使用卫康醇或其衍生物治疗抗酪氨酸激酶抑制剂的恶性肿瘤的方法
文献发布时间:2023-06-19 10:55:46
本申请是申请日为2013年6月24日、申请号为201380042796.0、发明名称为“使用卫康醇、二乙酰二脱水卫矛醇、二溴卫矛醇或类似物或其衍生物治疗具有基因多型性或AHI1失调或突变患者的抗酪氨酸激酶抑制剂的恶性肿瘤的方法”的中国专利申请的分案申请。
本申请案声明主张D.M.Brown等人于2013年5月17日所申请的美国临时申请案号61/824,672的利益,标题为"使用卫康醇、二乙酰二脱水卫矛醇、二溴卫矛醇或类似物或其衍生物治疗具有基因多型性患者的抗酪氨酸激酶抑制剂的恶性肿瘤的方法",其内容以引用方式并入本文中;以及声明主张D.M. Brown等人于2012年6月26日所申请的美国临时申请案号61/664,279的利益,标题为"使用卫康醇、二乙酰二脱水卫矛醇、二溴卫矛醇或类似物或其衍生物治疗具有基因多型性患者的抗酪氨酸激酶抑制剂的恶性肿瘤的方法",其内容以引用方式并入本文中。
技术领域
本发明涉及使用卫康醇(dianhydrogalactitol)、二乙酰二脱水卫矛醇(diacetyldianhydrogalactitol)、二溴卫矛醇(dibromodulcitol)或类似物或其衍生物,治疗具有基因多型性(genetic polymorphisms)患者的抗酪氨酸激酶抑制剂 (tyrosine-kinase-inhibitor resistant)的恶性肿瘤的方法、治疗以 AHI1(abelson-helperintegration site-1)基因的表现调控抗性的恶性肿瘤的方法,或治疗三阴性乳癌(triple-negative breast cancer)的方法,以及用于治疗具有基因多型性、以所述AHI1基因的表现调控抗性的恶性肿瘤或三阴性乳癌患者的抗酪氨酸激酶抑制剂的恶性肿瘤的医药组合物。
背景技术
在病患出现一些认为由致癌基因激酶的活性驱动的恶性肿瘤中,酪氨酸激酶抑制剂(Tyrosine Kinase Inhibitors,TKIs)的使用对于有效治疗反应是可靠的(P.A.
然而,TKIs用于治疗许多类型的恶性肿瘤(以前被认为通过化学疗法是不可治愈的)已被证明是一样有效的,有显著比例的病患对于TKI化学疗法是有抗性的。许多的这些患者为东亚血统,表明可能引起TKI化学疗法的抗性的基因变异的存在。
因此,在TKI化学疗法抗性的病患中迫切需要可以治疗恶性肿瘤的治疗方法及药物组合物。
此外,还有其他的恶性肿瘤,特别是包含但不限于慢性淋巴细胞性白血病(chronic lymphocytic leukemia,CLL),其与所述AHI1基因是相关的,特别是与所述AHI1基因的突变或失调。所述AHI1基因是一编码具有WD40重复序列及SH3区域的模蛋白的基因。在此基因的基因丛位上前病毒的插入与恶性肿瘤的发展是相关的,可能是所述基因的截短形式的表现(X.Jiang等人,“Ahi-1、一新颖的编码具有WD40重复序列及SH3区域的模蛋白的基因,是Ahi-1及Mis-2前病毒插入的靶向目标”,J.Virol.76:9046-9059(2002),并通过引用将其包括在内)。费城染色体阳性(Ph
因此,迫切需要改进方法以治疗与AHI1基因的突变或异常调控相关的恶性肿瘤,特别是白血病。
此外,三阴性乳癌是以肿瘤为特征的乳癌的一种形式,所述肿瘤没有表现雌激素受体(estrogen receptor,ER)、黄体激素受体(progesterone receptor,PR) 或HER-2基因。因为这些癌症对内分泌疗法或许多靶向药物没有反应,所以这种形式的乳癌代表一重要的临床挑战。目前对于三阴性乳癌的治疗策略包含许多化学疗法试剂,比如蒽环类、紫杉烷、易莎平(ixabepilone)及铂剂,同时包含选择的生物制剂,并可能包含抗EGFR药物。
然而,还迫切需要改进方法以治疗三阴性乳癌。
发明内容
