药物的缩酮衍生物及其制备方法、药物组合物和用途
文献发布时间:2023-06-19 09:44:49
本申请要求2019年8月2日向中国国家知识产权局提交的,专利申请号为201910713864.0,发明名称为“药物的缩酮衍生物及其制备方法、药物组合物和用途”在先申请的优先权。该申请的全文通过引用的方式结合于本申请中。
技术领域
本发明属于前药化合物技术领域,具体涉及药物的缩酮衍生物及其制备方法、药物组合物和用途。
背景技术
前药是一种重要的药物化合物衍生物。现已报道的前药衍生物包括载体前体药物和生物前体药物。其中,载体前体药物是指具有活性的化合物与其运输作用的载体通过共价键结合,在体内通过简单的水解作用卸掉载体,由活性化合物发挥药理作用。载体前体药物与母体化合物相比往往活性微弱或无活性。对于载体的结构,多是亲脂性,要求对生物体无害,且能及时释放活性化合物。市场上口服青霉素类药物往往采用载体前药的方式来提高生物利用度;生物前体药物不同于载体前体药物,活性物质不用与载体暂时性结合,而是通过自身分子结构的改变来发挥作用。生物前体药物本身通常没有活性,但其体内代谢物具有活性。一些非甾体抗炎药(如舒林酸sulindac)就是基于这样的思路设计的。
随着前药衍生广泛用于改良药物研发,其在新药研发中的地位也越来越重要。据统计,2008-2017年,共计30个美国FDA批准的新药属于前药化合物,约为FDA批准药物总量的10%。其中,在2017年美国FDA批准的新药中,前药化合物占比为约17%(Nat.Rev.DrugDiscov.2018,17(8):559-587)。
在药物化合物中,含羟基(包括酚羟基、羧羟基和醇羟基)的药物约占小分子药物的51%(https://pubs.acs.org.ccindex.cn/doi/pdf/10.1021/cc9800071),其中包括紫杉烷类、糖皮质类激素、前列腺素类似物、抗生素、抗病毒类和萜类等几大类药物的大多数分子。成酯是前药设计中最常用的修饰手段。已上市的前药中,大约49%是由酶的水解产生活性的。例如,水溶性差的药物的羟基常被设计成磷酸酯/磷酸盐,以提高其水溶性,更适于口服或注射给药。羟基的碳酸酯和氨基甲酸酯类前药可能对酶的稳定性更好。
中国专利申请CN107353399A和CN103285400A公开了酸敏感的聚合物前药,其中使用缩醛键将药物分子与聚合物链接,以改变药物分子在体内循环时的释放。然而,其前药释放时产生的分子片段可能具有某些不利于人体的性质,或者其在生理环境或体内酸性环境下的释放特性仍有待进一步改善(Louage,Benoit,Mies J.Van Steenbergen,Lutz Nuhn,Martijn DP Risseeuw,Izet Karalic,Johan Winne,Serge Van Calenbergh,WimE.Hennink,and Bruno G.De Geest."Micellar paclitaxel-initiated RAFT polymerconjugates with acid-sensitive behavior."ACS Macro Letters 6,no.3(2017):272-276;Zhai,Yinglei,Xing Zhou,Lina Jia,Chao Ma,Ronghua Song,Yanhao Deng,XueyaoHu,and Wei Sun."Acetal-linked paclitaxel polymeric prodrug based onfunctionalized mPEG-PCL diblock polymer for ph-triggered drug delivery."Polymers 9,no.12(2017):698.)。
为此,有必要进一步开发新的前体药物,其可以有利于前药代谢、释放特性的改善,和/或降低其释放分子的生理毒性或其他副作用。
发明内容
为改善上述技术问题,本发明提供如下式(I)所示的化合物、其消旋体、立体异构体、互变异构体或它们药学上可接受的盐:
其中,D选自将含有至少一个羟基的药物化合物去掉其一个羟基上的氢原子后得到的基团;
R
R
R
R
每个R
每个R
每个R
每个R
每个R
每个R
每个Y独立地选自化学键、-O-、-S-或无取代或任选被一个或多个R
M
根据本发明的实施方案,其中,D选自将含有至少一个羟基的药物化合物去掉其一个羟基上的氢原子后得到的基团,其中所述含有至少一个羟基的药物化合物可以选自包括但不限于下列的药物化合物:
紫杉烷类药物(例如紫杉醇、多烯紫杉醇、卡巴他赛);
糖皮质类激素(例如氢化可的松、地塞米松、强的松、曲安奈德、醋酸曲安奈德、莫米松、睾酮、雌二醇、乙炔雌二醇、利奈孕酮);
前列腺素类似物(例如他氟前列素、拉坦前列素、曲伏前列素、比马前列素、乌诺前列酮异丙酯);
帕利哌酮;
青蒿素类似物(如双氢青蒿素);
核苷类似物(吉西他滨、三氟尿苷、碘苷、氟尿苷、卡培他滨、扎西他滨、阿糖胞苷);
抗病毒类药物(更昔洛韦、阿昔洛韦、恩曲他滨、齐多夫定、拉米夫定、替诺福韦、恩替卡韦);
抗生素(多柔比星);
纳曲酮;
罗替戈汀;
雷帕霉素、他克莫司;
曲前列环素;
安舒法辛;
依托泊苷;常山酮;
康普瑞汀;
SN-38;
雷帕霉素;
氟维司群;
阿比特龙;
他汀类药物(辛伐他汀和洛伐他汀);
止疼药(叔丁啡、羟吗啡酮、纳布啡、左啡诺、氢吗啡酮、布托啡诺);
IDO抑制剂NLG919;和
萜类药物;
当所述含有至少一个羟基的药物化合物包含两个或更多个羟基时,所述基团D中的至少另一个羟基可以进一步去掉氢原子后与-CR
根据本发明的实施方案,所述药物化合物选自包括但不限于下列含有至少一个羟基的化合物:紫杉醇、多烯紫杉醇、卡巴他赛、地塞米松、他氟前列素、帕利哌酮、双氢青蒿素、吉西他滨、齐多夫定、睾酮、依托泊苷、NLG919、常山酮、康普瑞汀、SN-38、雷帕霉素、氟维司群、阿比特龙、或辛伐他汀。
根据本发明的实施方案,R
根据本发明的实施方案,R
根据本发明的实施方案,R
根据本发明的实施方案,R
或者,-O-R
根据本发明的实施方案,PDLLA可任选地通过去掉一个羟基上的氢原子后与其他基团键合。
根据本发明的实施方案,PEG可任选地通过去掉一个或两个羟基上的氢原子后与其他基团键合。
所述R
根据本发明的实施方案,所述PEG和PDLLA的分子量相同或不同,彼此独立地选自100~5000,例如150~3000,如200、300、400、500、600、700、800、900、1000、1100、1200、1300、1400、1500、1600、1700、1800、1900、2000、2100、2200、2300、2400、2500、2600、2700、2800、2900、3000。
