(例如,被指示为1、2、3、4、5、6...m’-1、m’的数个区域)是被选择的区域。所述过滤允许资料的明确(categorical)选择或拒绝。所述明确选择或拒绝是基于使用数个参考健康对象的,数个可能的系统性误差的已建立的分析。
所述经选择的资料接着被变换,以进一步将资料维度减少至经缩减的维度资料(例如,元件660)。在一些实施例中,经变换的资料660可以使用来自所有所述经选择的基因组区域的资料被产生,但资料的维度可以是大幅减少的。举例而言,来自超过20000个不同的基因组区域的资料可以被变换为少数数个数值。在一些实施例中,一单一数值可以被产生。在此,处于少数数个数值或一单一数值的型态的所述经缩减维度资料,是在上文中描述的可以做为全基因组特征的所述经缩减的维度特征。
在一些实施例中,来自元件655的经筛选的定序资料可以根据由所述定序资料代表的所述断片尺寸而被分类为数个子群组。举例而言,多个分位数(每个对应到一尺寸或尺寸范围)可以被衍生,而非对绑定于一特定区域的所有序列读数的一单一计数。举例而言,对应到140至150个碱基的数个断片的数个序列读数将被与对应到150至160个碱基的数个断片的数个序列读数分开分组。因此,额外的详细及精细的调校可以在所述资料被用于分类前被作出。
如例示在图6D中的,多个类型的资料可以被用于分类680,包括但不限于来自没有维度缩减的经选择/经过滤基因组区域的资料、经缩减的资料、经缩减及经分类的资料等。
当前的方法及系统提供相对于先前已知方法的优势。举例而言,分类是使用可以容易地自原始定序资料衍生的数个量值完成。当前方法及系统并不需要建构染色体特定分段地图,因此去除了用于产生这些地图的耗时过程。并且,当前方法允许计算机存储空间的更有效率的使用,因为所述方法不再需要供大型的分段地图的存储。
3.3.1.示例1:分类分数及Z分数的比较
图6E绘示将当前方法(分类分数)与一先前已知的分段方法(z分数)进行比较的一表格。所述资料展示,总体而言所述分类分数的预测力横跨所有阶段的乳癌样本而一贯地高于所述z分数的预测力。
图6F绘示使用所述分类分数法的改进的预测力,对于所有类型的癌症(顶端)可被观察到。对早期癌症的预测力是改善的及对晚期癌症的预测力格外优良(底部)。
4.甲基化计算机分析
4.1.甲基化特征
再次简要地参照图1B至1D,所述甲基化计算机分析140D接收由所述甲基化定序化验136产生的数个序列读数及基于所述数个序列读数决定数个甲基化特征156的数值。一般地,在本领域中已知的任何已知的基于甲基化的定序方式可以被用以评估遍及所述基因组或遍及一基因检测组合的数个CpG位点的甲基化状态(例如,参见美国专利第2014/0080715号及USSN第16/352,602号,上述两者通过引用被并入本文中)。甲基化特征156的数个示例包括下列中的任何特征:低甲基化计数的量值、过甲基化计数的量值、每个CpG位点的低甲基化分数、每个CpG位点的过甲基化分数、基于数个过甲基化分数的排名、基于数个低甲基化分数的排名及在一个或更多个CpG位点的异常甲基化(例如,低甲基化或过甲基化)断片的存在或不存在。数个甲基化特征可以是一断片甲基化模式,所述断片甲基化模式可以例如,通过计算满足一组标准的数个断片而被决定,或由可以是由与此模型分别或共同被训练的一分离的机器学习模型决定。
低甲基化计数的所述量值及过甲基化计数的所述量值分别意指低甲基化及过甲基化的cfDNA断片的总数量。低甲基化及过甲基化cfDNA断片使用数个甲基化状态向量而被辨识,如与图7A中的程序700关联地于下文中被描述的。
给定数个断片的低甲基化或过甲基化的存在,每个CpG位点的所述低甲基化分数及每个CpG位点的过甲基化分数与癌症机率的一估计相关。一般地,每个CpG位点的所述低甲基化分数及每个CpG位点的所述过甲基化分数是基于与低甲基化或过甲基化的所述CpG位点重叠的数个甲基化状态向量的计数。
基于过甲基化分数的所述排名及基于低甲基化分数的所述排名可以意指数个甲基化状态向量的排名,数个甲基化状态向量的所述排名是基于它们的相关的低甲基化/过甲基化分数。在一实施例中,所述排名意指数个断片的排名,数个断片的所述排名是基于在每个断片中的数个甲基化状态向量的一最大低甲基化/过甲基化分数。
在一CpG位点的一异常甲基化(例如,低甲基化或过甲基化)断片的存在或不存在对每个CpG位点可以被表示为一个0(不存在)或1(存在)数值。在一些面向中,一异常甲基化断片的所述存在/不存在是对提供最多信息增益的数个CpG位点被决定。举例而言,在一些情况中,约3000个CpG位点被辨识为提供信息增益及因此,对此特定特征有约3000个特征数值。
4.2.甲基化计算机分析总览
图7A是根据一实施例,描述用于辨识来自一对象的数个异常甲基化的断片的一过程700的一流程图。过程700的一示例在图7B中被视觉化地例示,及在下文中参考图7A被进一步地描述。在过程700中,一处理系统自所述对象的数个cfDNA断片产生710A甲基化状态向量。一甲基化状态向量包含在所述数个cfDNA断片中的所述数个CpG位点及所述数个CpG位点的甲基化状态(甲基或未甲基化)。举例而言,一甲基化状态向量可以被表示为 对于一给定甲基化状态向量,所述处理系统列举710B在所述甲基化状态向量中具有相同起始CpG位点及相同长度(亦即,CpG位点的集合)的甲基化状态向量的所有可能。因此每个甲基化状态可能是甲基化或未甲基化,在每个CpG位点仅有两个可能状态,及因此甲基化状态向量的独特的可能的计数依赖于2的次方,而使长度n的一甲基化状态向量将与甲基化状态向量的2 所述处理系统通过评估一健康控制组资料结构,计算710C对所述辨认的起始CpG位点/甲基化状态向量长度而言,观察到甲基化状态向量的每个可能的机率。在一实施例中,计算观察到一给定的可能的所述机率使用一马克夫链机率,以建模所述联合机率计算,所述联合机率计算将在下文中参考图7B而被更详细地描述。在其它实施例中,不同于马可夫链机率的计算方法被用以决定观察到甲基化状态向量的每个可能的所述机率。 为了产生一健康控制组资料结构,所述处理系统将衍生自在所述健康控制组中的数个个体的cfDNA的数个甲基化状态向量细分为数串(strings of)CpG位点。在一实施例中,所述处理系统细分所述甲基化状态向量,而使作为结果的数个串皆小于一给定的长度。举例而言,长度11的一甲基化状态向量可以被细分为数个串,小于或等于3的长度将造成9个长度3的串、10个长度2的串及11个长度1的串。在另一示例中,长度7的一甲基化状态向量被细分为长度小于或等于4的串将导致4个长度4的串、5个长度3的串、6个长度2的串及7个长度1的串。若一甲基化状态向量短于所述特定串长度或与所述特定串长度长度相同,则所述甲基化状态向量可以被变换为含有所述向量的所有CpG位点的一单一串。 所述处理系统通过数算对于在所述向量中的每个可能的CpG位点及甲基化状态的可能而言,存在所述控制组中,具有所述特定CpG位点作为串中的第一CpG位点及具有甲基化状态的该可能的串的数量,而纪录(tallies)所述数个串。举例而言,在一给定的CpG位点,及考虑3的串长,有2^3或8个可能的串配置。在该给定CpG位点,对于所述8个可能的串配置中的每个,所述处理系统纪录在所述控制组中,每个甲基化状态向量可能发生多少次。继续此示例,这可能涉及对在所述参考基因组中的每个起始CpG位点x,纪录下述量值: 所述处理系统使用每个可能的所述经计算机率,计算710D所述甲基化状态向量的一p值分数。在一实施例中,这包括辨识与正被考虑的所述甲基化状态向量符合的可能相对应的的所述经计算的机率。特定地,这是与所述甲基化状态向量具有相同一组CpG位点,或类似地具有相同的起始CpG位点及长度的可能性。所述处理系统加总具有少于或等于所述经辨认的机率的机率的任何可能的所述数个经计算的机率,以产生所述p值分数。 此p值代表在所述健康控制组中观察到所述断片的所述甲基化状态向量或更加不可能的其它甲基化状态向量的所述机率。因此,一低p值分数,大致对应于在一健康个体中罕见的一甲基化状态向量,及造成所述断片被标记为相对于所述正常控制组异常甲基化。一高p值分数一般地关联于在一健康对象中,在一相对概念上被预期存在的一甲基化状态向量。举例而言,若所述健康控制组是一非癌症组,一低p值指示所述断片相对于所述非癌症组而言是异常甲基化的,及因此可能指示在所述检测对象中的癌症的存在。 如上,所述处理系统对数个甲基化状态向量中的每个计算p值分数,所述数个甲基化状态向量中的每个代表在所述检测样本中的一cfDNA断片。为了辨认所述数个断片中的哪一个是异常甲基化的,所述处理系统可以基于数个甲基化状态向量的p值分数过滤710E所述数个甲基化状态向量的所述集合。在一实施例中,过滤是通过将所述p值分数与一阀值相比较及仅保留低于所述阀值的那些断片而执行。此阀值p值分数可以是在0.1、0.01、0.001、0.0001或类似的数量级上。 