本发明是关于在TKI化学疗法抗性的病患中提供一用于治疗恶性肿瘤的选择性治疗途径的方法及药物组合物。
本发明的一方面是提供一恶性肿瘤的治疗方法,其中所述恶性肿瘤的特征是对至少一酪氨酸激酶抑制剂(TKI)有抗性,原因为:(1)一基因中的至少一突变,其编码一蛋白,所述蛋白是一至少一TKI的标靶;或(2)于一原生型或突变状态中的至少一额外的基因的存在,其编码一给予至少一TKI的治疗效果的抗性的产物,所述方法包含:给予一烷基化己醣醇衍生物的治疗有效量。在一选择方案中,其中所述编码给予至少一TKI的治疗效果的抗性的产物的于原生型或突变状态中的至少一额外的基因为AHI-1。在另一选择方案中,所述对至少一TKI的抗性是由于一突变在ABL1蛋白的激酶区域中,所述ABL1蛋白是属于一TKIs的标靶的BCR-ABL融合蛋白的一部分。所述方法可以进一步包含:给予一BH3类似物的治疗有效量至目标对象的步骤。一选择方案,所述方法可以进一步包含:给予一STAT5抑制剂、一JAK2抑制剂、一Src抑制剂或两个或多个激酶抑制剂的组合的治疗有效量的步骤。
本发明的另一方面是提供一于患有具有一赋予TKIs抗性的生殖细胞缺失多型性的恶性肿瘤的目标对象中的恶性肿瘤的治疗方法,其包含:给予一治疗试剂的治疗有效量至目标对象以治疗所述恶性肿瘤的步骤,所述治疗试剂是选自于由卫康醇、卫康醇的衍生物或类似物、二乙酰二脱水卫矛醇、二乙酰二脱水卫矛醇的衍生物或类似物、二溴卫矛醇,以及二溴卫矛醇的衍生物或类似物所组成的群组。
所述恶性肿瘤可以是慢性骨髓性白血病(chronic myelogenous leukemia, CML)或非小细胞肺癌(non small cell lung carcinoma,NSCLC)。在另一选择方案中,所述恶性肿瘤可以是三阴性乳癌。
本发明的另一方面是提供一于患有与AHI1基因的突变或异常调控相关的恶性肿瘤的目标对象中的恶性肿瘤的治疗方法,其包含:给予一治疗试剂的治疗有效量至目标对象以治疗所述恶性肿瘤的步骤,所述治疗试剂是选自于由卫康醇、卫康醇的衍生物或类似物、二乙酰二脱水卫矛醇、二乙酰二脱水卫矛醇的衍生物或类似物、二溴卫矛醇及二溴卫矛醇的衍生物或类似物所组成的群组。在此选择方案中,所述恶性肿瘤可以是慢性骨髓细胞性白血病。所述方法可以进一步包含:给予一调节所述AHI1基因或所述AHI1蛋白的表现或活性的药剂的治疗有效量。
本发明的另一方面是提供一种结合筛选及治疗的方法,其结合:生殖细胞缺失多型性的筛选,以及如果所述生殖细胞缺失多型性被发现存在时于一目标对象中TKIs抗性的恶性肿瘤的治疗。
一般地,此方法包含下面步骤:
(1)于一具有恶性肿瘤的目标对象中筛选所述生殖细胞缺失多型性;以及
(2)如果所述生殖细胞缺失多型性被发现存在于所述具有恶性肿瘤的目标对象中,给予一治疗试剂的治疗有效量至目标对象以治疗所述恶性肿瘤,所述治疗试剂是选自于由卫康醇、卫康醇的衍生物或类似物、二乙酰二脱水卫矛醇、二乙酰二脱水卫矛醇的衍生物或类似物、二溴卫矛醇,以及二溴卫矛醇的衍生物或类似物所组成的群组。
这些方法可以进一步包含:于一患有一生殖细胞缺失多型性存在的恶性肿瘤的目标对象中给予(1)一BH3类似物;或(2)一BH3类似物及一抗酪氨酸激酶治疗试剂两者的治疗有效量的步骤。
合适的BH3类似物包含但不限于:
(1)胜肽;
(2)修饰胜肽;
(3)三砒啶基拟胜肽类;
(4)对苯酰胺基拟胜肽类;
(5)苯甲酰脲基拟胜肽类;
(6)欧巴妥拉(obatoclax);
(7)TW37;
(8)一个TW37的类似物或衍生物;
(9)(-)棉子酚;
(10)棉子酚衍生物;
(11)异恶唑啉衍生物;
(12)A-385358;
(13)一个A-385358的类似物或衍生物;
(14)ABT-737;
(15)一个ABT-737的类似物或衍生物;
(16)ABT-263;
(17)一个ABT-263的类似物或衍生物;
(18)TM-1206;以及
(19)一个TM-1206的类似物或衍生物。
一特别优选的BH3类似物是ABT-737。