根据本发明的实施方案,所述R
根据本发明的实施方案,所述R
其中
本发明还提供式(I)化合物、其消旋体、立体异构体、互变异构体或它们药学上可接受的盐的制备方法,包括将式D-H化合物与式(I-1)化合物反应得到式(I)所示的化合物:
其中,D、R
R
例如,R
根据本发明的实施方案,所述反应可以在惰性气氛中进行,例如在氮气氛中进行。
根据本发明的实施方案,所述反应可以在催化剂的存在下进行,例如在质子酸如对甲苯磺酸、吡啶对甲苯磺酸、1,2-二氯乙酸中的至少一种的存在下进行。
根据本发明的实施方案,当药物D-H中含有多个羟基时,可以采用常规的方法先对反应位点的OH进行保护,再与式(I-1)所示的化合物反应;待反应完成后再采用常规的方法脱去保护基,得到式(I)所示的化合物。
根据本发明的实施方案,所述反应还包括式(I-1)所示化合物的制备,包括如下方法中的至少一种:
方法一:在非极性溶剂中,缩酮化合物在三氟甲磺酸硅氧醚类化合物(TMSOTf等)以及有机碱(如Et
方法二:当R
方法三:在酸催化下(对甲苯磺酸等)缩酮化合物加热消除一分子醇得到:
其中,R
根据本发明的实施方案,所述非极性溶剂可以选自例如卤代烃、醚类、芳烃类溶剂,例如二氯甲烷(DCM)、四氢呋喃(THF)、甲苯中的一种、两种或更多种。
本发明还提供式(I-1)所示的化合物:
其中,R
本发明还提供一种药物组合物,其包含治疗有效量的式(I)所示的化合物、其消旋体、立体异构体、互变异构体或它们药学上可接受的盐。
根据本发明的实施方案,所述药物组合物还可任选包含药学上可接受的辅料,例如载体、赋形剂。作为实例,所述辅料可以选自下列中的一种或多种:崩解剂、助流剂、润滑剂、稀释剂或填充剂、粘合剂、着色剂。
根据本发明的实施方案,所述药物组合物可以为药物制剂,其可以选自包括但不限于注射液、纳米粒、胶束制剂等的剂型。
本发明还提供式(I)所示的化合物、其消旋体、立体异构体、互变异构体或它们药学上可接受的盐在制备药物中的用途。
根据本发明的实施方案,所述药物具有与上文所述含有至少一个羟基的药物化合物相同的用途,例如其可为紫杉烷类药物、糖皮质类激素、前列腺素类似物、帕利哌酮、青蒿素类似物、核苷类似物、抗生素、抗病毒类药物、纳曲酮、罗替戈汀、雷帕霉素、他克莫司、曲前列环素、安舒法辛、依托泊苷、常山酮、康普瑞汀、SN-38、雷帕霉素、氟维司群、阿比特龙、他汀类药物、止疼药、IDO抑制剂NLG919或萜类药物。
本发明还提供所述式(I)所示的化合物、其消旋体、立体异构体、互变异构体或它们药学上可接受的盐,或者所述药物组合物在预防或治疗疾病或病征中的用途,所述疾病为或病征上文所述含有至少一个羟基的药物化合物能够治疗的疾病或病征,例如为给予紫杉烷类药物、糖皮质类激素、前列腺素类似物、帕利哌酮、青蒿素类似物、核苷类似物、抗生素、抗病毒类药物、纳曲酮、罗替戈汀、雷帕霉素、他克莫司、曲前列环素、安舒法辛、依托泊苷、常山酮、康普瑞汀、SN-38、雷帕霉素、氟维司群、阿比特龙、他汀类药物、止疼药、IDO抑制剂NLG919或萜类药物时可以减轻或缓解的那些疾病或病征。
本发明还提供所述式(I)所示的化合物、其消旋体、立体异构体、互变异构体或它们药学上可接受的盐,或者所述药物组合物用于改善上文所述含有至少一个羟基的药物化合物的物理性质、化学性质或药学性质的用途。
本发明还提供所述式(I)所示的化合物、其消旋体、立体异构体、互变异构体或它们药学上可接受的盐,或者所述药物组合物用于改善上文所述含有至少一个羟基的药物化合物的代谢或释放特性的用途。
本发明还提供所述式(I)所示的化合物、其消旋体、立体异构体、互变异构体或它们药学上可接受的盐,或者所述药物组合物用于改善上文所述含有至少一个羟基的药物化合物在水或有机溶剂中的溶解度的用途。
本发明还提供所述式(I)所示的化合物、其消旋体、立体异构体、互变异构体或它们药学上可接受的盐,或者所述药物组合物用于改善上文所述含有至少一个羟基的药物化合物的药效或副作用的用途。
有益效果
1)本发明的前药可显著改善原起始药物的物理化学性质,如解决原起始药物如紫杉烷类药物等在水中溶解度差的问题。亲水性缩酮提高了药物化合物在水中的溶解度,提高了药物在体内的抗肿瘤效果,甚至可以降低原起始药物制剂的某些辅料(如紫杉烷注射剂中的辅料)所引发的毒副作用;另外,脂溶性的缩酮前药使得丰富药物制剂的种类成为可能,且其药理作用(如体内抗肿瘤效果)将显著优于市售制剂。
2)本发明的前药可改善原起始药物在有机溶剂中的溶解度,例如方便了糖皮质类固醇纳米制剂的制备,提高了体内治疗效果。
3)本发明的脂溶性前药可降低原起始药物在水溶液中的溶解度,从而用于实现针对慢性病治疗药物(如本文例举的糖皮质类激素,前列腺素类似物,帕利哌酮,双氢青蒿素,核苷类似物和抗病毒类药物等)的长效缓释。
术语定义和说明
除非另有说明,本申请说明书和权利要求书中记载的基团和术语定义,包括其作为实例的定义、优选的定义、实施例中具体化合物的定义等,可以彼此之间任意组合和结合。这样的组合和结合后的基团定义及化合物结构,应当属于本申请说明书记载的范围内。
本申请说明书和权利要求书记载的数值范围,当该数值范围被定义为“整数”时,应当理解为记载了该范围的两个端点以及该范围内的每一个整数。例如,“0~10的整数”应当理解为记载了0、1、2、3、4、5、6、7、8、9和10的每一个整数。“0~200”的整数应当理解为记载了0、1、2、3、4、5、6、7、8、9、10、11、……、100、101、102、103、104、105、106、107、108、109、110、111、……、190、191、192、193、194、195、196、197、198、199、200的每一个整数。
当该数值范围被定义为“数”时,应当理解为记载了该范围的两个端点、该范围内的每一个整数以及该范围内的每一个小数。
术语“卤素”指F、Cl、Br和I。换言之,F、Cl、Br和I在本说明书中可描述为“卤素”。
术语“C
术语“C
术语“C
术语“C
术语“3-20元杂环基”意指饱和的一价单环或双环烃环,其包含1-5个独立选自N、O和S的杂原子,优选“3-10元杂环基”。术语“3-10元杂环基”意指饱和的一价单环或双环烃环,其包含1-5个,优选1-3个选自N、O和S的杂原子。所述杂环基可以通过所述碳原子中的任一个或氮原子(如果存在的话)与分子的其余部分连接。特别地,所述杂环基可以包括但不限于:4元环,如氮杂环丁烷基、氧杂环丁烷基;5元环,如四氢呋喃基、二氧杂环戊烯基、吡咯烷基、咪唑烷基、吡唑烷基、吡咯啉基;或6元环,如四氢吡喃基、哌啶基、吗啉基、二噻烷基、硫代吗啉基、哌嗪基或三噻烷基;或7元环,如二氮杂环庚烷基。任选地,所述杂环基可以是苯并稠合的。所述杂环基可以是双环的,例如但不限于5,5元环,如六氢环戊并[c]吡咯-2(1H)-基环,或者5,6元双环,如六氢吡咯并[1,2-a]吡嗪-2(1H)-基环。含氮原子的环可以是部分不饱和的,即它可以包含一个或多个双键,例如但不限于2,5-二氢-1H-吡咯基、4H-[1,3,4]噻二嗪基、4,5-二氢噁唑基或4H-[1,4]噻嗪基,或者,它可以是苯并稠合的,例如但不限于二氢异喹啉基。根据本发明,所述杂环基是无芳香性的。
