一种检测体细胞突变的方法及装置
文献发布时间:2023-06-19 09:43:16
技术领域
本发明属于体细胞突变检测领域,具体涉及一种快速检测体细胞突变的方法及装置。
背景技术
人类基因组中存在各种各样的突变,包括核苷酸突变(SNV)、插入删除(InDel)等。其中相当一部分跟肿瘤的形成和发展息息相关。通过基因组测序,从测序的序列数据中,快速准确地鉴定出这些突变,对肿瘤的研究以及治疗有非常大的帮助。
随着测序技术的飞速发展,测序成本的迅速下降。肿瘤基因组研究中,由原来的小区域捕获(panel)测序,到全外显子组(exome)测序,再往全基因组(wgs)或者超高深度UMI测序方向发展。数据量几何级增长。而且样本量也从原来的肿瘤数个配对样本到几十,几百甚至上千发展。临床分析对分析速度和分析成本也有所要求。但现有的大部分软件和方法都出现得较早,而且大都面向科研小样本量处理,无法进行大数据量分析。
发明内容
本发明提供一种检测体细胞突变的方法及装置。
根据第一方面,一种实施例中提供一种检测体细胞突变的方法,包括:
候选突变提取步骤,包括根据比对文件中的彩色德布鲁恩图、比对信息中的至少一种,从待测样本、相应的对照样本的测序数据中提取候选突变;
过滤步骤,包括对提取的候选突变进行过滤,去除不符合判定条件的突变;
计算步骤,包括根据过滤的候选突变,计算质量特征值,根据所述质量特征值,对候选突变集进行过滤,得到突变检测结果。
根据第二方面,一种实施例中提供一种检测体细胞突变的装置,包括:
候选突变提取模块,用于根据比对文件中的彩色德布鲁恩图、比对信息中的至少一种,从待测样本、相应的对照样本的测序数据中提取候选突变;
过滤模块,用于对提取的候选突变进行过滤,去除不符合判定条件的数据;
计算模块,用于根据过滤的候选突变,计算质量特征值,根据所述质量特征值,对候选突变集进行过滤,得到检测结果。
根据第三方面,一种实施例中提供一种计算机设备,包括:存储器,用于存储程序;
处理器,用于通过执行所述存储器存储的程序以实现如第一方面所述的方法。
根据第三方面,一种实施例中提供一种计算机可读存储介质,包括程序,所述程序能够被处理器执行以实现如第一方面所述的方法。
依据上述实施例的检测体细胞突变的方法及装置,计算步骤之前先进行过滤,极大降低候选突变的数量,加快后续分析速度。
附图说明
图1显示为根据实施例1的方法检测的样本全外显子测序数据中体细胞突变位点计算得到的TMB指标与标准品TMB指标的关系曲线图。
图2显示为根据现有的mutect2检测的样本全外显子测序数据中体细胞突变位点计算得到的TMB指标与标准品TMB指标的关系曲线图。
图3显示为本实施例1的方法检测的样本靶向捕获测序数据中体细胞突变位点计算得到的TMB指标与标准品TMB指标的关系曲线图。
图4显示为根据现有的mutect2检测的样本靶向捕获测序数据中体细胞突变位点计算得到的TMB指标与标准品TMB指标的关系曲线图。
具体实施方式
下面通过具体实施方式结合附图对本发明作进一步详细说明。其中不同实施方式中类似元件采用了相关联的类似的元件标号。在以下的实施方式中,很多细节描述是为了使得本申请能被更好的理解。然而,本领域技术人员可以毫不费力的认识到,其中部分特征在不同情况下是可以省略的,或者可以由其他元件、材料、方法所替代。在某些情况下,本申请相关的一些操作并没有在说明书中显示或者描述,这是为了避免本申请的核心部分被过多的描述所淹没,而对于本领域技术人员而言,详细描述这些相关操作并不是必要的,他们根据说明书中的描述以及本领域的一般技术知识即可完整了解相关操作。
另外,说明书中所描述的特点、操作或者特征可以以任意适当的方式结合形成各种实施方式。同时,方法描述中的各步骤或者动作也可以按照本领域技术人员所能显而易见的方式进行顺序调换或调整。因此,说明书和附图中的各种顺序只是为了清楚描述某一个实施例,并不意味着是必须的顺序,除非另有说明其中某个顺序是必须遵循的。
本文中为部件所编序号本身,例如“第一”、“第二”等,仅用于区分所描述的对象,不具有任何顺序或技术含义。
本文中,肿瘤突变负荷(TMB)是指每百万碱基中被检测出的,体细胞基因编码错误、碱基替换、基因插入或缺失错误的总数。
第一方面,在一实施例中,提供一种检测体细胞突变的方法,包括:
候选突变提取步骤,包括根据比对文件中的彩色德布鲁恩图、比对信息中的至少一种,从待测样本、相应的对照样本的测序数据中提取候选突变;