所述酪氨酸激酶抑制剂可以是但不限于伊马替尼(imatinib)、伯舒替尼(bosutinib)、尼罗替尼(nilotinib)或达沙替尼(dasatinib)。一特别优选的酪氨酸激酶抑制剂是伊马替尼。在另一选择方案中,所述酪氨酸激酶抑制剂可以是但不限于厄洛替尼(erlotinib)、阿法替尼(afatinib)或达可替尼(dacomitinib)。
本发明的另一方面是提供一种用以使用于治疗一抗TKI肿瘤的烷基化己醣醇衍生物的投药的功效增加和/或副作用减少的方法,所述方法包含下面步骤:
(1)确认烷基化己醣醇衍生物的投药的功效和/或副作用发生相关的至少一因子或参数,所述烷基化己醣醇衍生物是用于治疗一抗TKI肿瘤;以及
(2)修改所述因子或参数,用以使所述烷基化己醣醇衍生物的投药的功效增加和/或副作用减少,所述烷基化己醣醇衍生物是用于治疗所述抗TKI肿瘤。
通常,在本方法中,所述因子或参数是选自于由下列所组成的群组:
(1)剂量调整;
(2)投药的途径;
(3)投药的时间表;
(4)使用适应症;
(5)病程阶段的选择;
(6)其他适应症;
(7)选择病患;
(8)病患/疾病表现型;
(9)病患/疾病基因型;
(10)前/后治疗准备;
(11)毒性管理;
(12)药物动力学/药效学监测;
(13)并用药;
(14)化学敏化作用;
(15)化学增效作用;
(16)后治疗病患管理;
(17)选择性医疗/治疗支持;
(18)原料药产品改良;
(19)稀释剂系统;
(20)溶剂系统;
(21)赋形剂;
(22)剂型;
(23)剂量套组及包装;
(24)药物传递系统;
(25)药物共轭形式;
(26)化合物类似物;
(27)前驱物;
(28)多重药物系统;
(29)生物治疗加强作用;
(30)抗生物治疗调控;
(31)放射线疗法增强;
(32)新颖作用机制;
(33)选择性靶细胞群疗法;以及
(34)使用一增加其活性的药剂。
本组合物的另一方面是提供一种用以使不理想给药疗法的功效增加和/ 或副作用减少的组合物,所述不理想给药疗法为使用一烷基化己醣醇衍生物用于治疗一抗TKI肿瘤,所述组合物包含一选自于由下列所组成的群组的选择方案:
(i)一修饰烷基化己醣醇衍生物或者一烷基化己醣醇衍生物或一修饰烷基化己醣醇衍生物的衍生物、类似物或前驱物的治疗有效量,其中相较于一未修饰烷基化己醣醇衍生物,所述修饰烷基化己醣醇衍生物或者所述修饰烷基化己醣醇衍生物的衍生物、类似物或前驱物具有对于一抗TKI肿瘤或与AHI1 基因的突变或异常调控相关的恶性肿瘤的治疗增加疗效或减少副作用;
(ii)一组合物包含:
(a)一烷基化己醣醇衍生物、一修饰烷基化己醣醇衍生物或者一烷基化己醣醇衍生物或一修饰烷基化己醣醇衍生物的衍生物、类似物或前驱物的治疗有效量;以及
(b)至少一附加治疗试剂、受到化学敏化作用的治疗试剂、受到化学增效作用的治疗试剂、稀释剂、赋形剂、溶剂系统或药物传递系统,其中相较于一未修饰烷基化己醣醇衍生物,所述组合物具有对于一抗TKI肿瘤或与AHI1基因的突变或异常调控相关的恶性肿瘤的治疗增加疗效或减少副作用;
(iii)并入于一剂型之中的一烷基化己醣醇衍生物、一修饰烷基化己醣醇衍生物或者一烷基化己醣醇衍生物或一修饰烷基化己醣醇衍生物的衍生物、类似物或前驱物的治疗有效量,其中相较于一未修饰烷基化己醣醇衍生物,并入于所述剂型之中的一烷基化己醣醇衍生物、一修饰烷基化己醣醇衍生物或者一烷基化己醣醇衍生物或一修饰烷基化己醣醇衍生物的衍生物、类似物或前驱物具有对于一抗TKI肿瘤或与AHI1基因的突变或异常调控相关的恶性肿瘤的治疗增加疗效或减少副作用;
(iv)并入于一剂量套组及包装之中的一烷基化己醣醇衍生物、一修饰烷基化己醣醇衍生物或者一烷基化己醣醇衍生物或一修饰烷基化己醣醇衍生物的衍生物、类似物或前驱物的治疗有效量,其中相较于一未修饰烷基化己醣醇衍生物,并入于所述剂量套组及包装之中的一烷基化己醣醇衍生物、一修饰烷基化己醣醇衍生物或者一烷基化己醣醇衍生物或一修饰烷基化己醣醇衍生物的衍生物、类似物或前驱物具有对于一抗TKI肿瘤或与AHI1基因的突变或异常调控相关的恶性肿瘤的治疗增加疗效或减少副作用;以及