术语“C
术语“5-20元杂芳基”应理解为包括这样的一价单环、双环或三环芳族环系:其具有5~20个环原子且包含1-5个独立选自N、O和S的杂原子,例如“5-14元杂芳基”。术语“5-14元杂芳基”应理解为包括这样的一价单环、双环或三环芳族环系:其具有5、6、7、8、9、10、11、12、13或14个环原子,特别是5或6或9或10个碳原子,且其包含1-5个,优选1-3各独立选自N、O和S的杂原子并且,另外在每一种情况下可为苯并稠合的。特别地,杂芳基选自噻吩基、呋喃基、吡咯基、噁唑基、噻唑基、咪唑基、吡唑基、异噁唑基、异噻唑基、噁二唑基、三唑基、噻二唑基、噻-4H-吡唑基等以及它们的苯并衍生物,例如苯并呋喃基、苯并噻吩基、苯并噁唑基、苯并异噁唑基、苯并咪唑基、苯并三唑基、吲唑基、吲哚基、异吲哚基等;或吡啶基、哒嗪基、嘧啶基、吡嗪基、三嗪基等,以及它们的苯并衍生物,例如喹啉基、喹唑啉基、异喹啉基等;或吖辛因基、吲嗪基、嘌呤基等以及它们的苯并衍生物;或噌啉基、酞嗪基、喹唑啉基、喹喔啉基、萘啶基、蝶啶基、咔唑基、吖啶基、吩嗪基、吩噻嗪基、吩噁嗪基等。
除非另有说明,杂环基、杂芳基或亚杂芳基包括其所有可能的异构形式,例如其位置异构体。因此,对于一些说明性的非限制性实例,吡啶基或亚吡啶基包括吡啶-2-基、亚吡啶-2-基、吡啶-3-基、亚吡啶-3-基、吡啶-4-基和亚吡啶-4-基;噻吩基或亚噻吩基包括噻吩-2-基、亚噻吩-2-基、噻吩-3-基和亚噻吩-3-基。
上述对术语“烷基”,如“C
本发明的术语“惰性气氛”应当理解为对反应呈惰性的气氛,包括但不限于选自氦(He)、氖(Ne)、氩(Ar)、氪(Kr)、氙(Xe)、氮中一种、两种或更多种的气氛。
附图说明
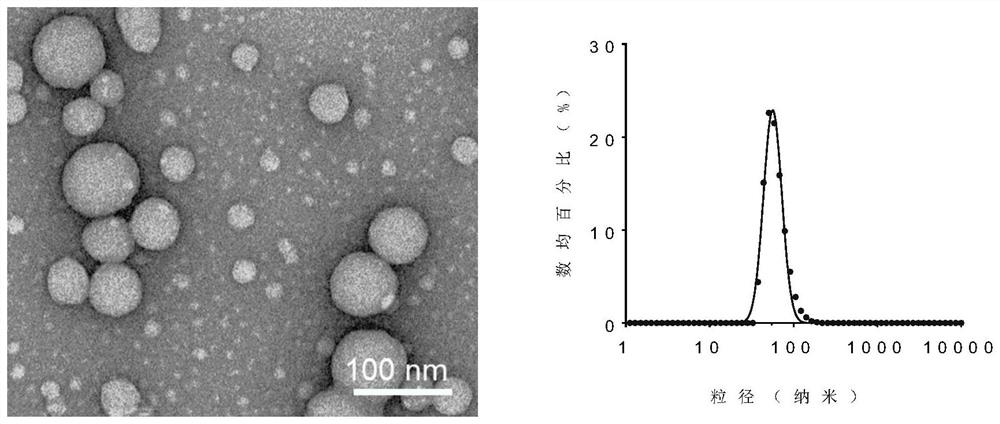
图1为Oleyl-K-7-PTX/DSPE-PEG
图2为mPEG
图3为Oleyl-K-7-PTX/DSPE-PEG
图4为mPEG-PDLLA-K-2'-PTX胶束和自制市售Genexol紫杉醇胶束制剂对MDA-MB-231荷瘤鼠肿瘤的抑制效果图。剂量:10mg/kg紫杉醇;箭头所指时间点为注射时间点。
图5为mPEG
图6为SA-K-DEX/DSPE-PEG
图7为尾静脉注射SA-K-DEX/DSPE-PEG
图8为大鼠关节炎治疗实验结束后关节的MicroCT图像。
图9为尾静脉注射4小时后大鼠关节内地塞米松的分布。剂量:相当于1.0mg/kg地塞米松。
图10为SA-K-DEX纳米晶的光学显微镜图。
图11为大鼠关节内注射SA-K-DEX纳米晶对关节炎的治疗效果图。剂量:相当于2.5mg/kg地塞米松;箭头所指时间点为注射时间点。
图12为大鼠关节内注射SA-K-DEX纳米晶后血液中地塞米松浓度测试。剂量:相当于2.5mg/kg地塞米松。
图13为SA-K-TAF/DSPE-PEG
图14为新西兰兔眼内压变化图。剂量:相当于他氟前列素10μg/只;箭头所指时间点为注射时间点。
图15为大鼠肌肉注射Oleyl-K-PAL/DSPE-PEG
图16为大鼠肌肉注射SA-K-DHA/DSPE-PEG
图17为SA-K-5'-GEM细胞毒性测试结果。
图18为大鼠肌肉注射SA-K-5’-AZT/DSPE-PEG
图19为大鼠血液注射mPEG
图20为β-葡萄糖-K-依托泊苷注射液和自制市售依托泊苷制剂Toposar对A549荷瘤鼠肿瘤的治疗效果。箭头所指时间点为注射时间点。剂量为15mg/kg依托泊苷。
具体实施方式
下文将结合具体实施例对本发明的技术方案做更进一步的详细说明。应当理解,下列实施例仅为示例性地说明和解释本发明,而不应被解释为对本发明保护范围的限制。凡基于本发明上述内容所实现的技术均涵盖在本发明旨在保护的范围内。
除非另有说明,以下实施例中使用的原料和试剂均为市售商品,或者可以通过已知方法制备。
液相色谱仪:安捷伦科技有限公司,Agilent 1260Infinity II;核磁:瑞士布鲁克公司,AV400;多功能酶标仪:Bio Tek,Synergy 4。
如无特殊说明,如下实施例中与商用制剂活性对比中涉及的对照组均指不施用任何药物组。
实验动物来源:裸鼠:北京维通利华实验动物技术有限公司,balb/c-Nude,6-8周,雌性,20g;成年SD大鼠:北京维通利华实验动物技术有限公司,9-11周,雌性,200-250g;新西兰兔2.5-3kg。实验用鼠饲养于SPF级实验环境中,兔子饲养于普通清洁环境中。
实施例1:己醇异丙烯基醚的合成
参照文献方法制备(Killian,D.B.,G.F.Hennion,and J.A.Nieuwland."ThePreparation of Someα-Unsaturated Ethers from 2,2-Dimethoxyalkanes1."Journalof the American Chemical Society 57.3(1935):544-545.)。
在250mL圆底烧瓶中加入120g 2,2-甲氧基辛烷,0.05g对甲苯磺酸,加热至回流。回流反应4h后,减压蒸馏得到产物己醇异丙烯基醚。
实施例2:(1-甲氧基乙烯基)苯的合成
参照文献方法制备(Pine S H,Zahler R,Evans D A,et al.Titanium-mediatedmethylene-transfer reactions.Direct conversion of esters into vinyl ethers[J].Journal of the American Chemical Society,1980,102(9):3270-3272)。
将1mmol苯基乙酸甲酯溶于2ml甲苯和四氢呋喃混合溶剂(3:1)中,将体系冷却至-40℃,随后向其中缓慢滴加1.1mmol Tebbe试剂的甲苯溶液(0.55M),在此温度下继续反应30min,随后升至室温继续反应1.5h,反应完全后将体系冷却至-10℃,向其中加入1ml 15%氢氧化钠水溶液淬灭,随后升至室温,乙醚萃取,无水硫酸钠干燥,过滤,浓缩,硅胶柱层析分离提纯得无色液体产物。
实施例3:胆固醇异丙烯基醚的合成