过滤步骤,包括对提取的候选突变进行过滤,去除不符合判定条件的突变;
计算步骤,包括根据过滤的候选突变,计算质量特征值,根据所述质量特征值,对候选突变集进行过滤,得到突变检测结果。
在一实施例中,比对文件中的比对信息可以是BAM文件中的alignment信息。
在一实施例中,对彩色德布鲁恩图比对、比对信息(如CIGAR字符串、MD字符串等)比对这两种方法检测到的候选突变进行快速过滤,极大降低候选突变的数量,加快后续分析速度。
在一实施例中,合并候选突变集,使用外部数据库,例如gnomAD胚系突变数据库,对突变集进行过滤。极大降低候选突变中的胚系突变的含量,显著加快后续分析速度,而且有效降低体细胞突变的假阳性。
在一实施例中,候选突变提取步骤中,包括将待测样本、对照样本的下机数据比对到参考基因组上,获得比对文件,然后根据比对文件中的彩色德布鲁恩图、比对信息中的至少一种,从成对的待测样本、对照样本的比对文件中提取候选突变。下机数据可以经过常规过滤后,再比对到参考基因组上,也可以不经过常规过滤,直接将下机数据比对到参考基因组上,优选地,下机数据先经过常规过滤,再比对到参考基因组上。
在一实施例中,所述比对文件选自BAM文件、CRAM文件、SAM文件中的至少一种。CRAM文件存储空间比较小,BAM文件存储空间比较大,可根据需要,通过samtools等软件进行格式转换。
在一实施例中,根据彩色德布鲁恩图提取候选突变的方法包括:合并待测样本的测序数据与对照样本的测序数据,比对到参考基因组上,通过局部序列组装,得到彩色德布鲁恩图,通过判断图路径中的颜色,找出非参考基因组特有的路径,提取出候选突变;
在一实施例中,根据比对文件中的比对信息提取候选突变的方法包括:通过分析待测样本、相应的对照样本的测序数据与参考基因组中的比对信息差异,提取出候选突变。
在一实施例中,所述比对文件中的比对信息选自CIGAR字符串、MD字符串中的至少一种。上述字符串包含错配信息,便于后续提取候选突变。
在一实施例中,所述比对文件中的比对信息为CIGAR字符串时,根据CIGAR字符串提取候选突变的方法包括:通过分析待测样本、对照样本的测序数据与参考基因组序列中的CIGAR字符串差异,提取出候选突变。
在一实施例中,过滤步骤中,包括根据序列支持数、序列频率等等中的至少一种进行过滤,去除不符合判定条件的突变。
在一实施例中,过滤步骤中,包括根据待测样本测序数据的序列支持数、相应的对照样本测序数据的的序列支持数、待测样本测序数据的序列频率、相应的对照样本测序数据的序列频率进行过滤,去除不符合判定条件的突变。
在一实施例中,所述过滤步骤包括,遍历所述候选突变提取步骤获得的候选突变中的突变,计算每一个突变位置上,待测样本测序数据中支持突变的测序序列频数AD、相应的对照样本测序数据中支持突变的测序序列频数NAD、待测样本测序数据中测序序列频率VAF、相应的对照样本的正常突变等位基因频率NVAF,根据AD、NAD、VAF和NVAF过滤候选突变;
在一实施例中,VAF=Allele Depth/Total Depth,其中,Allele Depth为支持突变的读长(也可表示为reads)的数量,Total Depth为该突变位点测序深度;也可以表示为VAF=Read Count/Total Count,其中,Read Count为支持突变的reads的数量,TotalCount为该突变位点测序深度。VAF(Variant Allele Frequency,突变等位基因频率)是指突变等位基因相对于所有等位基因的频率。
在一实施例中,所述根据AD、NAD、VAF和NVAF过滤候选突变,具体包括,根据AD、NAD、VAF和NVAF与各自阈值的大小关系,移除不符合判定条件的突变。
在一实施例中,所述根据AD、NAD、VAF和NVAF过滤候选突变,具体包括,移除候选突变中AD小于AD阈值的突变,或者移除NAD大于NAD阈值的突变,或者移除VAF小于VAF阈值的突变,或者移除NVAF大于NVAF阈值的突变。
在一实施例中,所述AD阈值为3,所述NAD阈值为3,所述VAF阈值为0.01,所述NVAF阈值为0.01。此处仅仅是示例性列举,本领域技术人员也可以根据需要设定其他阈值。
在一实施例中,过滤步骤中,还包括对待测样本、相应的对照样本中的胚系突变进行过滤。
