一种富马酸丙酚替诺福韦的工业合成方法
文献发布时间:2023-06-19 09:38:30
技术领域
本发明属于药物合成领域,具体涉及一种富马酸丙酚替诺福韦的工业合成方法。
背景技术
富马酸丙酚替诺福韦(TAF)是一种新型核苷类逆转录酶抑制剂(NRTI),该药是吉利德已上市药物Viread(富马酸替诺福韦二吡呋酯,TDF)的升级版。在临床试验中,已被证明在低于TDF十分之一剂量时,就具有非常高的抗病毒疗效,同时表现出更好的安全性,具有改善的肾功能和骨骼安全参数。
CN103842366B专利中公开了式TAF化合物的制备方法,如下式所示:
该专利中,式III中间体的生产周期较长,式IV中间体操作步骤繁琐,可操作性差,反应条件苛刻,设备要求高,生产周期较长,不利于大规模工业化生产。
现有技术中,有专利CN107793452A和CN106478725B等公开适于大规模工业化生产富马酸丙酚替诺福韦的制备工艺,通过改变反应溶剂及纯化方法等使其适用于工业化生产的同时提高其中间体替诺福韦单苯酯和丙酚替诺福韦的纯度。但是上述技术内容还存在反应周期过长的问题以及其中间体纯度并没有显著的提高。
因此,本领域渴望寻求一种适合工业化生产的并且其中间体纯度有显著提高的富马酸丙酚替诺福韦的制备工艺。
发明内容
本发明的目的在于提供一种生产周期短、操作简单并且适用于工业化生产的富马酸丙酚替诺福韦的制备工艺,以替诺福韦为起始物料,通过特殊的磷手性原子合成、纯化分离制备得到高纯度的富马酸丙酚替诺福韦。
本发明的技术方案中,提供了富马酸丙酚替诺福韦即9-[(R)-2-[[(S)-[[(S)-1-(异丙氧基羰基)乙基]氨基]苯氧基氧膦基]甲氧基]丙基]腺嘌呤半富马酸盐(式TAF化合物)的制备方法,其中包括以下步骤。
1、替诺福韦(PMPA)在碱性条件下与亚磷酸三苯酯反应制备得到替诺福韦单苯酯即式Ⅲ:
该步骤的反应溶剂为N,N-二甲基甲酰胺(DMF)或二甲基亚砜(DMSO)反应温度为90-100℃,反应时间为15-18h。选择高沸点溶剂,可大大缩短其反应时间,原研专利中反应时间大于48小时,而本反应只需要15-18小时即可以完成反应;
作为优化,步骤(1)还包括纯化步骤,具体为:反应结束后降温至室温,加入纯化水,水相用乙酸乙酯洗涤,在20~30℃之间水相用浓盐酸调节pH至2~3,降温至0~10℃,析晶2~3h。
进一步的优化,所述纯化步骤还包括二次纯化步骤:具体为,将析晶固体离心,滤饼用pH=1.5的稀盐酸淋洗2次后用乙酸乙酯搅洗1~2h,离心,所得固体产物60~65℃真空干燥24~26h,得到白色固体替诺福韦单苯酯。
该步骤经过二次纯化,可达到HPLC纯度99%以上。
2、替诺福韦单苯酯(式Ⅲ)通过二氯亚砜氯化,然后与L-丙氨酸异丙酯盐酸盐反应制备得到丙酚替诺福韦即式Ⅳ:
具体的:将替诺福韦单苯酯和反应溶剂加入反应瓶中,先升温至内温75~80℃,再滴入二氯亚砜,反应15~18h,得到替诺福韦单苯酯的酰氯化物;在-25~-20℃温度下将L-丙氨酸异丙酯盐酸盐一次性加入替诺福韦单苯酯的酰氯化物溶液中,并采用三乙胺调节pH值至4-7,升温至室温反应2~3h。