参照文献方法制备(Dujardin,G.,Rossignol,S.,&Brown,E.(1995).Efficientmercury-free preparation of vinyl and isopropenyl ethers of chiral secondaryalcohols andα-hydroxyesters.Tetrahedron letters,36(10),1653-1656)。具体方法如下:
向反应瓶中加入胆固醇10mmol,二氯甲烷25mL,2-甲氧基丙烯60mmol,氮气保护下向其中加入催化剂对甲苯磺酸(p-TSA)0.05mmol,点板监测反应完全后向其中加入0.5mL三乙胺终止反应,除溶剂,硅胶柱层析分离得到缩酮保护的胆固醇。
向反应瓶中加入缩酮保护的胆固醇1mmol,四氢呋喃5mL,DIPEA 1.3mmol,冷却至-20℃,缓慢向其中滴加三氟甲磺酸三甲基硅酯1mmol,恢复至室温,搅拌反应过夜。反应完全后,向其中加入3mL 1M氢氧化钠水溶液,分液,水相用二氯甲烷萃取,合并有机相,水洗,饱和食盐水洗,无水硫酸钠干燥,过滤,浓缩,硅胶柱层析分离得到无色液体产物。
实施例4:β-2,3,4,6-四苄基-D-吡喃葡萄糖异丙烯基醚的合成
在500mL两口瓶中加入β-1-氯代-2,3,4,6-四苄基-D-吡喃葡萄糖(24.3mmol)、双乙酰基甲基汞和氯仿,回流条件下反应。得目标产物,黄色固体,8.9g,收率70.5%。
实施例5:己醇-2'-紫杉醇缩丙酮偶合物(HE-K-2'-PTX)
氮气保护下向反应瓶中加入紫杉醇(0.1mmol,1eq),二氯甲烷1.5mL,己醇异丙烯基醚(0.6mmol)(己醇异丙烯基醚参考文献(Killian,D.B.,G.F.Hennion,andJ.A.Nieuwland."The Preparation of Someα-Unsaturated Ethers from 2,2-Dimethoxyalkanes1."Journal of the American Chemical Society 57.3(1935):544-545.)的方法制备),随后向其中加入催化剂二氯乙酸(0.05eq)。TLC监测原料紫杉醇反应完全后,向其中加入200μL三乙胺终止反应,浓缩,硅胶柱层析分离得白色固体产物HE-K-2'-PTX,产率80.3%。
实施例6:油醇-7-紫杉醇缩丙酮偶合物(Oleyl-K-7-PTX)的合成
其中,Oleyl代表油烯基。
氮气保护下向反应瓶中加入PTX-2'-TBS(0.1mmol,1eq),二氯甲烷1.5mL,油醇异丙烯基醚(0.6mmol)(油醇异丙烯基醚参考文献Pine S H,Zahler R,Evans D A,etal.Titanium-mediated methylene-transfer reactions.Direct conversion of estersinto vinyl ethers[J].Journal of the American Chemical Society,1980,102(9):3270-3272中记载的方法制备),随后加入催化剂二氯乙酸(0.05eq),TLC监测原料PTX-2'-TBS反应完全后向其中加入200μL三乙胺终止反应,浓缩,所得白色固体产物Oleyl-K-7-PTX-2'-TBS直接投入下一步。
氮气保护下向四氟乙烯反应瓶其中加入上一步粗品Oleyl-K-7-PTX-2'-TBS(0.1mmol),四氢呋喃0.5mL,加入TBAF(0.15mmol),室温下搅拌过夜,原料反应完全后向其中加入5mL饱和碳酸氢钠水溶液,分液,水相用DCM萃取(3×5mL),合并有机相,水洗,饱和食盐水洗,无水硫酸钠干燥,过滤,浓缩,硅胶柱层析分离得白色固体产物Oleyl-K-7-PTX,两步产率50.2%。
实施例7:聚乙二醇单甲醚
氮气保护下向反应瓶中加入PTX-2'-TBS(0.1mmol,1eq),二氯甲烷1.5mL,聚乙二醇单甲醚(分子量2000g/mol)异丙烯基醚(0.6mmol)(聚乙二醇单甲醚异丙烯基醚参考文献Pine S H,Zahler R,Evans D A,et al.Titanium-mediated methylene-transferreactions.Direct conversion of esters into vinyl ethers[J].Journal of theAmerican Chemical Society,1980,102(9):3270-3272中的方法制备),随后加入催化剂二氯乙酸(0.05eq),TLC监测原料PTX-2'-TBS反应完全后向其中加入200μL三乙胺终止反应,浓缩,所得白色固体产物mPEG
氮气保护下向四氟反应瓶其中加入上一步粗品mPEG
实施例8:甲醇-2'-紫杉醇缩苯乙酮偶合物(Ph-K-2'-PTX)的合成
氮气保护下向反应瓶中加入紫杉醇(0.1mmol,1eq),二氯甲烷1.5mL,1-甲氧基乙烯基苯(0.6mmol)(1-甲氧基乙烯基苯参考文献:Tebbe,F.N.,Parshall,G.W.,&Reddy,G.D.(1978).Olefin homologation with titanium methylene compounds.Journal of theAmerican chemical society,100(11),3611-3613中的方法制备)。随后向其中加入催化剂二氯乙酸(0.005mmol)。TLC监测原料紫杉醇反应完全后,向其中加入200μl三乙胺终止反应,浓缩,硅胶柱层析分离得白色固体产物Ph-K-2'-PTX,产率70%。
实施例9:己醇-2'-多烯紫杉醇缩丙酮(HE-K-2'-DOC)偶合物的合成
氮气保护下向反应瓶中加入多烯紫杉醇(0.1mmol,1eq),二氯甲烷1.5mL,己醇异丙烯基醚(0.6mmol)(己醇异丙烯基醚参考文献Killian,D.B.,G.F.Hennion,andJ.A.Nieuwland."The Preparation of Someα-Unsaturated Ethers from 2,2-Dimethoxyalkanes1."Journal of the American Chemical Society 57.3(1935):544-545中的方法制备),随后向其中加入催化剂二氯乙酸(0.05eq)。TLC监测原料多烯紫杉醇反应完全后,向其中加入200μL三乙胺终止反应,浓缩,硅胶柱层析分离得白色固体产物HE-K-2'-DOC,产率64%。
实施例10:聚乙二醇单甲醚