图7B是根据一实施例的一示例性p值分数计算的一示图715。为了计算给定一检测甲基化状态向量720A的一p值分数,所述处理系统取该检测甲基化状态向量720A,及列举710B甲基化状态向量的数个可能。在此例示性示例中,所述检测甲基化状态向量720A是 所述处理系统计算710C数个甲基化状态向量的被列举的数个可能的机率720B。因为甲基化是有条件地依赖于附近的CpG位点的甲基化状态,计算观察到一给定的甲基化状态向量的可能的所述机率的一个方法是使用马可夫链模型。一般地,一甲基化状态向量,比如 P( 马可夫链模型可以被用于使每个可能的所述条件机率的计算更有效率。在一实施例中,所述处理系统选择一马可夫链阶层k,所述马可夫链阶层k对应于在所述条件机率计算中要考虑多少在所述向量(或窗口)中的先验的CpG位点,而使所述条件机率被建模为P(S 为了计算对甲基化向量的一可能的每个经马可夫链建模机率,所述处理系统存取所述控制组的资料结构,特别是数个CpG位点及状态的各种串的计数。为了计算P(M [#of 所述计算可以通过应用一先验分布而额外实施所述数个计数的一平滑化。在一实施例中,所述先验分布是如在拉普拉斯平滑化中的一均匀先验。作为此实施例的一示例,一常数被加到上述等式的分子及另一常数(例如,两倍于在所述分子中的所述常数)被加到上述等式的分母。在其它实施例中,一演算法技术,比如聂氏平滑法(Knesser-Neysmoothing)被使用。 在所述例示中,上文中表示的公式被应用到覆盖位点23至26的所述检测甲基化状态向量720A。一旦所述经计算的机率720B被完成,所述处理系统计算710D一p值分数720C,所述p值分数720C加总数个机率,所述数个机率少于或等于符合所述检测甲基化状态向量720A的甲基化状态向量的可能的机率。 在一实施例中,计算机率及/或p值分数的计算机负担可以通过缓存至少一些计算而被进一步减少。举例而言,所述分析系统可以将数个甲基化状态向量(或其窗口)的可能的机率的计算缓存于暂时或永久记忆体中。若其它断片具有相同的CpG位点,缓存所述可能机率允许p分数数值的有效率的计算,而不需要重新计算潜在的可能机率。最终,所述处理系统可以为与来自向量(或其窗口)的一组CpG位点相关联的数个甲基化状态向量的可能中的每个计算p值分数。所述处理系统可以缓存所述p值分数,供用于决定包括所述相同CpG位点的其它断片的所述p值分数。一般地,具有相同CpG位点的甲基化状态向量的可能的所述p值分数可以被用于决定来自所述同一组CpG位点的所述可能的不同的一个的所述p值分数。 再次参考图7A,在一实施例中,所述处理系统使用710F一滑动窗口以决定甲基化状态向量的可能及计算p值。所述处理系统仅对连续的数个CpG位点的一窗口列举可能及计算p值,而非对整个甲基化状态向量列举可能及计算p值,其中所述窗口在(CpG位点的)长度上比至少一些断片短(否则,所述窗口便无济于事)。所述窗口长度可以是静态的、使用者决定的、动态的或另外选择的。 在为大于所述窗口的一甲基化状态向量计算p值时,所述窗口自所述向量中第一个CpG位点开始,辨识在所述窗口中的,连续一组来自所述向量的CpG位点。所述分析系统计算包括所述第一CpG位点的所述窗口的一p值分数。所述处理系统接着将所述窗口“滑动”到所述向量中的第二个CpG位点,及为所述第二窗口计算另一p值分数。因此,对于尺寸l的一窗口及甲基化向量长度m,每个甲基化状态向量将产生m–l+1个p值分数。在完成对所述向量的每个部份的所述p值计算后,来自所有滑动窗口的最低的p值分数被采取为所述甲基化状态向量的所述总体p值分数。在另一实施例中,所述处理系统合计所述数个甲基化状态向量的所述p值分数以产生一总体p值分数。 使用所述滑动窗口有助减少甲基化状态向量的被列举的可能的数量及若非如此则需要被执行的,对应的机率计算。示例机率计算被展示在图7B中,但一般地,甲基化状态向量的可能的数量随着所述甲基化状态向量的尺寸而呈2的次方指数增加。为了给出一现实的例子,断片可能具有多于54个CpG位点。所述处理系统可以对所述断片使用(举例而言是)尺寸5的一窗口,导致对所述甲基化状态向量的50个窗口中的每个执行50个p值计算,而非计算2^54(约1.8×10^16)个可能的机率以产生一单一p值分数。所述50个计算中的每个列举所述甲基化状态向量的2^5(32)个可能,总共导致50×2^5(1.6×10^3)个可能性计算。这导致对异常断片的准确辨识缺乏有意义命中的,要被执行的计算的一大量减少。 所述处理系统可以对异常断片的所述集合执行任何种类及/或可能性的额外分析。一额外分析自所述经过滤集合辨识710G数个低甲基化断片或数个过甲基化断片。低甲基化或过甲基化的数个断片可以被界定为一特定长度的数个CpG位点(例如,多于3、4、5、6、7、8、9、10个等)的数个断片,所述断片分别具有高百分比的甲基化CpG位点(例如,多于80%、85%、90%、或95%、或在50%至100的范围内的任何其它百分比)或高百分比的未甲基化CpG位点(例如,多于80%、85%、90%、或95%、或在50%至100的范围内的任何其它百分比)。描述于下文中的图7C,例示用于基于数个异常甲基化断片的集合,辨识一基因组的这些异常甲基化部分的一示例性过程。虽然图7C的数个步骤描述使用来自数个训练群组的异常甲基化断片训练一分类器的过程,特定步骤(例如,步骤730A至730E)也可以意指产生在步骤710G中被辨识的异常甲基化断片的集合的数个特征,比如甲基化特征。 在各种实施例中,所述处理系统可以辨识与一特定CpG位点重叠的一异常甲基化断片的一存在或不存在。如上文中所描述的,一CpG位点的一异常甲基化断片的所述存在/不存在可以作为一甲基化特征。作为一示例,若一过甲基化或低甲基化断片与一特定CpG位点重叠,则所述CpG位点可以被指定一个1的数值,指示一异常甲基化断片的存在。相反地,所述CpG位点可以被指定一个0的数值,指示在所述CpG位点处缺乏一异常甲基化断片。在各种实施例中,所述处理系统辨认提供最多信息增益的数个CpG位点及决定在提供最多信息增益的所述数个CpG位点处,一异常甲基化断片的所述存在/不存在。举例而言,对于每个CpG位点,给定一高度甲基化或未甲基化断片是否与该CpG位点重叠的知识,所述处理系统计算对所述标签(癌症对非癌症)的信息增益。所述处理系统基于每个CpG位点的所述信息增益选择居首的k个CpG位点及可以决定一异常甲基化断片在所述数个信息最丰富的CpG位点中的每个的所述存在/不存在。 一替代分析将一经训练的分类模型应用710H到数个异常断片的所述集合上。在下文中描述的所述经训练的分类模型可以意指展示于图1C中的所述预测性癌症模型170B。换言之,所述经训练的分类模型可以是一单一化验预测模型,所述单一化验预测模型输出基于数个全基因组特征152的一癌症预测190B。 所述经训练的分类模型可以被训练以辨识任何可以自所述数个甲基化状态向量被辨识的感兴趣的情况。一般地,所述经训练的分类模型可以应用数个分类技术中的任何一个。在一实施例中,所述分类器是一非线性分类器。在一特定实施例中,所述分类器是应用具有一高斯径向基函数核(Gaussian radial basis function(RBF)kernel)的一L2-正规化函数核逻辑回归(L2-regularized kernel logistic regression)的一非线性分类器。 在一实施例中,所述经训练的分类模型是一个二元分类器,所述二元分类器基于获得自具有癌症的一对象队列(cohort)的数个cfDNA断片的甲基化状态而被训练,及可选地基于获得自不具有癌症的一健康对象队列的数个cfDNA断片的甲基化状态而被训练,及接着被用以基于异常甲基化状态向量分类一检测对象可能具有癌症或不具有癌症的可能性。在进一步的实施例中,不同的分类器可以使用已知具有特定癌症(例如,乳癌、肺癌、前列腺癌等)的对象队列而被训练,以预测一检测对象是否具有那些特定癌症。 在一实施例中,所述分类器是基于来自过程710G的,关于过/低甲基化区域的信息,及是如同下文中参考图7C所述地被训练。 另一额外的分析计算来自一对象的所述数个异常断片一般性地指示癌症,或指示特定类型的癌症的所述对数胜算比。所述对数胜算比可以通过以非癌症的一机率(亦即,一减掉为癌症的一机率)为底,取为癌症的所述机率的一比率的对数值而被计算,上述两个机率皆如由被应用的分类模型所决定的。 图7D至7F展示横跨不同癌症阶段的来自各种对象的各种癌症的图表,所述图表描绘根据参考上方的图7A而被描述的过程而被辨识的所述数个异常断片的所述对数胜算比。此资料是经由检测超过1700个临床上可评估的对象而获得,其中超过1400个对象被过滤,包括近600个不具有癌症的对象及稍超过800个具有癌症的对象。在图7D中的第一张图表750A展示横跨三个不同级别:非癌症;I/II/II期;及IV期的所有癌症案例。