(v)受到一原料药产品改良的一烷基化己醣醇衍生物、一修饰烷基化己醣醇衍生物或者一烷基化己醣醇衍生物或一修饰烷基化己醣醇衍生物的衍生物、类似物或前驱物的治疗有效量,其中相较于一未修饰烷基化己醣醇衍生物,受到所述原料药产品改良的所述烷基化己醣醇衍生物、一修饰烷基化己醣醇衍生物或者一烷基化己醣醇衍生物或一修饰烷基化己醣醇衍生物的衍生物、类似物或前驱物具有对于一抗TKI肿瘤或与AHI1基因的突变或异常调控相关的恶性肿瘤的治疗增加疗效或减少副作用。
所述组合物比如可以包含:一并用药,包含(除其他选择方案外):一BH3 类似物、一受到化学敏化作用的治疗试剂、一受到化学增效作用的治疗试剂、一原料药产品改良、一稀释剂、一溶剂系统、一赋形剂、一剂型、一剂量套组及包装、一药物传递系统、一治疗试剂的改质、一前驱物系统或一多重药物系统。
附图说明
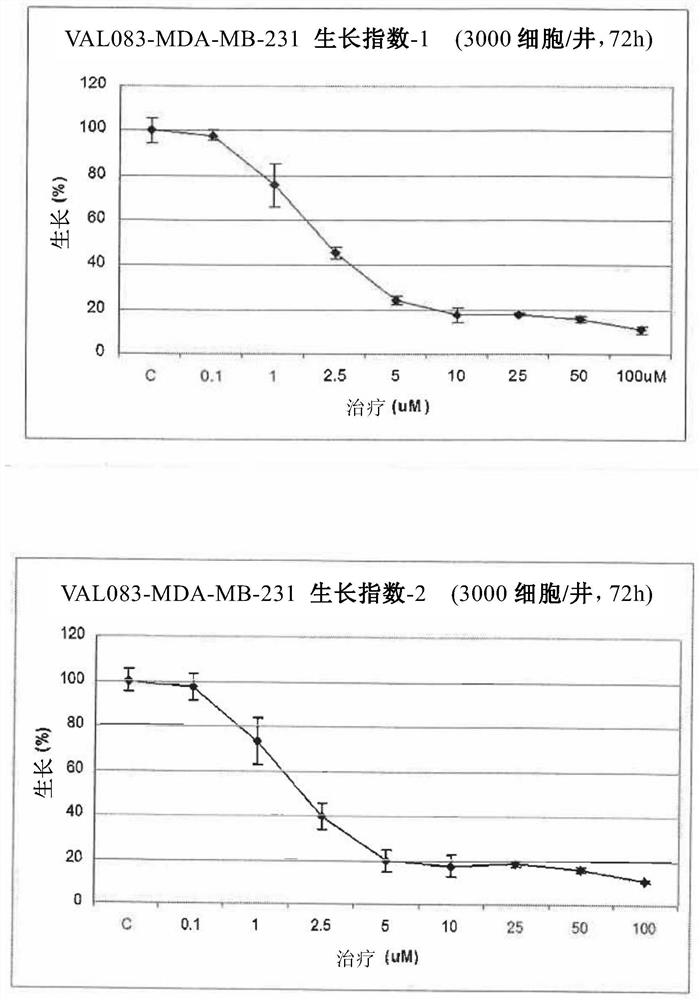
图1是于MDA-MB-231癌细胞生长上卫康醇的作用的曲线图(卫康醇浓度0.1~100μM、3000细胞数目/孔(cells/well)、72小时(二重复))。
图2是于HC C1143癌细胞生长上卫康醇的作用的曲线图(卫康醇浓度0.1 ~100μM、3000cells/well、72小时(二重复))。
图3是于K562癌细胞生长上卫康醇的作用的曲线图(卫康醇浓度0.1~ 100μM、2000cells/well、72小时(二重复))。
图4是于K562-Ahi-1癌细胞生长上卫康醇的作用的曲线图(卫康醇浓度 0.1~100μM、2000cells/well、72小时(二重复))。
具体实施方式
本发明提供多个在对TKI化学疗法有抗性的病患中可治疗恶性肿瘤的方法及药物组合物,特别在抗性是由于基因多型性的病患上。本发明进一步提供多个可治疗与AHI1基因的突变或异常调控相关的恶性肿瘤(特别是白血病) 的方法及药物组合物。本发明进一步提供多个可治疗三阴性乳癌的方法及药物组合物。
最新的研究已证实TKI化学疗法抗性至少部分是由于影响对TKI的细胞凋亡反应的基因多型性。
明确地说,这些多型性包含但不必限于在基因BCL2L11中的多型性(也称为BIM),其编码一BCL-2家族成员的单一BH3区段蛋白。所述单一BH3 区段蛋白活化细胞死亡是通过抵抗所述BCL2家族(BCL2、BCL2-like 1 (BCL-XL,也称为BCL2L1)、骨髓细胞白血病序列-1(myeloid cell leukemia sequence 1,MCL1)及BCL2相关蛋白A1(BCL2-related proteinA1,BCL2A1)) 的促活成员,或通过连接至所述促细胞凋亡BCL2家族成员(BCL2关联X蛋白(BCL2-associated X protein,BAX)及BCL2-拮抗/杀伤因子 (BCL2-antagonist/killer1,BAK1)),而直接地活化其促细胞凋亡功能;促细胞凋亡功能的活化将会导致细胞死亡(R.J.Youle及A.Strasser,“BCL-2蛋白家族:抵抗活化介导细胞死亡”,Nat.Rev.Mol.Cell.Biol.9:47-59(2008),并通过引用将其包括在内)。
以前还曾表明,几个激酶驱动的癌症,比如CML及EGFR NSCLC,可以通过丝裂原活化蛋白激酶1(MAPK-1)-依赖性磷酸化通过抑制BIM转录,亦可通过靶向蛋白酶体降解的BIM蛋白来维持一存活优势。在所有这些恶性肿瘤中,TKIs需要BIM上调节以诱导癌细胞的细胞凋亡,且BIM表现的抑制足以赋予体外(in vitro)TKIs抗性(J.Kuroda等人,“Bim及Bad介导Bcr/Abl