氮气保护下向反应瓶中加入卡巴他赛(0.1mmol,1eq),二氯甲烷1.5mL,TES单保护的己二醇异丙烯基醚(0.6mmol)(TES单保护的己二醇异丙烯基醚参考文献Tebbe,F.N.,Parshall,G.W.,&Reddy,G.D.(1978).Olefin homologation with titanium methylenecompounds.Journal of the American chemical society,100(11),3611-3613中的方法制备),随后向其中加入催化剂二氯乙酸(0.05eq)。TLC监测原料卡巴他赛反应完全后,向其中加入200μL三乙胺终止反应,浓缩,所得白色固体直接投入下一步。
取15mL四氟乙烯反应瓶,氮气保护下向其中加入上一步粗品(0.1mmol),四氢呋喃5mL,加入TBAF(0.15mmol),室温下搅拌过夜,原料反应完全后向其中加入5mL饱和碳酸氢钠水溶液,分液,水相用DCM萃取(3×5mL),合并有机相,水洗,饱和食盐水洗,无水硫酸钠干燥,过滤,浓缩,硅胶柱层析分离得白色固体产物C
氮气保护下向反应瓶中加入上一步产物C
实施例11:聚乙二醇单甲醚-聚乳酸-2'-紫杉醇缩丙酮偶合物(mPEG-PDLLA-K-2'-PTX)的合成
在100mL Schlenk瓶中加入紫杉醇(1mmol,1eq),TES单保护的己二醇异丙烯基醚(6mmol),置换氮气后,加入二氯甲烷(25mL)搅拌溶解,加入二氯乙酸(0.05eq)。原料反应完全后向体系中加入三乙胺,随后脱溶,柱层析得化合物PTX-K-C6-OTES,白色固体,收率:36%。
在10mL Schlenk管中加入PTX-K-C6-OTES(0.36mmol),加入THF(2mL)搅拌溶解,随后加入TBAF四氢呋喃溶液(0.40mmol)。反应结束后,加入饱和碳酸氢钠稀释体系,乙酸乙酯溶解后,水、饱和氯化钠萃取,无水硫酸钠干燥,滤液脱溶,柱层析得白色固体PTX-K-C6-OH,收率71%。
在25mL Schlenk管中加入端基为羧酸的mPEG
参考实施例5-11的方法,由上述药物制备表1-3中的紫杉烷缩酮衍生物。
实施例12:紫杉烷类缩酮前药偶合物的水解速度测试
37.0℃下,将20μL待测前药(上述实施例制备的前药)的乙腈溶液(7.5mM)加入到3mL水解缓冲液(pH 5.0,50mM,含有40%V乙腈)中。在1min,3min,5min,15min,0.5h,1h,2h,4h,8h,12h,24h,48h快速取100μL水解样品并加入100μL水解终止缓冲液(pH 8.0,200mM,含有40%V乙腈)终止水解,混合均匀后进液相色谱测定水解原料和水解产物含量,拟合水解曲线,计算水解半衰期,平行测试4次。测试结果如表1-3所示。
由表1-表3可以看出,紫杉烷类缩酮前药偶合物具有很明显的酸敏感性质,在pH5.0条件下半衰期最短为3mins,最长可达到29838mins,因此可以通过分子设计达到控制紫杉烷类药物酸敏感程度的目的。
对于紫杉醇不同反应位点水解活性不同,紫杉醇7位反应活性的要明显优于2'位。同时,发现相同反应位点水解活性与结构有密切关系,有以下规律:缩酮上的取代基为环状结构时,其水解活性要优于链状结构;与缩酮相连的碳原子为二级碳原子反应活性要优于一级碳;连接吸电子基团水解活性降低。
实施例13:紫杉烷类偶合物前药的细胞毒性测试
细胞培养条件:DMEM高糖培养基,10%FBS,1%P/S,37℃,5%CO
实验前一天将MDA-MB-231细胞以3000/孔密度种入96孔板。实验当天,将上述实施例制备的前药溶解于DMSO,用培养基将药物稀释至不同浓度,加入96孔板(n=4)。待细胞与上述实施例制备的前药在培养箱继续孵育3天后,将培养基换为100μL新鲜培养基+10μLCCK-8溶液。继续孵育2h后,测450nm的OD值,并按CCK-8说明书计算IC
由表1-3可以看出本发明制备的前药对于癌细胞具有和紫杉醇相近的杀伤效果。同时,细胞毒性数据与体外水解实验结果相吻合,说明本发明通过利用酸敏感缩酮键构建的前药具有很高的利用价值。
实施例14:亲水性紫杉烷缩酮偶合物前药注射液的制备
对于水溶性缩酮前药mPEG
实施例15:亲脂性紫杉烷缩酮偶合物前药纳米粒的制备
以Oleyl-K-7-PTX为例:准确称取7mg Oleyl-K-7-PTX和3mg DSPE-mPEG
实施例16:两亲性紫杉烷缩酮偶合物前药胶束的制备
以mPEG-PDLLA-K-2'-PTX为例,将5mg两亲性紫杉烷缩酮偶合物前药溶于500μL丙酮,搅拌下缓慢滴加入到含5.0mL pH 7.4PB缓冲液(10mM)的25mL圆底烧瓶中。室温旋蒸15min以去除丙酮,超速离心(10K MWCO),将mPEG-PDLLA-K-2'-PTX胶束浓缩至0.5mL制备前药胶束。HPLC计算胶束中的药物浓度后使用。
实施例17:紫杉烷类偶合物前药制剂体内抗肿瘤评价
从北京维通利华实验动物技术有限公司购入4-6周龄、体重16-20g的雌性balb/c-nu裸鼠,饲养于SPF级实验动物房。
在裸鼠右侧腋下皮下植入5×10
待A2780肿瘤体积长至2000mm
mPEG
Oleyl-K-7-PTX/DSPE-PEG
mPEG-PDLLA-K-2'-PTX胶束和自制市售紫杉醇胶束Genexol制剂(剂量以紫杉醇计为10mg/kg)的测试结果如图4所示。由图4可知,本发明制备的mPEG-PDLLA-K-2'-PTX胶束在同等剂量下(10mg/kg紫杉醇)其抑瘤活性显著优于商用药。
mPEG
实施例18:硬脂醇-地塞米松缩丙酮偶合物(SA-K-DEX)的合成
将地塞米松(1mmol)和硬脂醇异丙烯基醚(6mmol,1eq)(硬脂醇异丙烯基醚参考文献:Pine S H,Zahler R,Evans D A,et al.Titanium-mediated methylene-transferreactions.Direct conversion of esters into vinyl ethers[J].Journal of theAmerican Chemical Society,1980,102(9):3270-3272中记载的方法制备)置于Schlenk瓶中,置换体系内气体为氮气。加入5mL四氢呋喃溶解,之后加入二氯乙酸(0.2eq),25℃反应。TLC跟踪反应,待反应完成后,加入100μL三乙胺淬灭反应。旋干溶剂后硅胶柱纯化,得白色粉末,产率为92%。
参考实施例18中的方法,以上述药物为起始物,制备表4的糖皮质类激素缩酮衍生物。
实施例19:地塞米松缩酮偶合物前药的水解速度测试