IV期的所述癌症对数胜算比显著大于I/II/II期及非癌症的癌症对数胜算比。在图7D中的第二张图表750B展示横跨所有癌症分期及非癌症的乳癌案例,随着癌症的进展阶段,有一较小的对数胜算比增加。图7E中的第三张图表750C展示数个乳癌亚型。明显地,亚型HER2+及TNBC更为铺展开来,而HR+/HER2–较集中接近至约1。图7E中的第四张图表750D展示横跨所有癌症阶段及非癌症的肺癌案例,所述案例具有经过肺癌的数个渐进阶段的稳定进展。第五张图表750E展示横跨所有癌症阶段及非癌症的大肠直肠癌案例,再次展示经过大肠直肠癌的数个渐进阶段的稳定进展。图7F中的第六张图表750F展示横跨癌症的所有阶段及非癌症的前列腺癌案例。此示例不同于先前例示的多数示例,仅第IV期相较于其它阶段I/II/II及非癌症是显著不同的。 4.3.过/低甲基化区域及一分类器 图7C是根据一实施例,描述基于数个cfDNA断片的甲基化状态训练一分类器的一过程725的一流程图。所述过程存取数个样本的两个训练群组:一非癌症群组及一癌症群组,及获得700包含每个组中的数个样本的所述异常断片的一非癌症组甲基化状态向量及一癌症组甲基化状态向量。所述异常断片可以根据举例而言,图7A的所述过程而被辨识。 所述过程判定730A对每个甲基化状态向量,所述甲基化状态向量是否是低甲基化或过甲基化的。在此,若至少一些数量的CpG位点具有一特定状态(分别为甲基化或未甲基化)及/或具有一阀值百分比的位点处于所述特定状态(再次地,分别为甲基化或未甲基化),所述过甲基化或低甲基化标签被指派。如上文所界定的,若cfDNA断片具有至少五个CpG位点是未甲基化或甲基化的及(逻辑上的且)高于80%的所述断片CpG位点是未甲基化或甲基化的,则所述cfDNA断片分别被辨识为低甲基化或过甲基化的。低甲基化的总断片数(例如,低甲基化计数的量)及过甲基化的总断片数(例如,过甲基化计数的量)可以作为甲基化特征。 在一替代实施例中,所述过程考量所述甲基化状态向量的数个部分,及判定所述部分是否是低甲基化或过甲基化的,及可以区辨该部分是低甲基化或过甲基化的。此替代方案解析遗失的甲基化状态向量,所述遗失的甲基化状态向量在尺寸上是大型的,但含有至少一个密集低甲基化或过甲基化的区域。这界定低甲基化及过甲基化的过程可以被应用在图7A的步骤710G中。 所述过程对所述基因组中的每个CpG位点产生730B一低甲基化分数及一过甲基化分数。如已在上文中讨论的,所述基因组中的每个CpG位点的所述低甲基化分数及过甲基化分数可以作为一甲基化特征。为了在一给定CpG位点处产生两个分数,所述分类器在该CpG位点处采取四个计数:(1)与所述CpG位点重叠的,被标示为低甲基化的癌症集合的(甲基化状态)向量的计数;(2)与所述CpG位点重叠的,被标示为过甲基化的癌症集合的向量的计数;(3)与所述CpG位点重叠的,被标示为低甲基化的非癌症集合的向量的计数;及(4)与所述CpG位点重叠的,被标示为过甲基化的非癌症集合的向量的计数。此外,所述过程可以将每个群组的这些计数标准化,以计入所述非癌症群组及所述癌症群组间的群组大小的差异。 为了在一给定的CpG位点处产生730C所述低甲基化分数,所述过程采取(1)与(1)及(3)加总的比例。类似地,所述过甲基化分数是通过取(2)与(2)及(4)的一比例而被计算。此外,这些比例可以使用如上文所讨论的一额外的平滑化技术而被计算。鉴于来自所述癌症集合的数个断片的低甲基化或过甲基化的存在,所述低甲基化分数及所述过甲基化分数与癌症机率的一估计有关。 所述过程对每个异常甲基化状态向量产生730C一总计低甲基化分数及一总计过甲基化分数。所述总计过甲基化及低甲基化分数,是基于在所述甲基化状态向量中的所述数个CpG位点的所述过甲基化及低甲基化分数而被决定。在一实施例中,所述总计过甲基化及低甲基化分数是分别被指定为在每个状态向量中的所述数个位点的最大过甲基化及低甲基化分数。然而,在替代的实施例中,所述总计分数可以基于平均值、中位数、最大值或其它计算,所述计算使用在每个向量中的数个位点的过/低甲基化分数。 所述过程接着排名730D对象的所有甲基化状态向量,所述排名730D是按所述数个甲基化状态向量的总计低甲基化分数及按所述数个甲基化状态向量的总计过甲基化分数,造成每个对象两组排名。在特定实施例中,所述过程基于所述最大低甲基化分数或最大过甲基化分数排名数个断片,从而造成每个对象两组排名。这两组排名,其是基于数个过甲基化分数的所述排名及基于数个低甲基化分数的数个排名,可以作为甲基化特征。所述过程自所述低甲基化排名选择数个总计低甲基化分数及自所述过甲基化排名选择数个总计过甲基化分数。以所述被选择的数个分数,所述分类器对每个对象产生730E产生一单一特征向量。在一实施例中,被选择自两个排名的所述数个分数是以一固定排序被选择,所述固定排序对所述数个训练群组的每个中的每个对象的每个被产生的特征向量而言是相同的。作为一示例,在一实施例中,所述分类器自每个排名中选取第一、第二、第四、第八及第十六个总计过甲基化分数,及对每个总计低甲基化分数也相同,及将这些分数写在该对象的所述特征向量中。在步骤730F,所述过程训练730F一个二元分类器区辨所述癌症及非癌症训练组的特征向量。所述经训练的分类器可以在需要时,举例而言,在图7A的步骤710H被应用。 5.共通化验特征 再次参考图1B至1D,在各种实施例中,所述数个全基因组特征152、数个小变异特征154及数个甲基化特征156中的一个或更多个,也可以包括一个或更多个共通化验特征。共通化验特征意指可以无论所执行的物理化验(例如,全基因组定序化验132、小变异定序化验134、甲基化定序化验136)而被判定的特征。 共通化验特征可以对预测癌症提供有用信息。作为一示例,共通化验特征可以是所述cfDNA115的一特征。作为另一示例,共通化验特征可以是所述gDNA(例如,WBC DNA120)的一特征。举例而言,cfDNA或gDNA的一特征可以是在一样本中的cfDNA或gDNA的一总量。在罹癌个体中的肿瘤经常制造较高水平的DNA,及因此,DNA(例如,cfDNA)的总量对产生一癌症预测可以提供有用信息。作为另一示例,cfDNA的一特性可以是肿瘤衍生核酸的一总浓度。作为又一示例,cfDNA的一特性可以是获得自所述cfDNA样本的数个DNA断片的一平均或中位数断片长度。 6.基线分析及基线计算机分析 6.1.基线特征 再次参考图1B至1D,所述基线分析130及所述计算机分析140A判定数个基线特征150的数值。基线特征150的数个示例包括所述个体110的临床特征,比如年龄、体重、身体质量指数(BMI)、患者行为(例如,吸烟者/非吸烟者、每年吸烟包数(pack years smoked)、酒类摄取、缺乏身体活动)、家族史(例如,经由一问卷调查获得的家族史)、生理症状(例如,咳嗽、便血等)、解剖学观察(例如,乳房密度)及一外显生殖细胞系癌症的带原。在各种实施例中,所述个体110的数个临床特征可以经由一基线分析130直接被获得。换句话说,所述基线分析130可以直接产生数个基线特征150,而不需一基线计算机分析140A。 基线特征150的数个示例也可以包括衍生自生殖细胞系突变的一多基因风险分数。生殖细胞系突变可以包括与癌症相关联的已知生殖细胞系突变,比如ATM、BRCA1、BRCA2、CHEK2、PALB2、PTEN、STK11或TP53中的一者。在此,所述基线计算机分析140A可以执行所述数个步骤以决定由所述多基因风险分数代表的一特征数值。 6.1.基线分析及计算机分析工作流程 图8A绘示根据一实施例的,用于决定可以被用于将一患者分层的数个基线特征的一流程800。在步骤810A,与所述个体110相关联的数个临床特征被获得。作为一示例,数个临床特征可以作为所述个体110的一医学检查(例如,由一医学专家或一医师执行的一检查)的一结果被获得。 在步骤810B,所述个体的数个生殖细胞系突变被获得。在各种实施例中,经辨识的生殖细胞系突变自一物理化验对获得自所述个体的DNA的应用被获得。作为一示例,所述数个生殖细胞系突变经由所述全基因组定序化验132及所述全基因组计算机分析140B的应用而被辨识。特定地,所述全基因组定序化验132可以被应用到gDNA(例如,WBC DNA120),以产生数个序列读数,及所述全基因组计算机分析140B可以被应用以辨识存在的所述数个生殖细胞系突变。作为一示例,一生殖细胞系突变可以是在gDNA及cfDNA两者中共同地被发现的一拷贝数目变异,如关于所述拷贝数目畸变侦测工作流程而在上文中被讨论的(例如,段落3.2)。 在步骤810C,一多基因风险分数(PRS)基于被获得的所述生殖细胞系突变而对所述个体被决定。作为一示例,一PRS可以被表示为所述被辨识出的数个生殖细胞系突变的经加权总和。每个被指定到一生殖细胞系突变的权重可以经由训练资料被事前决定。