上述步骤选择先升温后投料二氯亚砜的方式,可避免杂质的产生,如果先投料后再升温,在升温的过程中容易产生非对映异构体杂质,降低光学纯度。
上述步骤选择L-丙氨酸异丙酯盐酸盐作为反应物替代常规反应物L-丙氨酸异丙酯(游离碱状态),是因为我方发现,游离碱L-丙氨酸异丙酯在进行游离过程中不稳定,会产生异丙醇,进而影响后续反应产生杂质,降低收率。
我方直接采用L-丙氨酸异丙酯盐酸盐进行反应,省略了游离步骤,并不会对后续反应产生影响,相反却提高了产物收率。
本发明的优选技术方案中,所述步骤(2)式Ⅲ与二氯亚砜反应溶剂为甲苯、二甲苯、四氢呋喃、乙腈、二氯甲烷等,优选甲苯。
步骤(2)还包括清洗和精制步骤。所述清洗步骤为将反应液中加入二氯甲烷搅拌,有机相依次用磷酸二氢钠、饱和氯化钠溶液洗涤,无水硫酸钠干燥,过滤,减压浓缩得到琥珀色油状物。
所述精制步骤包括两次精制步骤,第一次精制过程是将琥珀色油状物中加入混合溶剂中,升温溶解后降温析晶2~3h,过滤并干燥得到白色固体一,所述混合溶剂是甲苯和乙腈体积比10:1的混合物;第二次精制过程是将白色固体一投料反应瓶中,加入乙腈,升温至回流溶解,缓慢降温至0~5℃,析晶10~12h,过滤,干燥得到白色固体二。步骤(2) 制备得到的粗品Ⅳ通过二次结晶,能够有效除去非对应异构体,提高化合物Ⅳ的光学纯度。
3、丙酚替诺福韦即(式Ⅳ)通过与富马酸反应制备得到富马酸丙酚替诺福韦即式TAF:
步骤(3)所述的成盐工艺为:将式Ⅳ与0.49eq的富马酸以及乙腈加入至70~75℃溶解,趁热过滤,滤除不溶物,2h内降温至10~15℃,析晶2~3h,过滤、干燥得到TAF。
有益效果
1.所述步骤(1)采用了更高沸点的N-甲基吡咯烷酮替代了原研专利CN103842366B 中的乙腈,原研专利反应时间≥48h,本发明只需要15h就能反应完全,并且溶剂使用量较少,大大缩短了生产周期,节约了生产成本,减少了三废的处理,适合大规模工业生产;优化纯化步骤,通过二次纯化,使产物式Ⅲ纯度可达到99%以上。
2.所述步骤(2)制备得到的粗品Ⅳ磷手性中心S构型可达到88%以上,通过合适的甲苯/乙腈混合溶剂二次结晶,磷手性S构型光学纯度达到99.71%,而原研专利CN103842366B中磷手性S构型光学纯度为97.5%,
3.本发明制备的富马酸丙酚替诺福韦纯度大于99.90%,非对映异构体小于0.15%,其它单杂小于0.10%,具有较高的商业规模生产价值。
附图说明
图1实施例1中间体ⅢHPLC图谱。
图2实施例3中间体Ⅳ粗品(油状物)HPLC图谱。
图3实施例3中间体Ⅳ精制品HPLC图谱。
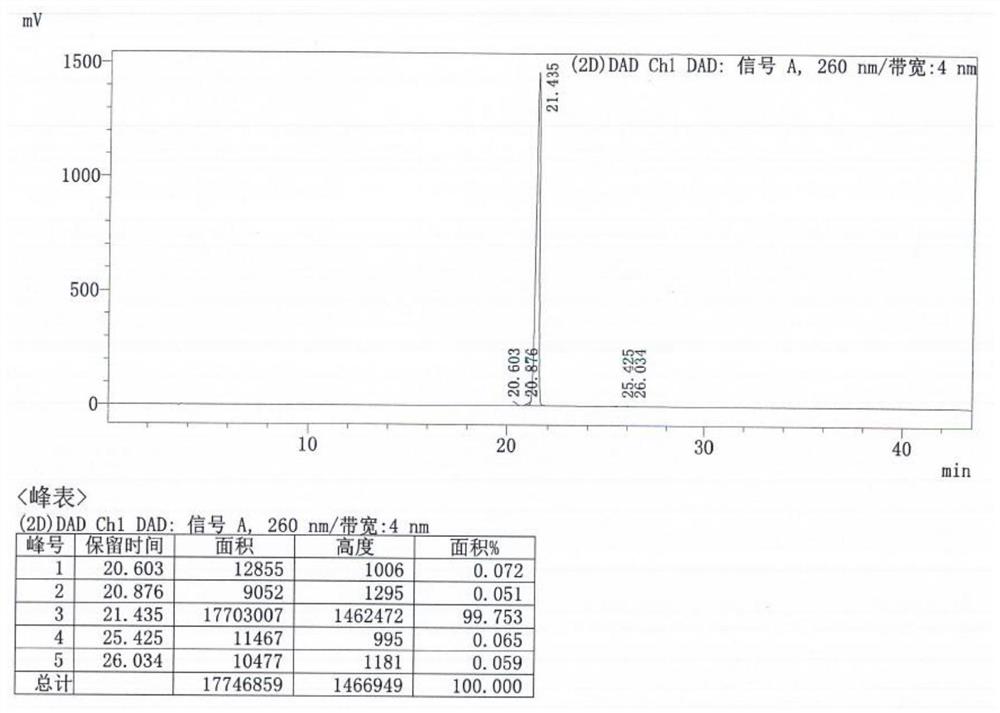
图4TAF HPLC图谱。
具体实施方式
下面通过具体实施例对本发明的技术方案进一步说明,这些实立为示例性的,不代表对本发明保护范围的限制。其他人根据本发明做出的一些非本质的修改和调整仍属于本发明的保护范围。