在一实施例中,过滤步骤中,对待测样本、相应的对照样本中的胚系突变进行过滤时,所使用的数据库包括但不限于gnomAD胚系突变数据库、千人基因组数据库、dbSNP数据库、ESP6500数据库等等中的至少一种。上述列举的数据库均为现有数据库。
在一实施例中,过滤步骤中,对待测样本、相应的对照样本中的胚系突变进行过滤时,根据阈值设定判定条件,将高于某阈值的突变作为胚系突变集合,检查胚系突变集与候选突变集是否有交集,如有交集,将交集的突变位点从候选突变集移除。
在一实施例中,所述根据阈值设定判定条件,具体包括根据等位基因频率AF与AF阈值的大小关系,判定胚系突变位点,得到胚系突变集合。
在一实施例中,所述根据阈值设定判定条件,具体包括选取等位基因频率AF大于AF阈值的突变作为胚系突变集合。
在一实施例中,所述AF阈值为0.001。此处的AF阈值仅仅是示例性列举,本领域技术人员可以根据需要设定其他AF阈值。
在一实施例中,计算步骤中,计算的质量特征值包括但不限于如下质量特征值中的至少一种:
1)待测样本和相应的对照样本测序数据中支持和非支持序列计数的费雪尔精确检测的P值;
2)待测样本和相应的对照样本测序数据中同一个位置上的等位基因突变数目;
3)待测样本和相应的对照样本测序数据中突变附近的短重复序列重复次数;
4)待测样本和相应的对照样本测序数据中的序列复杂度;
5)待测样本和相应的对照样本测序数据中支持和非支持突变的序列的比对质量值的秩和检验值;
6)待测样本和相应的对照样本测序数据中支持和非支持突变的序列的正负链计数的费学尔精确检验的P值;
7)待测样本和相应的对照样本测序数据中支持和非支持突变的序列的突变位置的秩和检验值;
8)待测样本和相应的对照样本测序数据中支持突变的序列的突变位置的碱基质量值中位数;
9)待测样本和相应的对照样本测序数据中支持突变的序列的突变位置的中位;
10)待测样本和相应的对照样本测序数据中支持突变的序列的比对质量值平方根;
11)待测样本和相应的对照样本测序数据中支持和非支持突变的序列计数;
12)待测样本和相应的对照样本测序数据中突变位置的测序深度;
13)待测样本和相应的对照样本中的突变的频率。
在一实施例中,待测样本、相应的对照样本来源于生物体,生物体也可称为个体、受试者,可以包括但不限于人体、动物体等等,动物体包括但不限于大鼠。在一优选的实施例中,待测样本、相应的对照样本来源于人体。
在一实施例中,所述待测样本包括但不限于肿瘤组织样本、血浆样本等等中的至少一种。
在一实施例中,所述对照样本包括但不限于血液白细胞样本、癌旁组织样本等等中的至少一种。
在一实施例中,所述相应的对照样本是指与所述待测样本属于同一个体来源的样本。
在一实施例中,所述待测样本、相应的对照样本的测序数据选自全基因组测序数据、全外显子测序数据、靶向捕获测序数据等等中的至少一种。
第二方面,在一实施例中,提供一种检测体细胞突变的装置,包括:
候选突变提取模块,用于根据比对文件中的彩色德布鲁恩图、比对信息中的至少一种,从待测样本、相应的对照样本的测序数据中提取候选突变;
过滤模块,用于对提取的候选突变进行过滤,去除不符合判定条件的数据;
计算模块,用于根据过滤的候选突变,计算质量特征值,根据所述质量特征值,对候选突变集进行过滤,得到检测结果。
第三方面,在一实施例中,提供一种计算机设备,包括:
存储器,用于存储程序;
处理器,用于通过执行所述存储器存储的程序以实现如第一方面所述的方法。
第四方面,在一实施例中,提供一种计算机可读存储介质,包括程序,所述程序能够被处理器执行以实现如第一方面所述的方法。
需要说明的是,待测样本、相应的对照样本的测序数据是离体样本的测序数据,因此,不是以有生命的人体或动物体为对象;并且,计算得到的突变检测结果只是中间结果,供后续的疾病诊断参考,属于中间参考信息,不是最终的诊断结果,在实际应用中,在利用本发明的方法检测体细胞突变,获得检测结果后,还需要结合受试者当前的主观感受症状、既往病史、家族遗传史等信息,才能得出最后的诊断结果或健康状况。单纯根据该突变检测结果是不能得到专利法意义上的诊断结果的。因此,本发明的技术方案不属于疾病的诊断方法,更不属于疾病的治疗方法。并且,本发明还可用于科研中相关疾病候选新药的筛选等其他非诊断、非治疗目的。
在一实施例中,提供一种快速检测体细胞突变的方法,包括:快速提取样本中的候选体细胞突变;使用支持突变的测序序列频数和频率进行快速过滤;使用胚系突变数据库进行快速过滤;包括突变质量分值计算在内的各种特征值计算;依据各种特征对突变进行过滤。解决了肿瘤配对样本体细胞突变检测速度慢的问题,同时具有较高的突变检测准确度和灵敏度,而且软件运算全程内存资源使用可控,对于全基因组测序等数据量巨大的情况,速度提升效果尤为明显。