作为一示例,一的所述PRS被计算为横跨每个个体i的所有风险基因座(loci)的对数(胜算比)的经加权总和: 一给定癌症的基因对数(胜算比):对数(OR)=(∑ 在步骤810D,可选地,对所述个体的一癌症预测可以基于所述数个基线特征而被提供,所述数个基线特征包括所述数个临床特征及所述多基因风险分数。在一实施例中,一癌症预测可以包括所述个体在一特定类别中的分层,所述分层是基于所述数个基线特征。所述个体的分层可以是有用的,因为所述个体的一癌症预测可以为所述个体被分类在其中的所述特定分层而被定制。一分层的一示例可以是发展出癌症的一风险(例如,高风险、中度风险、低风险)。 在一实施例中,所述数个基线特征的标准被用于将所述个体分层入一类别。作为一示例,一PRS的一标准可以是一阀值PRS百分位数(例如,数个个体的为首5、10、25、50%)。若所述个体被指定了高于所述阀值PRS百分位数的一PRS,则所述个体可以被分层在一第一群组中。相反地,若所述个体被指定了低于所述阀值PRS百分位数的一PRS,则所述个体可以被分层在一第二群组中。作为另一示例,一临床特征的一标准可以是癌症在一个体的家族史中的一存在或不存在。因此,若所述个体的家族史包括癌症的存在,则所述个体被指派进入一第一类别。相反地,若所述个体的家族史缺少癌症的存在,则所述个体被指派进入一第二类别。各种标准可以被联合地使用以进一步将数个个体分层入不同的类别中。 在一实施例中,所述癌症预测可以是基于所述数个基线特征的癌症的一存在或不存在。举例而言,给定所述PRS及一个或更多个临床特征,所述个体是否具有癌症的一预测可以被提供。 6.2.示例1:基于基线特征的癌症预测 图8B绘示数个逻辑回归预测性模型基于数个基线特征的不同组合,横跨不同类型的乳癌的表现。所关注的乳癌包括侵袭性乳癌(IBC)整体及基于激素受体状态的4个亚型的乳癌:(HR+/HER2-乳癌、HER2+/HR-乳癌、HER2+/HR+乳癌及三阴性乳癌(TNBC)。 许多基线特征对乳癌是有预测性的。不同的预测性模型预测不同类型的乳癌的存在的能力被测试。第一模型是基于作为步进逻辑回归的结果的所述变量选择、接收包括一PRS及乳房密度的数个基线特征的一第二模型、仅作为所述基线特征而接收PRS的一第三模型、及作为所述基线特征接收乳房密度的一第四模型。每个模型的所述表现作为曲线下面积(area under the curve,AUC)及留一交叉验证(LOOCV)结果两者被纪录。在图8B中,所述表现被表示为AUC/LOOCV。PRS及乳房密度强力地预测乳癌。作为数个基线特征接收PRS+乳房密度的所述模型一般地展现与基于步进逻辑回归的所述第一模型类似的表现,及比仅作为一特征接收PRS的所述模型及仅作为一特征接收乳房密度的所述模型表现更佳。对于一些癌症(例如,HER2+/HR-及HER2+/HR+),作为基线特征接收PRS+乳房密度的所述模型也比所述步进模型表现更佳。 7.预测性癌症模型的示例 除非另有说明,在此主题中被描述的数个所述预测性癌症模型中的每个经由一逻辑回归模型,基于一个或更多个类型的特征(例如,数个小变异特征、数个全基因组特征、数个甲基化特征及/或数个基线特征)产生一癌症预测。每个预测性癌症模型使用衍生自一循环游离基因组图谱(CCGA)研究(NCT02889978)的数个患者的第一子集的一组训练资料被训练,及随后使用衍生自所述CCGA研究的数个患者的一第二子集的一组测试资料被测试。 图9A绘示CCGA研究的数个实验参数。1792个患者被分类在所述训练集合中,以训练在不同计算机分析及预测性癌症模型中的所述模型,-而其它1008个患者作为一测试集合,被保持除外于所述训练。所述1792个患者相对于合格标准被评估,及不合格患者被自训练移除。总合而言,1726个患者的资料(981个癌症及577个非癌症)被用于训练在所述小变异工作流程中的所述数个模型、1720个患者的资料(975个癌症及576个非癌症)被用于训练所述全基因组工作流程的所述数个模型,及1699个患者的资料(964个癌症及568个非癌症)被用于训练所述甲基化工作流程的所述数个模型。 在所述训练集合中的所述1792个患者中,1399个患者通过了所述合格标准。所述1399个患者,其中841是罹癌的(所述841个中有437个表现有肿瘤组织)及558个是非癌症的,被用于使用一个10折交叉验证程序训练所述数个模型。所述1008个保持除外患者随后被用于测试所述数个模型的表现。图9B绘示用以为每个各自的预测性癌症模型决定数个特征的数值的数个实验细节(例如,基因检测组合,定序深度等)。除非另外说明,在下文中被描述的所述数个预测性癌症模型是使用来自所述循环游离基因组图谱(CCGA)研究的数个已知癌症类型被训练。如上文所描述的,所述CCGA样本集合包括下述癌症类型:乳癌、肺癌、前列腺癌、大肠直肠癌、肾癌、子宫癌、胰脏癌、食道癌、淋巴瘤、头颈癌、卵巢癌、肝胆癌、黑色素瘤、子宫颈癌、多发性骨髓瘤、白血病、甲状腺癌、膀胱癌、胃癌及肛门直肠癌。因此,除非另外说明,在下文中被描述的每个模型是一多癌症模型(或多癌症分类器),用于一种或更多种、两种或更多种、三种或更多种、四种或更多种、五种或更多种、十种或更多种或20种或更多种不同类型的癌症的侦测。在一些实施例中,所述一种或更多种癌症可以是一“高信号”癌症(定义为具有大于50%的5年癌症特定死亡率的癌症),比如肛门直肠癌、大肠直肠癌、食道癌、头颈癌、肝胆癌、肺癌、卵巢癌及胰脏癌,以及淋巴瘤及多发性骨髓瘤。高信号癌症倾向于更具侵略性及典型地在获得自一患者的检测样本中具有一高于平均的游离核苷酸浓度。 7.1.根据图1B的多重化验预测性癌症模型 图9C绘示根据展示于图1B中的实施例的,使用数个小变异特征、数个全基因组特征、及数个甲基化特征以预测癌症的存在的一预测性癌症模型的特异度及敏感度的一接收者操作特征(receiver operating characteristic,ROC)曲线。在此,所述预测性癌症模型是一惩罚性逻辑回归模型(penalized logistic regression model)及输出一分数(例如,一黏合分数(Bonded score))。所述黏合分数接着被用以预测癌症的存在或不存在。特定地,所述预测性癌症模型的所述数个小变异特征包括在一基因检测组合(共469个基因向量)中的每个基因的数个体细胞变异的一存在/不存在、所述预测性癌症模型的所述数个全基因组特征包括数个经缩减的维度特征(共5个经缩减的维度特征)、及所述预测性癌症模型的所述数个甲基化特征包括基于数个过甲基化分数的排名及基于数个低甲基化分数的排名。特别地,基于数个过甲基化分数的所述排名包括7个过甲基化断片(例如,排名1、排名2、排名4、排名8、排名16、排名32、及排名64的过甲基化断片)。类似地,基于数个低甲基化分数的所述排名包括7个低甲基化断片(例如,排名1、排名2、排名4、排名8、排名16、排名32、及排名64的低甲基化断片)。所述ROC曲线的所述总AUC是0.722。图9C绘示所述预测性癌症模型在一85%至100%特异度范围内的表现。在此示例中,所述ROC指示在一95%特异度的一约44%敏感度及在一99%特异度的一约36%敏感度。 7.2.根据图1C的单一化验预测性癌症模型 7.2.1.小变异化验预测性癌症模型 图10A绘示使用一第一组小变异特征预测癌症的存在的一预测性癌症模型的特异度及敏感度的一接收者操作特征(ROC)曲线。特定地,所述预测性癌症模型输出一分数,在下文中称作一“A_分数”,所述A_分数指示癌症的存在或不存在。所述ROC曲线的曲线下总面积(AUC)是0.697。鉴于目标是要于给定一组特异度(例如,95%或99%特异度)下达成一敏感度,图10A绘示所述预测性癌症模型在一85%至100%特异度范围内的表现。在此示例中,被提供至所述预测性癌症模型的所述数个小变异特征包括:数个体细胞变异的一总数及数个非同义突变的一总数。所述ROC曲线指示在一95%特异度的一约35%敏感度及在一99%特异度的一约19%敏感度。自99%特异度进展至95%特异度,所述ROC曲线非线性地增加,从而指示在此敏感度/特异度取舍中可能有真实阳性被侦测到。 图10B绘示使用一第二组小变异特征预测癌症的存在的一预测性癌症模型的特异度及敏感度的一ROC曲线。特定地,所述预测性癌症模型输出一分数,在下文中称作一变异基因分数,所述变异基因分数指示癌症的存在或不存在。所述ROC曲线的总AUC是0.664。图10B绘示所述预测性癌症模型在一85%至100%特异度范围内的表现。在此示例中,被提供至所述预测性癌症模型的所述数个小变异特征包括每个基因的数个体细胞变异的一AF。在此,每个基因的数个体细胞变异的所述AF代表在每个基因中的一体细胞变异的所述最大AF。因此,总数500个的每个基因的数个体细胞变异的所述最大AF的数值(对应于500个基因)作为数个特征数值被提供至所述预测性癌症模型。