在引起BIM的选择性剪接异构体的生长的BIM基因中,一项最新的发现已经发现了一缺失多型性,所述BIM是缺乏涉及细胞凋亡的促进期的重要 BH3区域。此多型性对CML及EGFR NSCLC细胞的TKI敏感性有深远的影响,如此已删除的对偶基因的一套足以使细胞本质上具有TKI抗药性。因此,此多型性作用于一显性效应以使这样的细胞对于TKI化学疗法具有抗性。此发现还包括结果:与不具多型性的个体相比,具多型性的个体对TKI反应有着显著地不佳。特别是,多型性的存在是与于TKI中对伊马替尼、TKI的较少反应程度有关,同时与于EGFR NSCLC中一具有EGFR TKI疗法的较短的无恶化存活期(progression-freesurvival,PFS)有关(K.P.Ng等人,“在癌症中一通常的BIM缺失多型性调和先天抗药性且对酪氨酸激酶抑制剂反应较差”,Nature Med.doi 10.138/nm.2713(March 18,2012),并通过引用将其包括在内)。
在CML中要识别这些新的TKI抗药性机制,执行双末端双标签的大量平行DNA定序以审视源自于目标对象的五个CML样品的基因组,所述目标对象是对具TKIs的治疗有敏感性或抗性。在所有CML样品中发现 BCR-ABL1易位,但在源自于完全缓解的病患的控制样品中没有被发现。
尽管对于所有抗TKI样品通常的各种结构变异被发现了,一种特别的变异被认为是特别显著的。这个变异发生在BIM基因的内含子2且被包含于一个相同的2903-bp缺失,所述相同的2903-bp缺失对于所有从有抗性的病患的三个样品是通常的;事实上,在所有三个表明为生殖细胞系及多型的病患中,这个变异是相同的。筛选中发现,在东亚血统的病患中此多型性显著频率的发生(但在非洲或欧洲血统的个体中是缺乏的)。
BIM基因的结构的检查表明,因为终止密码子及外显子(exon)3内多聚腺苷酸化信号的存在,所述外显子3的剪接(splicing)及外显子4的剪接发生在一相互排斥的方式。于CML细胞中所有可辨认的BIM转录体的定序已经证实外显子3及外显子4从未存在于相同的转录体。由于在外显子3的5’端上,其接近(107bp)于内含子外显子边界,推断如上所述的缺失多型性将会导致外显子3的优先剪接在外显子4之上。当一微基因被构建以评估缺失的是否存在,将会导致外显子3的选定的内含物在外显子4之上;在此模式系统中,对于外显子4之上的外显子3的内含物至少有五倍的偏好。于此模式系统中的结果是通过初级CML细胞的研究而证实的;对含多型性CML细胞显示相同的偏好,而一般的BIM转录没有受到多型性而影响。在由正常健康 HapMap的个体所得到的淋巴母细胞株中得到相似的结果,表明所述多型性具有一细胞系非依赖性反应。因此,这些结果表明,2.9-kb缺失区域是包含抑制BIM外显子3的剪接的顺式组件,其在包藏所述缺失的细胞中导致外显子3的优先剪接在外显子4之上。
促细胞凋亡的BH3区域被编码是完全通过BIM的外显子4(M.Adachi等人,“动力蛋白轻链连结单一BH3区段蛋白Bim异构体的命名法”,Cell Death Different.12:192-193(2005),并通过引用将其包括在内)。为了BIM的细胞凋亡功能,此区域是必需的(E.H Cheng等人,“防止BAX媒介细胞凋亡及 BAK媒介细胞凋亡的仅BCL-2、BCL-X(L)隔离BH3区域分子”,Mol.Cell.8: 705-711(2001);D.C.Huang及A.Strasser,“单一BH3区段蛋白:细胞凋亡的细胞死亡的基本启动”,Cell 103:839-842(2000),所有的这些皆通过引用将其包括在内)。这些观察结果说明一用于TKI抗药性的以往未知的机制。在此机制中,曝露于TKI后,含多型性CML细胞,并且,可以想象,其他携带此多型性或其他改变BIM剪接的多型性的恶性肿瘤细胞,将有利于含外显子-3的BIM转录体的表现在含外显子-4的BIM转录体之上,而导致含BH3 的BIM异构体的表现降低,且因此损害BH3-区域-依赖性细胞凋亡。为了证实这一点,一日本细胞株,KCL22(I.Kubonishi及I.Miyoshi,“在急性转化期中一由慢性骨髓性白血病的Ph1染色体阳性细胞株的建立”,Int.J.Cell Cloning 1:105-117(1983),并通过引用将其包括在内)其中包含测试2.9-kb缺失;证实相较于不具有所述缺失的细胞,从那行细胞表现出外显子3至外显子4转录体比率的增加。曝露于TKI后,这些KCL22细胞还表现出含有外显子4的转录体的诱导减少,同时表现出BIMEL蛋白及一主要的含BH3的 BIM异构体的浓度下降(M.Adachi等人.(2005))。