在一实施例中,提供的快速检测体细胞突变的方法包括如下步骤:
(1)快速提取样本中的候选体细胞突变,通过两种方法对候选突变进行提取。第一种方法是将肿瘤的测序数据和对照测序数据进行合并,同时比对到人类参考基因组序列,通过局部序列组装,组装成彩色德布鲁恩图。在图中,每一种颜色代表不同的数据来源,分别是肿瘤样本、对照样本和人类参考基因组。通过判断图路径中的颜色,找出非人类参考基因组特有的路径,可以提取出候选突变。并记录下候选突变在肿瘤样本和对照样本中的测序序列支持数。第二种方法是通过分析BAM/CRAM文件中的CIGAR提取出候选突变。CIGAR字符串记录了测序序列与参考基因组序列之间的差异,例如单碱基变换、碱基插入缺失等。同样记录下突变的测序序列支持数。这两种方法无先后顺序之分,可以先进行彩色德布鲁恩图分析,也可以先进行BAM/CRAM文件分析,也可以同时进行彩色德布鲁恩图分析、BAM/CRAM文件分析。
(2)使用支持突变的测序序列频数和频率进行快速过滤。遍历候选突变的集合中的突变,计算每一个突变位置上,肿瘤样本测序数据中支持突变的测序序列频数AD(AlleleDepth),以及相应的对照样本测序数据中支持突变的测序序列频数NAD(Normal AlleleDepth),以及肿瘤样本测序数据中测序序列频率(突变等位基因频率,Variant AlleleFrequency,VAF),以及相应的对照样本的normal variant allele frequency(NVAF,正常突变等位基因频率)。对于不符合判断条件的突变,例如,如果AD小于某个阈值,例如3;或者NAD大于某个阈值,例如3;或者VAF小于某个阈值,例如0.01;或者NVAF大于某个阈值,例如0.01,将被移除出候选突变集。判断过程为一票否决,不需要全部计算这4个坐标再作考虑,只要满足一个就被过滤。
其中,VAF=read count/total count,其中,read count为突变支持reads的数量,total count为该点测序深度;Total count为该点测序深度。VAF也可表示为AlleleDepth/Total Depth。
(3)使用胚系突变数据库进行快速过滤。从网上下载公开的人类遗传胚系突变数据库,选取allele frequency(AF,等位基因频率)大于一定阈值(例如0.001)的突变作为胚系突变集合。检查胚系突变集合与候选突变集里是否有交集,如有交集,将交集的突变位点移除出候选突变集。
步骤(2)与步骤(3)之间无特定的先后顺序之分,可以先进行步骤(2),再进行步骤(3),也可以先进行步骤(3),再进行步骤(2),也可以同时进行。由于步骤(3)是跟数据库比对,属于较为慢速的步骤,如果先进行步骤(3)可能会降低性能,因此,优选地,先进行步骤(2),再进行步骤(3)。
(4)突变特征值计算。
计算的突变特征值包括:
FET,肿瘤样本和相应的对照样本测序数据中支持与非支持序列计数,4个数值组成2x2表格,作为输入,进行费雪尔(Fisher)精确检测,计算P值。对P值进行Phred标准化(Phred scale)处理。其中:
FET(Phred scale Q)=-10*log10(P value)。
NALLE,肿瘤样本和相应的对照样本测序数据中同一个基因组位置上的等位基因突变数目。
HP,肿瘤样本和相应的对照样本测序数据中突变附近的短重复序列重复次数。查看突变位置左右,查看是否与突变的序列相同,如有,计数加1,直到左右再无相同序列。
SCS,肿瘤样本和相应的对照样本测序数据中的序列复杂度。计算突变位置左右拓展10bp的位置,对每一个种碱基进行计数,计算最高频数的两个碱基占总长度的百分比。
MQRankSum,遍历肿瘤样本和相应的对照样本测序数据中支持和非支持突变的序列,记录每一个序列的比对质量值,两组数据进行秩和检验计算P值。
FS,遍历肿瘤样本和相应的对照样本测序数据中支持和非支持突变的序列,记录每一个序列的正负链计数,4个数值组成2x2表格,作为输入,进行费雪尔精确检测,计算P值。P值进行Phred标准化(Phred scale)处理。