以下实例中式PMPA采购自荆门市帅邦化学科技有限公司,L-丙氨酸异丙酯盐酸盐采购自四川同晟生物医药有限公司,亚磷酸三苯酯采购自国药集团化学试剂有限公司,富马酸采购自阿拉丁试剂,其它试剂如无特别说明,均为普通市售产品。
缩写备注:
Ph
TEA:三乙胺
DMAP:4-二甲氨基吡啶
DMF:N,N-二甲基甲酰胺
实施例1式Ⅲ化合物的制备
将1.0Kg式PMPA、2.2L的DMF、704.3gTEA、425.2gDMAP和1619.7g Ph
实施例2式Ⅲ化合物的制备
将1.0Kg式PMPA、2.2L的DMSO、704.3gTEA、425.2gDMAP和1619.7g Ph
实施例3式Ⅳ化合物的制备
将800.0g式Ⅲ和6.4L甲苯加入10L反应瓶中,升温至内温75~80℃,滴入471.1g的SOCl
后处理过程:
反应液中加入4.8L二氯甲烷搅拌,有机相用10%磷酸二氢钠洗涤2次,饱和氯化钠溶液洗涤1次,无水硫酸钠干燥,过滤,滤液减压浓缩至无液体流出,得到琥珀色油状物HPLC纯度为88.94%,非对应异构体含量10.39%。
精制过程:
上述油状物中加入4L甲苯、400mL乙腈,升温至40~50℃溶解至清,缓慢降温至20~ 30℃,析晶2~3h,过滤,滤饼55~60℃真空干燥10~12h,得到350.0g白色固体。
将上350.0g白色固体投料3L反应瓶中,加入7倍(体积/质量=7)乙腈,升温至回流溶解,缓慢降温至0~5℃,析晶10~12h,过滤,滤饼55~60℃真空干燥10~12h,得到315.0g白色固体即式Ⅳ,HPLC纯度为99.71%,非对映异构体含量0.09%。
实施例4式Ⅳ化合物的制备
将800.0g式Ⅲ和6.4L四氢呋喃加入10L反应瓶中,升温至回流,滴入471.1g的SOCl
后处理过程:
反应液中加入4.8L二氯甲烷搅拌,有机相用10%磷酸二氢钠洗涤2次,饱和氯化钠溶液洗涤1次,无水硫酸钠干燥,过滤,滤液减压浓缩至无液体流出,得到琥珀色油状物HPLC纯度为86.6%,非对映异构体含量12.53%。
精制过程:
上述油状物中加入4L甲苯、400mL乙腈,升温至40~50℃溶解至清,缓慢降温至20~ 30℃,析晶2~3h,过滤,滤饼55~60℃真空干燥10~12h,得到342.0g白色固体。
将上329.0g白色固体投料3L反应瓶中,加入7倍(体积/质量=7)乙腈,升温至回流溶解,缓慢降温至0~5℃,析晶10~12h,过滤,滤饼55~60℃真空干燥10~12h,得到304.5g白色固体即式Ⅳ,HPLC纯度为99.65%,非对映异构体0.11%。
实施例5:TAF化合物的制备:
将300.0g式Ⅳ和36.5g富马酸装入5L反应瓶中,加入3L乙腈,升温至70~75℃,搅拌溶清后,趁热过滤,2~3h降温至10~15℃,搅拌析晶2~3h,过滤,滤饼用乙腈淋洗,60~65℃真空干燥20~24h,得到329.8g式TAF,收率98.0%,HPLC纯度99.98%,非对映异构体未检出。
- 一种富马酸丙酚替诺福韦的工业合成方法
- 一种富马酸丙酚替诺福韦片剂及其制备方法