符合这些发现,曝露于伊马替尼(尽管为有效的BCR-ABL1抑制)后, KCL22细胞对伊马替尼诱导细胞凋亡具有抵抗,且显示出损害细胞凋亡信号,如在BCR-ABL1-相关性讯息传递中减少所证实的。增加含外显子4的 BIM异构体及编码BH3(但没有含有外显子3)BIM异构体的表现后,KCL22 细胞还对细胞凋亡的诱发有高度敏感性。依次的,这说明于KCL22细胞中受损的伊马替尼诱导细胞凋亡可以通过一类BH3药物的增加而回复,所述类 BH3药物是通过结合及抑制促活BCL2家族成员而功能地模仿单一BH3区段蛋白(M.S.Cragg等人,“通过BH3类似物释放致癌基因激酶的抑制剂的能力”,Nat.Rev.Cancer 9:321-326(2009),并通过引用将其包括在内)。
这些BH3类似物的其中之一为ABT-737。ABT-737是4[4-[(4’–氯[1,1’- 联苯]-2-基)甲基]1-哌嗪基]-N-[[4-[[(1R)-3-二甲胺基)-1-[(苯硫基)甲基]丙基] 胺基]-3-硝基苯基]磺酰基]苯甲酰胺。ABT-737的结构式如下述式(I)所示。 ATB-737的活性被描述于M.F.van Delft等人,“如果中和Mcl-1,所述BH3 类似物ABT-737经由Bak/Bax靶向选择性Bcl-2蛋白且有效地诱发细胞凋亡”,Cancer Cell 10:398-399(2006)及被描述于M.F.Bruncko等人,“有效 Bcl-2及Bcl-xL的双抑制剂的研究”,J.Med.Chem.50:641-662(2007),这两者皆通过引用将其包括在内。
另外的BH3类似物包含但不限于ABT-737的类似物或衍生物,包含下面:(1)ABT-737的类似物,其中以氟、溴或碘取代键结至苯环的氯;(2) ABT-737的类似物,其中以低级烷基取代一或多个苯环的氢;(3)ABT-737的类似物,其中以低级烷基取代一或多个哌嗪基部分的氢;以及(4)ABT-737的类似物,其中以包含一或两个低级烷基团的另一部分取代二甲胺基部分,所述低级烷基团是被连接至二乙胺基部分的胺基。
其他的BH3类似物于本领域中为已知的,且被进一步描述于下面。
在KCL22中一BH3类似物的使用确实回复了伊马替尼诱导细胞凋亡。结果也证实,含有外显子3的转录体的抑制siRNA媒介基因表现对于伊马替尼不会敏化KCL22细胞,表明在TKI抗药性中含有外显子3的异构体可能没有扮演一重要作用。
在原始伊马替尼-敏感性K562 CML细胞的BIM基因中,锌指核酸酶(zinc fingernuclease,ZFN)所促进的基因靶向用来精确地重建缺失多型性。此靶向开始进行之前,这些K562细胞缺乏所述缺失多型性且对伊马替尼是敏感的;所述细胞通过启动细胞凋亡而对伊马替尼有反应。ZFN所促进的基因靶向后,在BIM剪接及表现中,然后对变化及TKI诱发细胞凋亡分析了所述细胞。生成次表现株(subclone)是用于缺失多型性的异质接合子(K562-BIM
在含细胞多型性使用或不使用具有伊马替尼的治疗中,再表现了最丰富的BIM异构体-BIMEL。类似于以ABT-737所看到的结果,在含缺失K562 细胞中,BIMEL的强迫表现还增强伊马替尼的能力而活化细胞凋亡。同样地,相较于不具有缺失的细胞,从具有缺失多型性的目标对象所得到的初级CML 细胞通过细胞凋亡对于伊马替尼诱导死亡的敏感性较低,而且含细胞缺失多型性的相对TKI抗药性可以通过所述BH3类似物ABT-737的增加而克服。