ReadPosRankSum,遍历肿瘤样本和相应的对照样本测序数据中支持和非支持突变的序列,记录突变在每一个序列上的位置。两组数据进行秩和检验,计算P值。
MBQV,遍历肿瘤样本和相应的对照样本测序数据中支持突变的序列,记录每一个序列上突变位置的碱基质量值,计算中位数。
MPOS,遍历肿瘤样本和相应的对照样本测序数据中支持突变的序列,记录每一个序列的突变位置与最近的序列末端的距离,计算中位数。
MQ,遍历肿瘤样本和相应的对照样本测序数据中支持突变的序列,记录每一个序列的比对质量值,计算平方根。
AD,肿瘤样本和相应的对照样本测序数据中支持和非支持突变的序列计数。
DP,肿瘤样本和相应的对照样本测序数据中突变位置的测序深度,为该位置的覆盖的测序序列计数。
AF,在肿瘤样本和对照样本测序数据中的突变的频率。AF=AD/DP。
调整以上各特征值的阈值,对候选突变集进行过滤。输出过滤前的突变集合以及过滤后的突变集合。
以下实施例中进行全外显子测序、靶向捕获测序时,采用的测序仪为IlluminaHiseq2000,测序方法参照仪器说明书进行。此处仅仅是示例性说明,也可以采用其他测序仪。
以下实施例中的比对软件仅仅是示例性说明,也可以采用其他比对软件将样本的测序数据比对到参考基因组上。
实施例1
本实施例中涉及的40个标准品样本(10对配对细胞系提取的DNA作为体外诊断试剂参考品,用于肿瘤突变负荷(TMB)检测产品的性能评价)均购自中国食品药品检定研究院,批号:360042-201901,具体信息参见表1。表1中,TMB-x-y%,x为样本编号,y为梯度编号,梯度是通过肿瘤细胞系与对照样本进行混样,形成的四个浓度梯度,肿瘤细胞含量具体包括1%、2%、5%、10%。例如,样本TMB-1-1%中,肿瘤细胞含量为1%。
每一对样本分别通过全外显子测序策略和靶向捕获测序技术进行建库测序,肿瘤样本全外显子测序深度约为700X,对照样本的全外显子测序深度为100X;肿瘤样本靶向捕获测序深度约为2000X,对照样本靶向捕获测序深度为400X。
表1标准品具体信息
下机数据预处理和基因组比对得到BAM格式的压缩比对文件,具体是通过BWA软件将肿瘤样本、相应的对照样本的下机数据比对到参考基因组上,得到BAM格式的比对文件,BAM文件为本实施例的方法的输入文件,分别通过传统的体细胞突变检测软件金标准mutect2和本实施例的方法进行体细胞突变检测,对比不同建库测序策略下,两种方法检测体细胞突变检测的资源消耗情况和运行速度快慢。
下机数据可以不用过滤,直接比对到参考基因组上,也可以先进行常规过滤,再比对到参考基因组上。本实施例中是将下机数据先进行常规过滤,包括通过碱基质量值(阈值标准:Q20>90%,Q30>85%)过滤掉碱基质量差的读段;GC含量(正常应为正态分布,无多个峰值),用于检测样品数据是否有其它来源DNA序列污染或接头序列的二聚体污染;检测有无AT、GC分离的现象,从而排除测序或建库的系统误差对后续生信分析造成的影响。去除接头序列、重复序列,再比对到参考基因组上。
以下为样本应用本实施例的方法进行体细胞突变检测的具体流程:
对于BAM文件中的测序序列,通过两种方法对候选突变进行提取。第一种方法是将肿瘤的测序数据(即待测样本数据)和对照测序数据进行合并,同时加入人类参考基因组序列,通过局部序列组装,组装成彩色德布鲁恩图。在图中,每一种颜色代表不同的数据来源,分别是肿瘤样本、对照样本和人类参考基因组。通过判断图路径中的颜色,找出非人类参考基因组特有的路径,可以提取出候选突变。并记录下候选突变在肿瘤样本和对照样本中的测序序列支持数。第二种方法是通过分析BAM文件中的CIGAR提取出候选突变。CIGAR字符串记录了测序序列与参考基因组序列之间的差异,例如单碱基变换,碱基插入缺失等。同样记录下突变的测序序列支持数。CIGAR字符串指示核苷酸序列与至少一个参考核苷酸序列的相似性和/或差异。
例如,某个样本突变提取结果如下:
#CHROM POS ID REF ALT 1 3640079.A G。
对两种方法检测到的候选突变进行快速过滤,过滤所依据的特征值包括突变的肿瘤测序序列的支持数、对照样本测序序列的支持数、肿瘤测序序列频率(支持数/该点总深度)、对照样本测序序列频率。极大降低候选突变的数量,加快后续分析速度。
本实施例中,过滤后,保留满足如下条件的数据:
肿瘤样本测序数据中支持突变的测序序列频数AD≥3;
对照样本测序数据中支持突变的测序序列频数NAD≤1;