将地塞米松缩酮偶合物前药溶于600μL乙腈,并与12.5mL pH 5.0的水解液(含0.1%吐温80)混合,最终地塞米松缩酮偶合物前药的浓度约为50μM。37.0℃摇床孵育前药水解(100rpm),在3min,6min,9min,15min,25min,30min,45min,1h,1.5h,2h,3h,6h快取100μL样品并加入100μL pH 8.0磷酸盐缓冲液(200mM),混合后置于4℃冰箱。最后,由HPLC测定水解半衰期和溶解度,检测结果如表4所示。
由表4可知,通过更换不同的R侧链,地塞米松缩丙酮前药显示出不同的物理化学性质,如表现出不同的溶解度和水解半衰期。增加脂肪侧链的碳链长度,可以显著增加前药在二氯甲烷中的溶解度。不同类型的侧链对前药的水解速度也有影响,通常来说,脂肪仲醇侧链要比脂肪伯醇侧链的水解速度要快,PEG侧链相对于脂肪伯醇侧链水解速度要快。因此本发明制备的地塞米松缩丙酮前药由于在有机溶剂中的水解速度和溶解度显著提高,更便于纳米制剂的制备,提高了体内治疗效果。
实施例20:SA-K-DEX/DSPE-PEG
将6mg SA-K-DEX和4mg DSPE-PEG
由图6可知,SA-K-DEX/DSPE-PEG
实施例21:SA-K-DEX/DSPE-PEG纳米粒体内药效评价
CIA大鼠模型的建立:180-200g雄性SD大鼠饲养一周适应环境。将2mg/mL牛II型胶原蛋白与不完全佐剂等体积混合,充分乳化,配制成胶原蛋白终浓度为1mg/mL的乳剂。初次免疫于大鼠尾基注射该乳剂200μL,7日后尾基注射乳剂100μL。初次免疫后10天大鼠足跖开始红肿。实验设置模型组(地塞米松磷酸注射液)、正常对照组、用药治疗组(SA-K-DEX/DSPE-PEG纳米粒),每组7只大鼠。
纳米粒给药方案:分别于初次免疫后第15、20、25天给药,对大鼠尾静脉注射纳米粒溶液(1mg/kg),第28天结束实验处死大鼠,并用MicroCT观察治疗效果。
治疗效果如图7和8所示。本实验所用大鼠CIA模型,大鼠后足足跖于初次免疫后第10天开始红肿,第15天时达到肿胀高峰。在三次尾静脉注射药物后,可以看到SA-K-DEX/DSPE-PEG
实施例22:SA-K-DEX/DSPE-PEG纳米粒关节分布
CIA大鼠单次分别注射SA-K-DEX/DSPE-PEG纳米粒和地塞米松磷酸注射液4小时后处死大鼠,取炎症关节。按照LC-MS测试的标准步骤处理样品,由LC-MS测定关节中的地塞米松药物浓度。为了测定地塞米松在关节中的整体浓度,需要将样品进行酸解,以释放地塞米松。
药物体内分布实验中LC-MS数据结果如图9所示。由图9可知,在发炎的踝关节处,地塞米松的整体含量和游离含量要高于地塞米松磷酸钠注射液组,具有显著性差异,这证实了纳米粒在发炎的关节部位具有富集效果,是SA-K-DEX药效提高的关键所在。
实施例23:SA-K-DEX纳米晶的制备
在2mL离心管中加入500mg氧化锆钢珠和300μL SA-K-DEX无水乙醇溶液(含3mgSA-K-DEX),随后加入600μL pH 9.0NaOH水溶液。离心管敞口放入4℃冰箱中,挥发乙醇约48h。
向剩余溶液中加入100μL含2%的吐温80水溶液,用涡旋仪搅拌(2700转/分钟)0.5h–6h,可获取尺寸可调的SA-K-DEX纳米晶。通过光学显微镜观察可大致观察纳米晶的形态和尺寸。
纳米晶的光线显微镜检测结果如图10所示。由图10可知,在光学显微镜下,SA-K-DEX纳米晶显示为针状晶体,长度在10-15μm之间。
搅拌结束后,将分散均匀的液体转移至一个离心管中。离心后,弃去上层清液。加入适量含有0.1%吐温80的水溶液。重复上述步骤3次后,纳米晶冻干后备用。使用前,用pH7.4PBS重悬,并使用涡旋分散均匀。
实施例24:地塞米松制剂体内药效评价
按实施例21造模,纳米晶给药方案:于初次免疫后第21天给药,对大鼠踝关节腔注射SA-K-DEX纳米晶(剂量以SA-K-DEX计2.5mg/kg),第50天结束实验处死大鼠。
测试结果如图11所示。由图11可知,在关节腔注射SA-K-DEX纳米晶后,治疗效果至少能在给药后28天内维持稳定。
实施例25:地塞米松纳米晶血液中地塞米松浓度的测定
在大鼠炎症关节中注射SA-K-DEX纳米晶(剂量以SA-K-DEX计:2.5mg/kg)的14、21和28天后,取血。按照血液样品LC-MS测试的标准步骤处理样品,由LC-MS测定血液中的地塞米松药物浓度。为了测定地塞米松在血液中的整体浓度,需要将样品进行酸解,以释放地塞米松。
对大鼠血浆中地塞米松浓度进行定期检测,LC-MS数据结果如图12所示。由图12可知,第14、21、28天仍能测到浓度在20ng/mL以上的总地塞米松,说明SA-K-DEX纳米晶能够在大鼠关节腔缓慢水解释放,显著延长了地塞米松在大鼠体内的滞留时间,提升了治疗效果。
实施例26:硬脂醇-他氟前列素缩丙酮偶合物(SA-K-TAF)的合成
将TBS-TAF(0.1mmol,1eq)和硬脂醇异丙烯基醚(0.6mmol)置于Schlenk瓶中,置换体系内气体为氮气。加入2mL四氢呋喃溶解,之后加入二氯乙酸(0.2eq),25℃反应。TLC跟踪反应,待反应完成后,加入100μL三乙胺淬灭反应。旋干溶剂后硅胶柱纯化,得无色油状物。
将此油状物用四氢呋喃溶解,加入四丁基氟化铵(0.08mmol),25℃搅拌反应。反应完全后,旋干溶剂后硅胶柱纯化,得无色油状物SA-K-TAF,产率为60%。
实施例27:胆固醇-他氟前列素缩丙酮偶合物(Chol-K-TAF)的合成
将TBS-TAF(0.1mmol)和胆固醇异丙烯基醚(0.6mmol,1eq)(胆固醇异丙烯基醚参考文献:Dujardin,G.,Rossignol,S.,&Brown,E.(1995).Efficient mercury-freepreparation of vinyl and isopropenyl ethers of chiral secondary alcohols andα-hydroxyesters.Tetrahedron letters,36(10),1653-1656中的方法制备)置于Schlenk瓶中,置换体系内气体为氮气。加入2mL四氢呋喃溶解,之后加入二氯乙酸(1.0eq),25℃反应。TLC跟踪反应,待反应完成后,加入100μL三乙胺淬灭反应。旋干溶剂后硅胶柱纯化,得无色油状物。将此油状物用四氢呋喃溶解,加入四丁基氟化铵(0.08mmol),25℃搅拌反应。反应完全后,旋干溶剂后硅胶柱纯化,得无色油状物Chol-K-TAF,产率为65%。
(d,J=6.3Hz,6H),1.19–1.04(m,8H),0.99(s,3H),0.91(d,J=6.5Hz,3H),0.86(m,6H),0.66(s,3H).