所述ROC曲线指示在一95%特异度的一约38%敏感度及在一99%特异度的一约31%敏感度。这代表与展示于图10A中的所述预测性癌症模型相比下的一改进。 图10C绘示使用一第三组小变异特征预测癌症的存在的一预测性癌症模型的特异度及敏感度的一ROC曲线。特定地,所述预测性癌症模型输出一分数,在下文中称作一排序分数,所述排序分数指示癌症的存在或不存在。所述ROC曲线的总AUC是0.672。图10C绘示所述预测性癌症模型在一85%至100%特异度范围内的表现。在此示例中,所述预测性癌症模型的所述数个小变异特征包括根据数个体细胞变异的AF的为首6个排名顺序。所述ROC曲线指示在一95%特异度的一约37%敏感度及在一99%特异度的一约30%敏感度。再次地,这代表与展示于图10A中的所述预测性癌症模型相比下的一改进。 7.2.2.全基因组预测性癌症模型 图10D绘示使用一组全基因组特征预测癌症的存在的一预测性癌症模型的特异度及敏感度的一ROC曲线。特定地,所述预测性癌症模型输出一分数,在下文中称作一WGS分数,所述WGS分数指示癌症的存在或不存在。所述ROC曲线的总AUC是0.706。图10D绘示所述预测性癌症模型在一85%至100%特异度范围内的表现。在此示例中,所述预测性癌症模型的所述数个全基因组特征包括遍及所述基因组的数个区间段的区间段分数及数个经缩减的维度特征(例如,基于PCA而被衍生的数值)。合计,37000个特征数值代表遍及所述基因组的数个区间段的区间段分数,而5至200个较低维度成分的数个特征数值作为输入被提供至所述癌症预测性模型。所述ROC曲线指示在一95%特异度的一约40%敏感度及在一99%特异度的一约33%敏感度。 7.2.3.甲基化预测性癌症模型 图10E绘示使用一第一组甲基化特征预测癌症的存在的一预测性癌症模型的特异度及敏感度的一ROC曲线。在此,所述预测性癌症模型是一惩罚性逻辑回归模型。特定地,所述预测性癌症模型输出一分数,在下文中称作二元分数,所述二元分数指示癌症的存在或不存在。所述ROC曲线的总AUC是0.719。图10E绘示所述预测性癌症模型在一85%至100%特异度范围内的表现。在此示例中,所述预测性癌症模型的所述数个甲基化特征包括基于数个过甲基化分数的排名及基于数个低甲基化分数的排名。特别地,基于数个过甲基化分数的所述排名包括7个过甲基化断片(例如,排名1、排名2、排名4、排名8、排名16、排名32、及排名64的过甲基化断片)。类似地,基于数个低甲基化分数的所述排名包括7个低甲基化断片(例如,排名1、排名2、排名4、排名8、排名16、排名32、及排名64的低甲基化断片)。所述ROC曲线指示在一95%特异度的一约45%敏感度及在一99%特异度的一约36%敏感度。 图10F绘示使用一第二组甲基化特征预测癌症的存在的一预测性癌症模型的特异度及敏感度的一ROC曲线。特定地,所述预测性癌症模型输出一分数,在下文中称作WGBS分数,所述WGBS分数指示癌症的存在或不存在。所述ROC曲线的总AUC是0.724。图10F绘示所述预测性癌症模型在一85%至100%特异度范围内的表现。在此示例中,所述预测性癌症模型的所述数个甲基化特征包括异常甲基化(例如,低甲基化或过甲基化)的断片在数个CpG位点处的存在或不存在。在此特定示例中,提供最多信息增益的所述数个CpG位点(例如,约3000个CpG位点)被辨识。因此,有共约3000个此特征的特征值,每个特征值是0或1,指示在一被辨识的CpG位点处是否有一异常甲基化断片。所述ROC曲线指示在一95%特异度的一约42%敏感度及在一99%特异度的一约35%敏感度。 图10G绘示使用一第三组甲基化特征预测癌症的存在的一预测性癌症模型的特异度及敏感度的一ROC曲线。特定地,所述预测性癌症模型输出一分数,在下文中称作一MSUM分数,所述MSUM分数指示癌症的存在或不存在。所述ROC曲线的总AUC是0.718。图10G绘示所述预测性癌症模型在一85%至100%特异度范围内的表现。在此示例中,所述预测性癌症模型的所述数个甲基化特征包括所述数个异常甲基化(例如,低甲基化或过甲基化)的断片在数个CpG位点处的存在或不存在的特征的所述平均值。在此示例中,有一平均值代表在上文中与图10F关联地被描述的共约3000个数值的平均。在此,所述数个数值是0或1,及指示在一感兴趣的CpG位点处是否有一异常甲基化断片。所述ROC曲线指示在一95%特异度的一约42%敏感度及在一99%特异度的一约32%敏感度。 7.2.4.单一化验预测性癌症模型的结果的加总 图10H绘示数个单一化验预测性癌症模型(例如,应用于衍生自所述小变异定序化验、全基因组定序化验及甲基化定序化验中的每一个的特征的数个预测性癌症模型)中每一个的表现。特定地,包括衍生自由所述小变异定序化验产生的数个序列读数的数个特征的所述预测性癌症模型,意指如在上文中于图10B中描述的,输出一变异基因分数的所述预测性癌症模型。包括衍生自由所述全基因组定序化验产生的数个序列读数的数个特征的所述预测性癌症模型,意指如在上文中于图10D中描述的,输出一WGS分数的所述预测性癌症模型。对衍生自由所述甲基化定序化验产生的数个序列读数的数个特征的所述预测性癌症模型,意指如在上文中于图10F中描述的,输出一WGBS分数的所述预测性癌症模型。 图10H包括一图表1000,所述图表1000绘示不同的预测性模型及非癌症/癌症间的量化交集。此外,图10H在所述图表下方绘示一矩阵1050,所述矩阵1050指示由一预测性癌症模型进行的,对癌症的辨识(例如,实心点)或未辨识(例如,空心点)。 如展示于图10H中的,534个已知非癌症患者及483个已知癌症患者使用所述数个预测性癌症模型中的每个被分析。在所述483个已知癌症患者中,所述三个预测性癌症模型中的每个在244个患者中成功辨认出癌症的存在。此外,所述全基因组预测性模型及所述甲基化预测性模型,但非所述小变异预测性模型,成功地在额外的27个患者中辨认出癌症的存在。在一个或更多个预测性模型成功辨认出的癌症间的进一步的交集被展示在矩阵1050中。 值得注意的是,有五个患者是临床上非癌症的,但所述数个预测性模型中的至少两个辨认出癌症的一存在。在这五个患者中,所述患者中的一个随后被临床诊断出癌症。额外的四个患者可能是伪阳性。 7.3.额外的单一化验预测性癌症模型 图10I绘示数个预测性癌症模型对横跨数个不同阶段的不同类型的癌症的表现。特定地,图10I绘示每个单一化验预测性癌症模型(例如,小变异预测性癌症模型、全基因组(WG)预测性癌症模型及甲基化预测性癌症模型)在一95%特异度的敏感度。在此,所述小变异预测性癌症模型意指包括非同义变异的所述总数的一特征及输出一A分数的所述预测性癌症模型。此小变异预测性癌症模型的一示例在上文中与图10A相关联地被描述。所述WG预测性癌症模型意指包括数个全基因组特征,包括遍及所述基因组的数个区间段的区间段分数,及数个经缩减的维度特征(例如,基于PCA而衍生的数个数值)的一预测性癌症模型。所述WG预测性癌症模型输出一WGS分数。此全基因组预测性癌症模型的一示例在上文中与图10D相关联地被描述。所述甲基化预测性癌症模型意指包括数个甲基化特征,包括异常甲基化(例如,低甲基化或过甲基化)的断片在数个CpG位点处存在或不存在,的所述预测性癌症模型。所述甲基化预测性癌症模型输出一WGBS分数及在上文中与图10F相关联地被描述。 图10I进一步划分出了数个预测性癌症模型对具有一大于25%的5年死亡率的癌症及对具有一小于25%的5年死亡率的癌症的表现。具有一大于25%的5年死亡率的癌症包括肛门直肠癌、子宫颈癌、大肠直肠癌、食道癌、胃癌、头颈癌、肝胆癌、肺癌、淋巴癌、多发性骨髓瘤、卵巢癌、胰管腺癌(pancreatic ductal adenocarcinoma)、肾癌及三阴性乳癌。具有一小于25%的5年死亡率的癌症包括膀胱癌、ER+乳癌、黑色素瘤、前列腺癌、甲状腺癌及子宫癌。如绘示于图10I中的,相较于所述数个预测模型在早期癌症(例如,I/II/III期癌症)的敏感度,所述数个预测模型对晚期癌症(例如,IV期癌症)的敏感度是增进的。此外,如绘示在图10I中的,在本文中描述的所述数个单一化验模型对具有大于25的5年死亡率的癌症,在95%特异度展现较高的敏感度。举例而言,在95%特异度,所述甲基化化验在具有高死亡率(大于25%的5年死亡率)的癌症(于I/II/III期)展现一60%侦测敏感度,相对于在具有低死亡率(小于25%的5年死亡率)的癌症(于I/II/III期)展现一13%侦测敏感度。 图10J至10L中的每张图绘示(除了展示于图10I中的所述小变异(在图10J至10L中的每张当中被称为非同义变异)、WG及甲基化预测性癌症模型外)额外的数个预测性癌症模型对不同类型的侵袭性癌症的表现。所述额外的数个预测性癌症模型包括作为输入接收年龄、吸烟习惯及癌症家族史的数值的一基线预测性模型,以及接收每个基因的数个体细胞变异的AF的所述特征的数个数值的一小变异预测性模型(称为AF变异)。