肿瘤样本测序数据中测序序列频率VAF≥0.005;
对照样本测序数据中测序序列频率NVAF≤0.01。
合并这两个候选突变集,使用外部数据库,本实施例具体为gnomAD胚系突变数据库(网址:http://gnomad.broadinstitute.org/),对突变集进行过滤,具体是筛选等位基因频率AF大于AF阈值的突变作为胚系突变集,检查胚系突变集与候选突变集是否有交集,如有交集,将交集的突变位点从候选突变集移除,本实施例的AF阈值为0.001。极大降低候选突变中的胚系突变的含量,显著加快后续分析速度,而且有效降低体细胞突变的假阳性。
计算突变的多个质量特征值,具体如下:
FET,肿瘤样本和相应的对照样本测序数据中支持与非支持序列计数,4个数值组成2x2表格,作为输入,进行费雪尔(Fisher)精确检测,计算P值。对P值进行Phred标准化(Phred scale)处理。其中:
FET(Phred scale Q)=-10*log10(P value)。
NALLE,肿瘤样本和相应的对照样本测序数据中同一个基因组位置上的等位基因突变数目。
HP,肿瘤样本和相应的对照样本中突变附近的短重复序列重复次数。查看突变位置左右,查看是否与突变的序列相同,如有,计数加1,直到左右再无相同序列。
SCS,肿瘤样本和相应的对照样本测序数据中的序列复杂度。计算突变位置左右拓展10bp的位置,对每一个种碱基进行计数,计算最高频数的两个碱基占总长度的百分比。
MQRankSum,遍历肿瘤样本和相应的对照样本测序数据中支持和非支持突变的序列,记录每一个序列的比对质量值,两组数据进行秩和检验计算P值。
FS,遍历肿瘤样本和相应的对照样本测序数据中支持和非支持突变的序列,记录每一个序列的正负链计数,4个数值组成2x2表格,作为输入,进行费雪尔精确检测,计算P值。P值进行Phred标准化(Phred scale)处理。
ReadPosRankSum,遍历肿瘤样本和相应的对照样本测序数据中支持和非支持突变的序列,记录突变在每一个序列上的位置。两组数据进行秩和检验,计算P值。
MBQV,遍历肿瘤样本和相应的对照样本测序数据中支持突变的序列,记录每一个序列上突变位置的碱基质量值,计算中位数。
MPOS,遍历肿瘤样本和相应的对照样本测序数据中支持突变的序列,记录每一个序列的突变位置与最近的序列末端的距离,计算中位数。
MQ,遍历肿瘤样本和相应的对照样本测序数据中支持突变的序列,记录每一个序列的比对质量值,计算平方根。
AD,肿瘤样本和相应的对照样本测序数据中支持和非支持突变的序列计数。
DP,肿瘤样本和相应的对照样本测序数据中突变位置的测序深度,为该位置的覆盖的测序序列计数。
AF,在肿瘤样本和对照样本测序数据中的突变的频率。AF=AD/DP。
突变结果和指标值信息输出到VCF文件中。
通过过滤程序对指标的信息进行判断,过滤VCF中的突变,得到最终VCF结果。
过滤时,各质量特征值的阈值如下表2所示。
表2
分别用本实施例的方法(数据结果中以Inhouse表示)、Mutect2两种方法对40个标准品样本的全外显子测序数据进行体细胞突变检测。每个标准品(如TMB-1-0%)中有很多位点,每个位点的突变频率不同,数据量较大,此处不再全部展示。
所需的具体资源及其运行速度参见下表3,从表3的结果可以发现,本实施例的方法比传统的mutect2不仅运行速度快了35倍,占用的平均内存也是mutect2的20倍。表3中,Inhouse是指本实施例的方法,Mutect2是指现有的软件。
表3
图1显示为根据本实施例的方法检测的全外显子测序数据体细胞突变位点计算得到的TMB指标与标准品TMB指标的关系曲线图,标准品TMB指标是已知的,横坐标是标准品的TMB值,纵坐标是本实施例的方法计算的TMB值,图1中Inhouse是指本实施例的方法;图2显示为根据现有的mutect2检测的全外显子测序数据中体细胞突变位点计算得到的TMB指标与标准品TMB指标的关系曲线图,横坐标是标准品的TMB值,纵坐标是mutect2计算的TMB值,图2中Mutect是现有的软件,具体是mutect2软件。从图1和图2的结果可以发现,采用本实施例的方法检测的体细胞突变位点计算得到的TMB指标,与标准品TMB的一致性达到0.8813,高于mutect2软件的结果,mutect2软件与标准品TMB的一致性仅仅为0.8774。