按照实施例26-27的方法,以上述药物为起始物,制备前列腺素类似物的缩酮衍生物。
实施例28:他氟前列素缩酮前药纳米粒的制备
将7mg SA-K-TAF和3mg DSPE-PEG
参考上述方法,还制备了Chol-K-TAF纳米粒。
实施例29:他氟前列素缩酮前药纳米粒结膜下注射对正常家兔眼压的影响
实验动物为正常眼压的新西兰兔(体重为3.5-4.5公斤),每组动物为2只。将新西兰兔麻醉,左眼滴加盐酸丙美卡因滴眼液,用27号针头无菌注射器将100μL SA-K-TAF/DSPE-PEG
测试结果如图14所示。由图14可知,SA-K-TAF/DSPE-PEG
实施例30:油醇-帕利哌酮缩丙酮偶合物(Oleyl-K-PAL)的合成
氮气保护下向反应瓶中加入帕利哌酮(0.1mmol,1eq),二氯甲烷1.5mL,二氯乙酸(0.1mol),室温下搅拌30min后,向其中加入油醇异丙烯基醚(0.6mmol),TLC监测原料帕利哌酮反应完全后向其中加入400μL三乙胺终止反应,浓缩,硅胶柱层析分离得白色固体产物Oleyl-K-7-PAL,产率83%。
实施例31:Oleyl-K-PAL/DSPE-PEG
准确称取7mg Oleyl-K-PAL和3mg DSPE-mPEG
实施例32:肌肉注射Oleyl-K-PAL/DSPE-PEG
在大鼠后腿肌肉中注射Oleyl-K-PAL/DSPE-PEG
测试结果如图15所示。由图15可知,Oleyl-K-PAL/DSPE-PEG
实施例33:硬脂醇-双氢青蒿素缩丙酮偶合物(SA-K-DHA)的合成
将双氢青蒿素(0.3mmol,1eq)和硬脂醇异丙烯基醚(1.8mmol)置于Schlenk瓶中,置换体系内气体为氮气。加入4mL四氢呋喃溶解,之后加入吡啶对甲苯磺酸(PPTS)(0.05eq),25℃反应。TLC跟踪反应,待反应完成后,加入100μL三乙胺淬灭反应。旋干溶剂后硅胶柱纯化,得白色粉末SA-K-DHA,产率为60%。
实施例34:SA-K-DHA/DSPE-PEG
准确称取7mg SA-K-DHA和3mg DSPE-mPEG
实施例35:肌肉注射SA-K-DHA/DSPE-PEG
在大鼠后腿肌肉中注射SA-K-DHA/DSPE-PEG
测试结果如图16所示。由图16可知,SA-K-DHA/DSPE-PEG
实施例36:硬脂醇-5'-吉西他滨缩丙酮偶合物(SA-K-5'-GEM)的合成
氮气保护下在反应瓶中加入0.44mmol Fmoc保护氨基的吉西他滨(0.44mmol)(Fmoc保护氨基的吉西他滨参考文献:Peter G.M.Wuts.(2014).Greene’s ProtectiveGroups in Organic Synthesis.John Wiley&Sons,Inc.(Fifth Edition)中的方法制备),硬脂醇异丙烯基醚(1.32mmol),4mL四氢呋喃,最后加入催化剂二氯乙酸(0.1eq),30℃反应。TLC监测反应结束后,加入10mL二氯甲烷和0.2mL DBU(1,8-二氮杂二环十一碳-7-烯)。TLC监测反应Fmoc脱保护完成后,加入100mL乙酸乙酯稀释,用饱和碳酸氢钠溶液、饱和食盐水洗涤,有机相用无水硫酸钠干燥浓缩后柱层析分离得白色蜡状固体SA-K-5'-GEM,产率77%。
实施例37:硬脂醇-5’-齐多夫定缩丙酮偶合物(SA-K-5'-AZT)的合成
氮气保护下在反应瓶中加入0.75mmol(1eq)齐多夫定,硬脂醇异丙烯基醚(2.25mmol),5mL四氢呋喃,最后加入二氯乙酸(0.1eq),30℃反应。TLC监测反应结束后,加入0.5mL三乙胺终止反应,浓缩后硅胶柱层析分离得白色蜡状固体SA-K-5'-AZT,产率78%。
按照实施例36-37方法,以下列药物分子,三氟尿苷、碘苷、氟尿苷、卡培他滨、拉米夫定、恩曲他滨、扎西他滨、阿糖胞苷、更昔洛韦、阿昔洛韦和多柔比星等为起始物,制备其缩丙酮前药。
实施例38:吉西他滨偶合物前药的细胞毒性测试
细胞培养条件:DMEM高糖培养基,10%FBS,1%P/S,37℃,5%CO
实验前一天将A2780或A549细胞以3000/孔密度种入96孔板。实验当天,将SA-K-5'-GEM溶解于DMSO,用培养基将药物稀释至不同浓度,加入96孔板(n=4)。待细胞与上述实施例制备的前药在培养箱继续孵育3天后,将培养基换为100μL新鲜培养基+10μL CCK-8溶液。继续孵育2h后,测450nm的OD值,并按CCK-8说明书计算IC
测试结果如图17所示。由图17可知,硬脂醇-5’-吉西他滨缩丙酮偶合物(SA-K-5'-GEM)具有比吉西他滨更强的抗肿瘤活性。
实施例39:SA-K-5'-AZT/DSPE-PEG
准确称取7mg SA-K-5'-AZT和3mg DSPE-mPEG
实施例40:肌肉注射SA-K-5'-AZT/DSPE-PEG
在大鼠后腿肌肉中注射SA-K-5'-AZT/DSPE-PEG
实施例41:聚乙二醇单甲醚-睾酮缩丙酮偶合物(mPEG
将聚乙二醇单甲醚(分子量2000g/mol)异丙稀基醚0.75mmol和对甲苯磺酸0.04mmol溶于20mL四氢呋喃,体系中加入0.1mmol睾酮,室温反应。待反应结束后,用400μLTEA淬灭。将体系旋干,纯化得到白色固体102mg,产率为46%。
根据实施例41方法,制备上列睾酮、雌二醇、乙炔雌二醇和利奈孕酮性激素类药的缩酮前药。
实施例42:大鼠尾静脉注射mPEG
在大鼠尾静脉注射mPEG
测试结果如图19所示。由图19可知,mPEG
实施例43:β-葡萄糖-K-依托泊苷缩丙酮偶合物(β-Glucose-K-ETP)的合成
在10mL Schlenk瓶中加入依托泊苷(0.1mmol)、β-2,3,4,6-四苄基-D-吡喃葡萄糖异丙烯基醚(0.6mmol)和二氯甲烷(3mL)。搅拌均匀后,将二氯乙酸的二氯甲烷溶液(0.02mmol)加入体系。在30℃下搅拌反应5小时。反应结束后,加入三乙胺将反应淬灭。体系脱溶,经硅胶柱层析得中间体,白色固体,41mg。在15mL氢化内管中加入中间体(41mg,0.4mmol),加入超干乙醇(1mL)搅拌均匀后,向体系中加入预先用超干乙醇除水的5%氢氧化钯碳(100mg)。拧紧氢化反应釜,充4个大气压反应96小时。拆釜后,将体系滤除钯碳,滤液脱溶,经硅胶柱层析得化合物β-Glucose-K-ETP,白色固体,26mg,收率:90%。