所述特定的小变异预测性模型(例如,AF变异)在上文中与图10B关联地被描述。 特定地,图10J绘示所述数个预测性癌症模型在95%特异度对癌症总体、乳癌、肺癌、前列腺癌、大肠直肠癌、子宫癌、肾癌及胰腺癌的表现。图10K绘示所述数个预测性癌症模型在95%特异度对食道癌、淋巴瘤、头/颈癌、卵巢癌、甲状腺癌、肝胆癌、子宫颈癌及多发性骨髓癌的表现。图10L绘示所述数个预测性癌症模型在95%特异度对黑色素瘤、膀胱癌、未知癌症、胃癌、肛门直肠癌、2个以上原发位置的癌症及其它癌症的表现。 图10M绘示数个预测性癌症模型对数个不同阶段的大肠直肠癌的表现。特定地,图10M绘示每个单一化验预测性癌症模型在一95%特异度的所述敏感度。在此,所述数个预测性癌症型中的每一个在早期(例如,I/II期)及晚期(例如,III/IV期)癌症展现一大于50%的敏感度。 图10N绘示额外(在展示于图10M中的所述非同义变异、WG及甲基化预测性癌症模型外)的数个预测性癌症模型对数个不同阶段的大肠直肠癌的表现。图10N绘示每个单一化验预测性癌症模型在一95%特异度的所述敏感度。 图10O绘示数个预测性癌症模型对数个不同阶段及数个不同类型的乳癌的表现。图10O绘示每个单一化验预测性癌症模型在一95%特异度的所述敏感度。在此,图10O展示数个预测性癌症模型的所述表现对不同类型的乳癌是不同的。举例而言,所述AF变异、非同义变异、WG及甲基化预测性癌症模型中的每个,相较于对HER2+及HR+/HER2-乳癌,对三阴性乳癌(TNBC)更为敏感。此外,比起对早期乳癌,预测性癌症模型对较晚期乳癌的所述敏感度是改善的。进一步地,如也被展示在图10L中的,在95%特异度,对TNBC的侦测敏感度比HER2+或HR+/HER2-各自更高(例如,使用所述甲基化化验,对TNBC62%分别对上39%及14%)。 图10P绘示数个预测性癌症模型对数个不同阶段及数个不同类型的肺癌的表现。图10P绘示每个单一化验预测性癌症模型在一95%特异度的所述敏感度。在此,图10P展示数个预测性癌症模型的所述表现对不同类型的肺癌是不同的。举例而言,比起对早期肺癌,预测性癌症模型对较晚期肺癌的所述敏感度是改善的。此外,一般地,所述非同义变异、WG及甲基化预测癌症模型中的每个相较于所述基线及AF变异预测性模型,对不同类型及不同阶段的肺癌展现出较高的敏感度。 7.4.根据图1D的多阶段,多化验预测性癌症模型 图11A绘示预测癌症的存在的一个二阶段预测性癌症模型的特异度及敏感度的一ROC曲线。在此,所述预测性癌症模型包括四个第一阶段模型(例如,展示于图1D中的数个预测性模型180),所述四个第一阶段模型各自是逻辑回归。所述预测性癌症模型进一步包括一第二阶段模型(例如,展示于图1D中的整体预测性模型185),其中所述第二阶段模型是一随机森林模型。 根据图1D,所述预测性癌症模型的一第一阶段包括分别分析数个全基因组特征、数个小变异特征及数个甲基化特征的分离的预测性模型180B、180C及180D。特定地,所述预测性模型180B是如与图10D相关联地被描述的,输出一WGS分数的一预测性模型。所述预测性模型180C是如与图10B相关联地被描述的,输出一变异基因分数的一预测性癌症模型及与图10C相关联地被描述的,输出一排序分数的一预测性癌症模型的一结合。所述预测性模型180D是如与图10E相关联地被描述的,输出一个二元分数的一预测性癌症模型、与图10F相关联地被描述的,输出一WGBS分数的一预测性癌症模型及与图10G相关联地被描述的,输出一MSUM分数的一预测性癌症模型的一结合。 所述预测性癌症模型的所述第二阶段是一总体预测性模型是被训练以使用所述WGS分数、变异基因分数、排序分数、二元分数;WGBS分数及MSUM分数中的每一个的数值输出一癌症预测。因此,在发展时,所述预测性癌症模型的所述第二阶段,作为输入,接收所述数个分数及输出对一个体110的一癌症预测,比如癌症在所述个体中的一存在/不存在。 所述ROC曲线的所述总AUC是0.722。图11A绘示所述预测性癌症模型在一85%至100%特异度范围中的表现。所述ROC曲线指示在一95%特异度的一约43%敏感度及在一99%特异度的一约34%敏感度。 7.5.示例多阶段,多化验预测性癌症模型 如在上文中于图1D中被描述的,一预测性癌症模型(例如,预测性癌症模型195)可以是一多阶段多化验预测性癌症模型(“多阶段模型”)。一多阶段模型包括两个或更多个第一阶段预测性癌症模型(例如,预测性癌症模型180A、180B、180C及180D),所述两个或更多个第一阶段预测性癌症模型中的每个产生对一对象的一癌症预测。所述数个第一阶段预测性癌症模型(“第一阶段模型”)中的每个自决定自获得自一个体(例如,个体110)的一检测样本的一个或更多个特征产生一癌症预测。 在一实施例中,每个第一阶段模型被配置以自一类似类型的数个特征(例如,来自一单一定序化验的数个特征)产生一癌症预测。举例而言,一第一个第一阶段模型可以使用数个小变异特征(例如,小变异特征154)产生对所述个体的一癌症预测,一第二个第一阶段模型可以使用数个甲基化特征(例如,甲基化特征156)产生一癌症预测等。在另一实施例中,每个第一阶段模型是配置以自一个或更多个类型的特征产生一癌症预测。举例而言,一第一个第一阶段模型可以使用数个小变异特征及数个甲基化特征产生对所述个体的一癌症预测,及所述第二个第一阶段模型可以使用数个全基因组特征(例如,全基因组特征152)及数个基线特征(例如,数个基线特征150)产生对所述个体的一癌症预测。 对于一多阶段模型,有许多可能的第一阶段模型的配置。举例而言,可能有两个、三个、四个、五个等的第一阶段模型被包括在一多阶段模型中。作为另一示例,可能有一个、两个、三个、四个、五个等类型的特征被用作对一第一阶段模型的一输入。 一多阶段模型包括一总体预测性模型(例如,总体预测性模型185),所述总体预测性模型产生一癌症预测(例如,癌症预测190)。所述总体预测性模型(“第二阶段模型”)输入所述数个第一阶段模型的所述癌症预测及产生所述癌症预测。所述第二阶段模型可以使用上文中描述的各种方法中的任何方法被训练。作为一示例,所述第二阶段模型可以是一随机森林模型,被训练以使用衍生自具有一已知的癌症诊断的数个检测序列的数个特征决定一癌症预测。 对于一多阶段模型,有许多可能的第二阶段模型的配置。举例而言,所述多阶段模型可以被训练以使用两个、三个、四个、五个等产生自所对应的第一阶段模型的癌症预测来产生一癌症预测。作为另一示例,一多阶段模型可以具有多于两个阶段。为了例示,两个第二阶段模型的所述结果可以被输入进一癌症预测模型(“第三阶段模型”),所述癌症预测模型被训练以决定对一个体的一癌症预测。 7.5.1.示例1:使用WGS及小变异特征的第一阶段模型 如上文中所述地,一多阶段模型的一示例包括两个第一阶段模型及一单一第二阶段模型。在此示例中,所述第一个第一阶段模型(“第一模型”)是一癌症预测模型,所述癌症预测模型配置以使用数个小变异特征决定对一个体的一癌症预测。输入到所述第一模型中的所述特征可以使用类似于在段落2“小变异计算机分析”中被描述的数个技术而被衍生。所述第一模型可以输出一分数(例如,一A分数),所述第一分数指示对所述个体的一癌症预测。段落7.2.1“小变异化验预测性癌症模型”描述可以被用作所述第一模型的一预测性癌症模型。图10A至10C例示一第一模型的所述预测性能力的数个示例。进一步地,在此示例中,所述第二个第一阶段模型(“第二模型”)是一癌症预测模型,所述癌症预测模型配置以使用数个WGS特征决定对一个体的一癌症预测。被输入所述第二模型的所述数个特征可以使用类似于在段落3,“全基因组计算机分析”中被描述的技术而被衍生。所述第二模型可以输出一分数(例如,一WGS分数),指示对所述个体的一癌症预测。段落7.2.2,“全基因组预测性癌症模型”描述可以被用作所述第二模型的一预测性癌症模型。图10D例示所述第二模型的所述预测性能力的一示例。最后,在此示例中,所述第二阶段模型(“结合模型”)是配置以使用所述第一模型及第二模型的所述癌症预测产生一癌症预测。在此,所述第二阶段模型使用一第二层正规化逻辑回归演算法,以决定一癌症预测,但可以使用其它演算法。 图10Q例示根据一第一示例实施例的,一多阶段模型的一ROC曲线图。图10R根据一示例实施例,以一不同视角例示展示于图10Q中的所述多阶段模型的所述ROC曲线图。所述多阶段模型是以上文描述的方式配置。特定地,所述多阶段模型包括(i)一第一模型,以使用数个小变异特征(在包含与癌症相关的数个基因的一针对性基因检测组合(GRAIL的专有针对性小变异检测组合)中的每个基因的最大等位基因频率)作为一输入,产生一癌症预测,(ii)一第二模型,以使用数个WGS特征(横跨包含与癌症相关的数个基因的一针对性基因检测组合(GRAIL的专有针对性小变异检测组合)的每个基因的拷贝数目z分数),产生一癌症预测,及(iii)一结合模型,以使用所述第一模型及所述第二模型的所述癌症预测(使用第二层正规化逻辑回归)产生一癌症预测。所述多阶段模型被应用到获得自数个个体的一群组的数个检测序列,以决定一癌症预测。所述数个检测序列包括液态癌症序列、非癌症序列及固态癌症序列。 所述ROC曲线绘图例示对于所述第一模型、所述第二模型及所述结合模型,所述真实阳性率作为所述伪阳性率的一函数。在所述ROC曲线图上的数条曲线提供所述数个癌症预测模型中的每个的所述预测能力的一表现(图10Q包括于99%特异度(展示为一方型)、98%特异度(展示为一个三角形)及95%特异度(展示为一圆形)的敏感度估计。值得注意的是,所述结合模型的所述预测能力比所述第一模型或所述第二模型中的任一个都更好。换句话说,在一多阶段癌症预测模型中使用两个癌症预测模型的所述预测性结果,提供比个别的癌症预测模型更好的预测能力。 表7.5.1.1.提供例示于图10Q及10R中的所述多阶段模型的所述预测能力的量化。所述表格表示所述第一模型、所述第二模型及所述结合模型的所述特异度、敏感度。所述敏感度的95%信赖区间的下界及上界也被展示于所述表格中。 在此,所述结合模型在所有特异度上胜过所述第一模型及所述第二模型。