这些结果已证实所述BIM缺失多型性通过远离含有BH3的异构体的偏移剪接损害对伊马替尼的细胞凋亡反应,且此偏差足以使CML细胞对于伊马替尼具有本质上的抗性。这些结果也已经证实在通过以BH3类似物治疗含细胞多型性中,对伊马替尼的细胞凋亡反应可以被回复,所述BH3类似物比如为ABT-737,但本发明并不局限于此。
如上所述,TKI疗法抗性是特别流行在东亚血统的目标对象的。因此,在具有CML的东亚目标对象中TKI反应上的缺失多型性的影响上,一回顾性分析已经被执行。此分析涉及一组具有由新加坡、马来西亚或日本同类者的慢性期CML的新诊断病患。在这些患者中,在具有及不具有缺失多型性的个体中,所述临床反应对于具有一伊马替尼的标准剂量(400mg/day)的第一线疗法进行了比较。临床反应是依照欧洲白血病网(EuropeanLeukemiaNet, ELN)准则分类的;根据所述ELN准则(其在BCR-ABL1转录体水平中包含从未实现一完整的细胞发生反应或3-log减少的目标对象),有抗性的个体被定义为“不理想反应”或“失败”,而敏感的个体是符合ELN定义的“最优选反应物”。在两个地理的同类者中,与控制组相比,具有缺失多型性的目标对象相较于敏感性疾病更有可能具有阻抗疾病;具有缺失多型性的病患之间的阻抗疾病相较于没有的那些,其整体比值比为2.94,表现出一显著的结果。相比之下,两组之间无显著差异是关于其他潜在预后的或混杂因子,包含由诊断至启动伊马替尼治疗的平均时间、在诊断上的Sokal分数或具有干扰素的前治疗。还要注意的是,大多数具有缺失多型性的有抗性的目标对象后来没有以伯舒替尼、尼罗替尼或达沙替尼来反应第二代TKI疗法,一与在细胞株中所观察的先天抗药性一致的发现,而且一表明缺失多型性所赋予的抗药性的发现不限于伊马替尼,但延伸到其他的TKI治疗试剂。
于CML中TKI抗药性与BCR-ABL1激酶区域中,体细胞突变的获得是最通常相关的,其于疾病的慢性期中可以发现高达50%的有抗性的个体(P.La Rosée及A.Hochhaus,“于慢性骨髓性白血病中伊马替尼的抗性:机制及临床意义”,Curr.Hematol.Malig.Rep.3:72-79(2008),并通过引用将其包括在内)。然而,以上所描述的所述缺失多型性为生殖细胞系,且足以引起体外(in vitro)先天TKI抗药性,即使在激酶-区域突变的缺乏中,预测具有生殖细胞缺失多型性的个体对于TKI疗法是有抗性的。在一个研究来验证这个假设中,所述目标对象被区分为下面三个临床组:(1)不具有一BCR-ABL1突变的抗药性;(2)具有一BCR-ABL1突变的抗药性;以及(3)敏感性。具有缺失多型性的个体,与没有的那些相比,其相较于相结合的第(2)及(3)组更可能是第(1) 组。在产生TKI疗法抗性中,此为缺失多型性的体内(invivo)作用的强而有力的证据。
在另一激酶驱动癌症(亦即EGFR NSCLC)中,BIM生物标志物的作用也已经被验证,其中在以EGFR抑制剂所治疗的病患中,于EGFR中敏感的突变预测高反应率(J.G.Paez等人,“在肺癌中EGFR的突变:吉非替尼疗法 (Gefitinib Therapy)临床反应的相关性”,Science304:1497-1500(2004);T.J. Lynch等人,“在对于吉非替尼的非小细胞肺癌的表皮生长因子受体潜在反应能力中活化突变”,N.Engl.J.Med.350:2129-2139(2004),这两者皆通过引用将其包括在内),且其中BIM表现对于TKI敏感性是必需的。EGFR抑制剂包含但不限于吉非替尼、厄洛替尼、西妥昔(cetuximab)、拉帕替尼、帕尼单抗(panitumumab)及凡德他尼(vandetanib)。一非小细胞肺癌的额外及相关方面为,它是特别常见于东亚国家,其中活化EGFR突变可以高达50%的 NSCLCs(西方国家则为15%),且在女性东亚非吸烟者之间是丰富的。