实施例44:β-葡萄糖-K-依托泊苷前药体内抗肿瘤评价
从北京维通利华实验动物技术有限公司购入4-6周龄、体重16-20g的雌性balb/c-nu裸鼠,饲养于SPF级实验动物房。
在裸鼠右侧腋下皮下植入5×10
β-Glucose-K-ETP治疗测试结果如图20所示,由图20可知β-Glucose-K-ETP注射液具有很好的安全性和药效,具有很好的抗肿瘤活性,其效果优于商用依托泊苷注射液。
实施例45:硬脂醇-NLG919缩丙酮偶合物(SA-K-NLG919)的合成
氮气保护下在反应瓶中加入1.0mmol NLG919,硬脂醇异丙烯基醚(6.0mmol),20mL二氯甲烷溶解,加入二氯乙酸(0.7eq),30℃反应2h。TLC监测反应结束后,加入0.2mL三乙胺终止反应,浓缩后硅胶柱层析分离得黏液状SA-K-NLG919,产率20%。
实施例46:硬脂醇-常山酮缩丙酮偶合物(SA-K-Halofuginone)的合成
在反应瓶中加入Fmoc保护氨基的常山酮(0.08mmol)(参考文献:Peter G.M.Wuts.(2014).Greene’s Protective Groups in Organic Synthesis.John Wiley&Sons,Inc.(Fifth Edition)),硬脂醇异丙烯基醚(0.48mmol),4mL四氢呋喃溶解,加入吡啶鎓对甲苯磺酸盐(0.05eq),30℃反应10min。TLC监测反应结束后,加入10mL二氯甲烷和0.08mL DBU(1,8-二氮杂二环十一碳-7-烯),室温下反应5min后,加入100mL乙酸乙酯稀释,用饱和碳酸氢钠溶液饱和食盐水洗涤,有机相用无水硫酸钠干燥。浓缩后柱层析分离得白色固体SA-K-Halofuginone,产率70%。
实施例47:硬脂醇-康普瑞汀缩丙酮偶合物(SA-K-CA4)的合成
取15ml Schlenk管,向其中加入原料康普瑞汀0.1mmol,二氯甲烷2.0ml,硬脂醇异丙烯基醚0.6mmol,搅拌下向其中加入1,2-二氯乙酸0.005mmol。室温下反应,点板检测反应完全后向其中加入300μl三乙胺终止,直接旋干,硅胶柱层析分离,得白色蜡状产物SA-K-CA4,产率73%。
实施例48:硬脂醇-7-乙基-10-羟基喜树碱缩丙酮偶合物(SA-K-SN-38)的合成
取15ml Schlenk管,向其中加入原料7-乙基-10-羟基喜树碱0.1mmol,二氯甲烷2.0ml,硬脂醇异丙烯基醚0.6mmol,搅拌下向其中加入1,2-二氯乙酸0.005mmol。室温下反应,点板检测反应完全后向其中加入300μl三乙胺终止,直接旋干,硅胶柱层析分离,得白色固体SA-K-SN-38,产率76%。
实施例49:硬脂醇-雷帕霉素缩丙酮偶合物(SA-K-Rap)的合成
取15ml Schlenk管,向其中加入原料雷帕霉素0.1mmol,二氯甲烷2.0ml,硬脂醇异丙烯基醚0.6mmol,搅拌下向其中加入1,2-二氯乙酸0.005mmol。室温下反应,点板检测反应完全后向其中加入300μl三乙胺终止,直接旋干,硅胶柱层析分离,得白色固体SA-K-Rap(混合物),产率46%。
实施例50:硬脂醇-氟维司群缩丙酮偶合物(SA-K-FUL)的合成
取15ml Schlenk管,向其中加入原料TBS保护的氟维司群0.1mmol,二氯甲烷2.0ml,硬脂醇异丙烯基醚0.6mmol,搅拌下向其中加入1,2-二氯乙酸0.005mmol。室温下反应,点板检测反应完全后向其中加入300μl三乙胺终止,直接旋干,随后向其中加入3ml四氢呋喃,加入四正丁基氟化铵1.0mmol,反应完全后,向其中加入10ml水,用乙酸乙酯萃取,收集有机相,饱和食盐水洗,无水硫酸钠,过滤,浓缩,硅胶柱层析分离,得白色固体SA-K-FUL,两步产率61%。
实施例51:硬脂醇-阿比特龙缩丙酮偶合物(SA-K-ABI)的合成
取15ml Schlenk管,向其中加入原料阿比特龙0.1mmol,二氯甲烷2.0ml,正十四烷氧基异丙烯基醚0.6mmol,搅拌下向其中加入1,2-二氯乙酸0.015mmol,室温下反应,点板检测反应完全后向其中加入300μl三乙胺终止,直接旋干,硅胶柱层析分离,得无色油状产物SA-K-ABI,产率83%。
实施例52:聚乙二醇单甲醚-辛伐他汀缩丙酮偶合物(mPEG1000-K-SIM)的合成
将辛伐他汀(0.1mmol)和mPEG1000异丙烯氧基醚(0.6mmol)置于Schlenk瓶中,加入2mL四氢呋喃溶解,之后加入二氯乙酸(0.2eq),25℃反应。TLC跟踪反应,待反应完成后,加入100μL三乙胺淬灭反应。旋干溶剂后反相柱纯化,得白色固体,产率为68%。
参考实施例52的方法,使用洛伐他汀为起始物,制备其缩丙酮前药。
表1.2'-紫杉醇缩丙酮前药水解半衰期和对MDA-MB-231细胞的半抑制浓度
表2.2'-紫杉醇缩酮前药水解半衰期和对MDA-MB-231细胞的半抑制浓度
表3.7-紫杉醇缩丙酮前药水解半衰期和对MDA-MB-231细胞的半抑制浓度
表4.地塞米松缩丙酮前药水解半衰期以及在二氯甲烷中的溶解度
发明人发现,与上述化合物的测试结果类似的是,相对于各起始药物,经由上述实施例制备得到的前药的溶解度可以得到显著有利的改善,并且其药理活性也显著优于市售的起始药物,从而在改善治疗效果和制剂种类方面取得预料不到的显著进步。
本文对本发明示例性的实施方案进行了说明。但是应当理解,本发明的保护范围不拘囿于上述实施方案。在本发明的精神和原则之内,所作出的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 药物的缩酮衍生物及其制备方法、药物组合物和用途
- 二苯氧基烷基胺衍生物和芳氧基烷基胺衍生物、药物组合物、所述药物组合物用于治疗、预防或抑制慢性肺炎性疾病的用途和用于治疗或预防所述疾病的方法