表7.5.1.1:一多阶段模型的癌症预测 图10S绘示根据一实施例,所述第一模型、所述第二模型及所述结合模型的敏感性作为敏感性的一函数的一比较。图10S是被包括在表7.5.1.1中的所述资料的一视觉化。 表7.5.1.2提供所述第一模型及所述结合模型的所述预测能力的另一量化。在此示例中,所述检测序列包括27个液态癌症检测序列、574个非癌症检测序列及890个固态癌症检测序列。在所述表格中,“真”表示一模型判定一阳性癌症预测及“伪”表示一模型判定一阴性癌症预测。在一癌症类型下的一数字表示具有所指示的预测的检测序列的数量。因此,举例而言,所述结合模型正确地将三个液态癌症检测序列辨认为“真”而所述第一模型错误地将那相同的三个液态癌症检测序列辨认为“伪”。
表7.5.1.2:对液态/非液态癌症的分类比较 7.5.2.示例2:使用WGS及甲基化特征的第一阶段模型 示例2是另一个具有一第一模型、一第二模型及一结合模型的多阶段模型。在此示例中,所述第一模型是配置以使用数个WGS特征决定对一个体的一癌症预测。输入到所述第一模型中的所述特征可以使用类似于在段落3,“全基因组计算机分析”中被描述的技术被衍生。所述第一模型可以输出一分数(例如,一WGS分数),指示对所述个体的一癌症预测。段落7.2.2,“全基因组预测性癌症模型”描述可以被用作所述第一模型的一预测性癌症模型。图10D例示所述第二模型的所述预测性能力的一示例。进一步地,在此示例中,所述第二模型是配置以使用数个甲基化特征决定对一个体的一癌症预测。输入所述第二模型的所述数个特征可以使用类似于在段落4,“甲基化计算机分析”中被描述的技术被衍生。所述第二模型可以输出一分数(例如,一M分数),指示对所述个体的一癌症预测。段落7.2.3,“甲基化预测性癌症模型”描述可以被用作所述第二模型的一预测性癌症模型。图10E至10F例示所述第二模型的所述预测性能力的数个示例。最后,在此示例中,所述结合模型是配置以使用所述第一模型及第二模型的所述癌症预测产生一癌症预测。 图10T至10U根据一第一示例实施例绘示一多阶段模型的ROC曲线图形。图10T显示一多阶段模型决定对一对象的一肺癌预测的能力,及图10U显示所述多阶段模型、所述第一模型及所述第二模型决定所述对象的一乳癌预测的能力。 所述多阶段模型以上文所述的方式被配置。特定地,所述多阶段模型包括(i)一第一模型,以使用数个WGS特征(如在段落3.1中描述的在所述读数计数的为首5个主成分上的主成分投影)作为一输入,产生一癌症预测,(ii)一第二模型,以使用数个甲基化特征(如在段落4.3中描述的过甲基化计数及/或低甲基化计数),产生一癌症预测,及(iii)一结合模型,使用所述第一模型及所述第二模型的所述癌症预测产生一癌症预测。所述多阶段模型被应用到获得自数个个体的一群组的数个检测序列,以决定一癌症预测。 所述ROC曲线绘图例示对于所述第一模型、所述第二模型及所述结合模型,所述敏感度作为所述特异度的一函数。在所述ROC曲线图上的数条曲线提供所述数个癌症预测模型中的每个的所述预测能力的一表现。在此案例中,所述结合模型的所述改进较展示于示例1中的所述多阶段模型小。因此,分析在所述ROC曲线图上的数条曲线中的每条的曲线下面积(“AUC”)是有用的。所述AUC是所述数个方法中的每个的所述总体预测能力的一测量方式,具有一较高的AUC一般而言代表一更加敏感及特定的模型。 表7.5.2.1.提供所述多阶段模型对不同类型的癌症决定一癌症预测的所述AUC。所述表格包括表示癌症的类型的一栏目及所述结合模型、所述第一模型及所述第二模型的所述AUC。在此,癌症预测的小改进见于肺癌及大肠直肠癌。
表7.5.2.1:液态/非液态癌症的分类比较 7.5.3.示例3:使用基线及甲基化特征的第一阶段模型 示例3是另一个具有一第一模型、一第二模型及一结合模型的多阶段模型。在此示例中,所述第一模型是配置以使用数个甲基化特征决定对一个体的一癌症预测。输入到所述第一模型中的所述特征可以使用类似于在段落4,“甲基化计算机分析”中被描述的技术被衍生。所述第一模型可以输出一分数(例如,一M分数),指示对所述个体的一癌症预测。段落7.2.3,“甲基化预测性癌症模型”描述可以被用作所述第二模型的一预测性癌症模型。图10E至10F例示所述第二模型的所述预测性能力的一示例。进一步地,在此示例中,所述第二模型是配置以使用数个基线特征决定对一个体的一癌症预测。被输入所述第二模型的所述数个特征可以使用类似于在段落6,“基线分析及基线计算机分析”中被描述的技术被衍生。所述第二模型可以输出一分数,指示对所述个体的一癌症预测。图8B例示所述第二模型当被应用到乳癌时的所述预测性能力的示例。最后,在此示例中,所述结合模型是配置以使用所述第一模型及第二模型的所述癌症预测产生一癌症预测。 图10V、图10W及图10X根据一示例实施例例示一多阶段模型的ROC曲线图形。图10V显示一多阶段模型在决定对具有一高信号性癌症的一个体的一癌症预测上的能力,图10W显示所述多阶段模型在决定对所述个体的一肺癌预测上的能力,及图10X显示所述多阶段模型在决定对所述个体的一HR-乳癌预测上的能力。 所述多阶段模型以上文所述的方式被配置。特定地,所述多阶段模型包括(i)一第一模型,以使用数个甲基化特征(如在段落4.3中描述的过甲基化计数及/或低甲基化计数),产生一癌症预测,(ii)一第二模型,以使用数个基线特征(包括性别、种族、身体质量指数(BMI)、酒类摄取、哺乳史、BiRAD乳房密度、吸烟史、二手烟及/或肺癌的直系家族史)产生一癌症预测,及(iii)一结合模型,使用所述第一模型及所述第二模型的所述癌症预测(使用逻辑回归)产生一癌症预测。所述多阶段模型被应用到获得自数个个体的一群组的数个检测序列,以决定一癌症预测。 所述ROC曲线绘图例示对于所述第一模型、及所述结合模型,所述敏感度作为所述特异度的一函数。在所述ROC曲线图上的数条曲线提供所述数个癌症预测模型中的每个的所述预测能力的一表现。第二模型未被例示,但表现得较所述结合模型差。对于图10V(高信号癌症),所述ROC曲线对所述第一阶段模型(基线特征)具有0.65的一曲线下面积(AUC),及对所述结合模型(基线及甲基化特征)具有0.85的一AUC。对于图10W(肺癌),所述ROC曲线对于所述第一阶段基线模型具有0.83的一曲线下面积(AUC),对所述第一阶段甲基化模型具有一0.89的一AUC,及对所述结合模型(基线及甲基化特征)具有0.93的一AUC。及对于图10X(HR-乳癌),所述ROC曲线对于所述第一阶段基线模型具有0.69的一曲线下面积(AUC),对所述第一阶段甲基化模型具有一0.75的一AUC,及对所述结合模型(基线及甲基化特征)具有0.83的一AUC。值得注意的是,所述结合模型的所述预测能力比所述第一模型或所述第二模型中的任一个都更好。换句话说,在一多阶段癌症模预测模型中使用两个癌症预测模型的所述预测性结果,提供比个别的癌症预测模型更好的预测能力。 7.6.示例单一阶段,多重化验预测性癌症模型 如上文中在图1B中描述的,图12A至12P绘示数个单一阶段预测性癌症模型(例如,预测性癌症模型160),所述单一阶段预测性癌症模型使用来自一个或更多个基于定序的化验的一个或更多个特征(例如,基线特征150、全基因组特征152、小变异特征154及/或甲基化特征156),以产生对一对象的一癌症预测。 图12A至12P中的每张图绘示根据展示于图1B中的所述实施例,使用一个或更多个小变异特征、全基因组特征、甲基化特征及或基线特征来预测癌症的存在的数个单一阶段预测性癌症模型,对在(先前描述的)所述CCGA研究中的所有癌症的所述特异度及敏感度的一接收者操作特征(ROC)曲线。