分别用本实施例的方法、Mutect2两种方法对40个标准品样本靶向捕获测序的数据进行体细胞突变检测,所需的具体资源及其运行速度参见下表4,表4中,Inhouse是指本实施例的方法,mutect2是现有的软件,从表4可以明显看出,本实施例的方法相对于传统的mutect2,不仅运行速度是传统的mutect2的90倍,占用的平均内存仅仅为mutect2的1/50。
表4
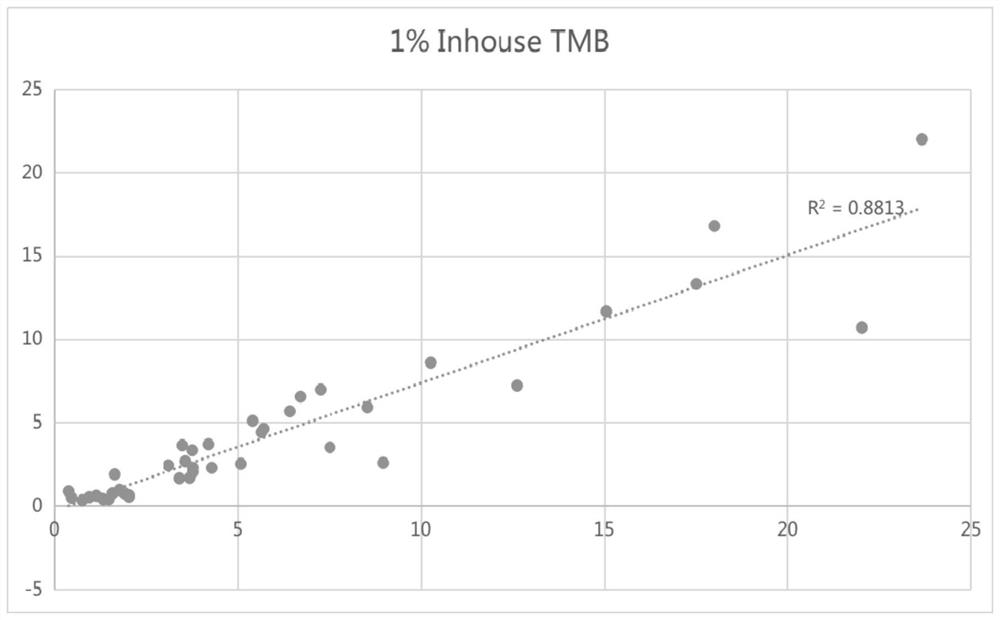
图3显示为根据本实施例的方法检测的靶向捕获测序数据中体细胞突变位点计算得到的TMB指标与标准品TMB指标的关系曲线图,标准品TMB指标是已知的,横坐标是标准品的TMB值,纵坐标是本实施例的方法计算的TMB值,1%是指只观察VAF≥1%的突变,图3中Inhouse是指本实施例的方法;图4显示为根据现有的mutect2检测的靶向捕获测序数据中体细胞突变位点计算得到的TMB指标与标准品TMB指标的关系曲线图,横坐标是标准品的TMB值,纵坐标是mutect2计算的TMB值,图4中Mutect是现有的软件,具体是mutect2软件。从图3、图4可以看出,根据本实施例的方法检测的靶向捕获测序数据体细胞突变位点计算得到的TMB指标,与标准品TMB的一致性达到0.9448,高于mutect2软件的结果(mutect2软件与标准品TMB的一致性仅仅为0.9012)。
从本实施例的数据结果可知,本实施例的方法是一种比传统方法更快速实现癌组织体细胞突变检测,靶向捕获测序的数据应用本实施例的方法进行体细胞突变检测,需要的资源消耗更少,速度更快。本实施例的方法组合多种过滤方法,降低假阳性的同时,保证了突变的检出率,可用于后续TMB等指标的准确计算。
实施例2
本实施例中涉及的肿瘤样本(即Cancer样本)及其对应的对照样本(即Normal样本,或称配对样本)购自罗氏制药公司(Roche)。Cancer样本为cfDNA标准品样本,Normal样本为VAF=0的样本,每个cfDNA标准品样本与相应的Normal样本为一对样本,共6对样本。cfDNA标准品样本为6个已知突变位点的不同突变等位基因频率(variant allelefrequency,VAF)样本和不同文库起始量的cfDNA样本,通过靶向捕获测序技术进行建库测序,测序深度为20000X。
下机数据预处理和基因组比对得到BAM格式的压缩比对文件,具体是通过BWA软件将肿瘤样本、相应的对照样本的下机数据比对到参考基因组上,得到BAM格式的比对文件,该文件为本实施例的方法的输入文件,通过本实施例的方法进行体细胞突变检测,与标准品已知的突变信息相对比,评估本实施例的方法检测突变的最低阈值和性能。
以下为样本应用本实施例的方法进行体细胞突变检测的具体流程:
对获取的BAM文件中的测序序列,通过两种方法对候选突变进行提取。
第一种方法是将肿瘤的测序数据和对照测序数据进行合并,同时比对到人类参考基因组序列上,通过局部序列组装,组装成彩色德布鲁恩图。在图中,每一种颜色代表不同的数据来源,分别是肿瘤样本、对照样本和人类参考基因组。通过判断图路径中的颜色,找出非人类参考基因组特有的路径,可以提取出候选突变,并记录下候选突变在肿瘤样本和对照样本中的测序序列支持数。
第二种方法是通过分析BAM文件中的CIGAR提取出候选突变。CIGAR字符串记录了测序序列与参考基因组序列之间的差异,例如单碱基变换、碱基插入缺失等。同样记录下突变的测序序列支持数。
对两种方法检测到的候选突变进行快速过滤,过滤所依据的特征值包括突变的肿瘤样本测序序列的支持数、对照样本测序序列的支持数、肿瘤样本测序序列频率(支持数/该点总深度)、对照样本测序序列频率。极大降低候选突变的数量,加快后续分析速度。
本实施例中,过滤后,保留满足如下条件的数据:
肿瘤样本测序数据中支持突变的测序序列频数AD≥1;
对照样本测序数据中支持突变的测序序列频数NAD≤4;
肿瘤样本测序数据中测序序列频率VAF≥0.00;
对照样本测序数据中测序序列频率NVAF≤0.2。
例如,某样本位点的筛选结果如下:
1 3640079.A G;
肿瘤样本测序数据中支持突变的测序序列频数AD=30;
对照样本测序数据中支持突变的测序序列频数NAD=1;
肿瘤样本测序数据中测序序列频率VAF=0.1;
对照样本测序数据中测序序列频率NVAF=0.01。
默认参数,该点保留。
1 3640079.A G;
肿瘤样本测序数据中支持突变的测序序列频数AD=2;