在此,被例示的所述数个预测性癌症模型中的每个包含一XGBoost模型,但可以是其它机器学习模型。特定地,所述预测性癌症模型的所述小变异特征包括两个特征中的一个:(1)在一基因检测组合(GRAIL的专有507癌症关联基因检测组合)中的数个基因中的每个的非同义体细胞变异的最大等位基因频率(MAF)(参见,例如,段落2.1;亦见图12A),或(2)遍及一基因检测组合(GRAIL的专有507癌症关联基因检测组合)中的所有基因的数个体细胞变异的顺序统计量,但可以包括其它特征。这些特征的范围可以在0至1间,大部分的值确切为0,及典型的非零数值是低于0.1(参见,例如,段落2.1;亦见图12B、12D、12F、12H、12J、12L、12N及12P)。所述预测性癌症模型的所述全基因组特征包括数个经缩减的维度特征(共5个经缩减的维度特征)(如在段落3.1中描述的),但可以包括其它特征。所述预测性癌症模型的所述甲基化特征包括基于数个过甲基化分数的排名及基于数个低甲基化分数的排名(如在段落4.1及4.3中描述的),但可以包括其它特征。特定地,基于数个过甲基化分数的所述排名包括7个过甲基化断片(例如,排名1、排名2、排名4、排名8、排名16、排名32、及排名64的过甲基化断片)。类似地,基于数个低甲基化分数的所述排名包括7个低甲基化断片(例如,排名1、排名2、排名4、排名8、排名16、排名32、及排名64的过甲基化断片)。所述基线分析包括下述临床特征的分析:BMI、年龄、吸烟期间的连续变量,及乳房组织密度、自陈综合健康、自陈运动强度的指示变量,但可以包括其它特征。数个指示变量被编码为0或1,及数个连续变量被用于它们被报告的范围内(参见,例如,段落6.1)。 如上文中所描述的,图12A例示应用来自已知与癌症风险相关联的数个基因的一针对性检测组合的数个小变异特征(例如,MAF)来预测癌症的存在的一个单一阶段预测性癌症模型,对在先前描述的所述CCGA研究中的所有癌症的所述特异度及敏感度的一接收者操作特征(ROC)曲线。所述ROC曲线指示在一98%特异度的一约29%敏感度、在一98%特异度的一约33%敏感度,及0.643的一曲线下面积。 图12B例示应用来自已知与癌症风险相关联的数个基因的一针对性检测组合的数个小变异特征(例如,排名顺序)来预测癌症的存在的一个单一阶段预测性癌症模型,对在先前描述的所述CCGA研究中的所有癌症的所述特异度及敏感度的一ROC曲线。所述ROC曲线指示在一37%特异度的一约32%敏感度、在一98%特异度的一约33%敏感度,及0.661的一曲线下面积。 图12C例示应用来自一甲基化化验的数个甲基化特征来预测癌症的存在的一个单一阶段预测性癌症模型,对在先前描述的所述CCGA研究中的所有癌症的所述特异度及敏感度的一ROC曲线。所述ROC曲线指示在一98%特异度的一约18%敏感度、在一98%特异度的一约36%敏感度,及0.685的一曲线下面积。 图12D例示应用数个小变异特征及数个甲基化特征来预测癌症的存在的一个单一阶段预测性癌症模型,对在先前描述的所述CCGA研究中的所有癌症的所述特异度及敏感度的一ROC曲线。所述ROC曲线指示在一98%特异度的一约28%敏感度、在一98%特异度的一约37%敏感度,及0.688的一曲线下面积。 图12E例示应用数个全基因组定序(例如,WGS)特征来预测癌症的存在的一个单一阶段预测性癌症模型,对在先前描述的所述CCGA研究中的所有癌症的所述特异度及敏感度的一ROC曲线。所述ROC曲线指示在一98%特异度的一约13%敏感度、在一98%特异度的一约31%敏感度,及0.644的一曲线下面积。 图12F例示应用数个小变异特征及数个全基因组定序(WGS)特征来预测癌症的存在的一个单一阶段预测性癌症模型,对在先前描述的所述CCGA研究中的所有癌症的所述特异度及敏感度的一ROC曲线。所述ROC曲线指示在一98%特异度的一约31%敏感度、在一98%特异度的一约36%敏感度,及0.668的一曲线下面积。 图12G例示应用数个WGS特征及数个甲基化特征来预测癌症的存在的一个单一阶段预测性癌症模型,对在先前描述的所述CCGA研究中的所有癌症的所述特异度及敏感度的一ROC曲线。所述ROC曲线指示在一98%特异度的一约32%敏感度、在一98%特异度的一约39%敏感度,及0.688的一曲线下面积。 图12H例示应用数个小变异特征、数个WGS特征及数个甲基化特征来预测癌症的存在的一个单一阶段预测性癌症模型,对在先前描述的所述CCGA研究中的所有癌症的所述特异度及敏感度的一ROC曲线。所述ROC曲线指示在一98%特异度的一约27%敏感度、在一98%特异度的一约38%敏感度,及0.693的一曲线下面积。 图12I例示应用数个基线特征来预测癌症的存在的一个单一阶段预测性癌症模型,对在先前描述的所述CCGA研究中的所有癌症的所述特异度及敏感度的一ROC曲线。所述ROC曲线指示在一98%特异度的一约3%敏感度、在一98%特异度的一约9%敏感度,及0.559的一曲线下面积。 图12J例示应用数个小变异特征及数个基线特征来预测癌症的存在的一个单一阶段预测性癌症模型,对在先前描述的所述CCGA研究中的所有癌症的所述特异度及敏感度的一ROC曲线。所述ROC曲线指示在一98%特异度的一约19%敏感度、在一98%特异度的一约33%敏感度,及0.702的一曲线下面积。 图12K例示应用数个甲基化特征及数个基线特征来预测癌症的存在的一个单一阶段预测性癌症模型,对在先前描述的所述CCGA研究中的所有癌症的所述特异度及敏感度的一ROC曲线。所述ROC曲线指示在一98%特异度的一约33%敏感度、在一98%特异度的一约39%敏感度,及0.718的一曲线下面积。 图12L例示应用数个小变异特征、数个甲基化特征及数个基线特征来预测癌症的存在的一个单一阶段预测性癌症模型,对在先前描述的所述CCGA研究中的所有癌症的所述特异度及敏感度的一ROC曲线。所述ROC曲线指示在一98%特异度的一约29%敏感度、在一98%特异度的一约38%敏感度,及0.704的一曲线下面积。 图12M例示应用数个WGS特征及数个基线特征来预测癌症的存在的一个单一阶段预测性癌症模型,对在先前描述的所述CCGA研究中的所有癌症的所述特异度及敏感度的一ROC曲线。所述ROC曲线指示在一98%特异度的一约24%敏感度、在一98%特异度的一约31%敏感度,及0.684的一曲线下面积。 图12N例示应用数个小变异特征、数个WGS特征及数个基线特征来预测癌症的存在的一个单一阶段预测性癌症模型,对在先前描述的所述CCGA研究中的所有癌症的所述特异度及敏感度的一ROC曲线。所述ROC曲线指示在一98%特异度的一约25%敏感度、在一98%特异度的一约37%敏感度,及0.704的一曲线下面积。 图12O例示应用数个WGS特征、数个甲基化特征及数个基线特征来预测癌症的存在的一个单一阶段预测性癌症模型,对在先前描述的所述CCGA研究中的所有癌症的所述特异度及敏感度的一ROC曲线。所述ROC曲线指示在一98%特异度的一约30%敏感度、在一98%特异度的一约38%敏感度,及0.712的一曲线下面积。 图12P例示应用数个小变异特征、数个WGS特征、数个甲基化特征及数个基线特征来预测癌症的存在的一个单一阶段预测性癌症模型,对在先前描述的所述CCGA研究中的所有癌症的所述特异度及敏感度的一ROC曲线。所述ROC曲线指示在一98%特异度的一约30%敏感度、在一98%特异度的一约37%敏感度,及0.710的一曲线下面积。 上文中例示的ROC曲线图例示了对运用如所述的一个或更多个特征的所述数个上述的癌症预测性模型中的每个,所述真实阳性率作为伪阳性率的一函数。所述数个ROC曲线图的曲线中的每个提供了所述数个癌症预测模型中的每个的所述预测能力的一代表。值得注意的是,包括两个或更多个、三个或更多个或四个特征的所述数个癌症预测性模型的所述预测力比基于一单一特征的所述数个癌症预测性模型更好。换言之,于在本文中所描述的癌症预测性模型中使用两个或更多个定序特征,导致具有比基于一单一特征的模型更好的癌症预测能力的一模型。 其它注意事项 上文中的数个实施例的详细描述涉及附随的附图,所述附图例示本揭示的特定实施例。具有不同结构及运作方式的其它实施例并不离开当前揭示的范围。“本发明”或类似的词汇是参考在本说明书中提出的,申请人的发明的许多替代方面或实施例的特定示例而被使用,及其使用或不使用皆不是意在限制申请人的发明的范围或权利要求的范围。本说明书仅是为读者的便利而被划分为数个段落。标题不应被视为限制本发明的范围。定义是意在作为本发明的描述的一部分。应被了解的是,本发明的各种细节可以被改变,而不脱离本发明的范围。此外,上文的描述仅是为了例示的目的,及非为了限制的目的。
- 用于癌症侦测的多重化验预测模型
- 应用于多重存取通讯系统的碰撞侦测方法与装置