对照样本测序数据中支持突变的测序序列频数NAD=2;
肿瘤样本测序数据中测序序列频率VAF=0.01;
对照样本测序数据中测序序列频率NVAF=0.1。
默认参数,该点被过滤掉。
合并两个候选突变集,使用外部数据库,本实施例具体为gnomAD胚系突变数据库(网址:http://gnomad.broadinstitute.org/),对突变集进行过滤,具体是筛选等位基因频率AF大于AF阈值的突变作为胚系突变集,检查胚系突变集与候选突变集是否有交集,如有交集,将交集的突变位点从候选突变集移除,本实施例的AF阈值为0.001。大大降低候选突变中的胚系突变的含量,加快后续分析速度和体细胞突变的假阳性。
计算突变的多个质量特征值,具体如下:
FET,肿瘤样本和相应的对照样本测序数据中支持与非支持序列计数,4个数值组成2x2表格,作为输入,进行费雪尔(Fisher)精确检测,计算P值。对P值进行Phred标准化(Phred scale)处理。其中:
FET(Phred scale Q)=-10*log10(P value)。
NALLE,肿瘤样本和相应的对照样本测序数据中同一个基因组位置上的等位基因突变数目。
HP,肿瘤样本和相应的对照样本测序数据中突变附近的短重复序列重复次数。查看突变位置左右,查看是否与突变的序列相同,如有,计数加1,直到左右再无相同序列。
SCS,肿瘤样本和相应的对照样本测序数据中的序列复杂度。计算突变位置左右拓展10bp的位置,对每一个种碱基进行计数,计算最高频数的两个碱基占总长度的百分比。
MQRankSum,遍历肿瘤样本和相应的对照样本测序数据中支持和非支持突变的序列,记录每一个序列的比对质量值,两组数据进行秩和检验计算P值。
FS,遍历肿瘤样本和相应的对照样本测序数据中支持和非支持突变的序列,记录每一个序列的正负链计数,4个数值组成2x2表格,作为输入,进行费雪尔精确检测,计算P值。P值进行Phred标准化(Phred scale)处理。
ReadPosRankSum,遍历肿瘤样本和相应的对照样本测序数据中支持和非支持突变的序列,记录突变在每一个序列上的位置。两组数据进行秩和检验,计算P值。
MBQV,遍历肿瘤样本和相应的对照样本测序数据中支持突变的序列,记录每一个序列上突变位置的碱基质量值,计算中位数。
MPOS,遍历肿瘤样本和相应的对照样本测序数据中支持突变的序列,记录每一个序列的突变位置与最近的序列末端的距离,计算中位数。
MQ,遍历肿瘤样本和相应的对照样本测序数据中支持突变的序列,记录每一个序列的比对质量值,计算平方根。
AD,肿瘤样本和相应的对照样本测序数据中支持和非支持突变的序列计数。
DP,肿瘤样本和相应的对照样本测序数据中突变位置的测序深度,为该位置的覆盖的测序序列计数。
AF,在肿瘤样本和对照样本测序数据中的突变的频率。AF=AD/DP。
突变结果和指标值信息输出到VCF文件中。
通过过滤程序对指标的信息进行判断,过滤VCF中的突变,得到最终VCF结果。
本实施例的过滤条件参数如下表5所示。
表5
检测结果如表6所示。
表6
从表6可以看出,Inhouse突变数目所在列为本实施例的方法所检测得到的突变数目,根据检测结果对比可知,本实施例的方法可实现cfDNA VAF 0.5%以上的突变检测,灵敏度高,假阳性低。
本领域技术人员可以理解,上述实施方式中各种方法的全部或部分功能可以通过硬件的方式实现,也可以通过计算机程序的方式实现。当上述实施方式中全部或部分功能通过计算机程序的方式实现时,该程序可以存储于一计算机可读存储介质中,存储介质可以包括:只读存储器、随机存储器、磁盘、光盘、硬盘等,通过计算机执行该程序以实现上述功能。例如,将程序存储在设备的存储器中,当通过处理器执行存储器中程序,即可实现上述全部或部分功能。另外,当上述实施方式中全部或部分功能通过计算机程序的方式实现时,该程序也可以存储在服务器、另一计算机、磁盘、光盘、闪存盘或移动硬盘等存储介质中,通过下载或复制保存到本地设备的存储器中,或对本地设备的系统进行版本更新,当通过处理器执行存储器中的程序时,即可实现上述实施方式中全部或部分功能。
以上应用了具体个例对本发明进行阐述,只是用于帮助理解本发明,并不用以限制本发明。对于本发明所属技术领域的技术人员,依据本发明的思想,还可以做出若干简单推演、变形或替换。
- 一种检测体细胞突变的方法及装置
- 一种肿瘤组织单样本体细胞突变检测方法及装置