因此,对于包藏TKI敏化EGFR突变的NSCLC细胞株进行搜寻,但搜寻为莫名的TKI抗药性(定义为缺乏任何已知的赋予次发抗药性的突变)。一个这样的细胞株,HCC2279,被鉴定,其显著地没有活化细胞凋亡(尽管为有效的EGFR抑制)。在HCC2279细胞中,所述缺失多型性的存在被证实;于 BIM功能上多型性的存在的作用也被确定。与没有多型性的细胞相比,相较于含外显子4(因此含有BH3)的BIM异构体,所述缺失导致含外显子-3的BIM 异构体的较大表现。尤其是,主要的外围血液单核细胞来自于具有EGFR NSCLC的目标对象,且有或没有所述缺失多型性,其还显示相同的结果(在具有多型性的细胞中,提高了含有外显子3的BIM异构体的表现)。曝露于 TKI后,HCC2279细胞还减少了含有外显子4的转录体及BIMEL蛋白的诱导,同时损害了通过聚ADP核糖聚合酶(PARP)切割测量的细胞凋亡信号的活化。符合所述观念,TKI抗药性是一含BH3的BIM蛋白的浓度下降的结果,如上所述的此类BH3药物ABT-737的增加增强了TKI诱发细胞凋亡信号及细胞死亡。为了确认于EGFR NSCLC中所述缺失多型性足以引起TKI 抗药性,所述缺失多型性被引入至TKI敏感性PC9细胞中。类似于关于 K562-BIM
在具有活化EGFR突变的NSCLC的目标对象中,一研究还被进行以确定与对EGFRTKIs的反应时间相关的缺失多型性是否存在。关于已知的预后因子,有或没有缺失多型性的个体并无差异,包含阶段(超过85%的目标对象为阶段IV)。然而,在具有缺失多型性的个体中,多型性的存在以中位数PFS 的6.6个月(相较于没有的那些的11.9个月)预测一明显较短的无恶化存活期 (PFS)。在使用Cox回归模型的多变量分析中,只有所述缺失多型性及所述 TKI抗药性外显子20突变的存在(J.Wu等人,“具有表皮生长因子外显子20 突变的肺癌与不好的吉非替尼治疗反应是相关的”,Clin.Cancer Res.14: 4877-4882(2008);H.Sasaki等人,“于日本肺癌中EGFR外显子20插入突变”,Lung Cancer 58:324-328(2007),这两者皆通过引用将其包括在内)成为较短的PFS用独立预后因子。
这些结果表明了下述本质,虽然癌症应根据它的体细胞后天驱动突变而归类,生殖细胞多型性对于标靶治疗可以直接地调节这样的癌症的反应,且可以强烈地影响临床结果。特别是,这些结果表明,一共同的BIM缺失多型性有助于分子定义病患之间所看到的反应的异质性,所述病患具有一以标靶治疗所治疗的特定类型的癌症。在不同的癌症中,这些结果也强调了单一生殖细胞多型性可以如何强烈地影响临床结果,所述癌症在这些恶性肿瘤内调解TKI敏感性中共享一个共同的或大体上共同的生物学且可能反应了BIM 的核心作用。这很可能包括其他的也取决于TKI敏感性的BIM表现的恶性肿瘤(P.M.Gordon及D.E.Fisher,“在c-KIT-依赖性胃肠道基质瘤细胞株内调解伊马替尼诱导细胞凋亡中的促凋亡因子BIM的作用”,J.Biol.Chem.285: 14109-14114(2010);B.Will等人,“在JAK2突变的人类红血球中通过Bim 介导JAK2抑制所诱发的细胞凋亡以及通过所述BH3类似物ABT-737提高 JAK2抑制所诱发的细胞凋亡”,Blood 115:2901-2909(2010),这两者皆通过引用将其包括在内)。
只有在东亚血统的个体中发现所述BIM缺失多型性。因此,有趣的是,在CML中,相较于欧洲及北美中的个体(26%),东亚中的个体(~50%)之中对于伊马替尼的不完证的细胞生成反应的高比例已经被报导。据估计,在~21%的东亚患者中,所述缺失多型性构成抵抗性;这或许可以解释,在某种程度上,于完整细胞生成反应率中的差异是在这两个世界人口之间观察到的。
如用于TKI抗药性的生殖系生物标志物,在含有后天的(亦即体细胞)突变的生物标志物之上,所述BIM缺失多型性还提供了几个优势。首先,所述 BIM缺失多型性在初期表现的时间可以用来预测个体在增加发展TKI抗药性的风险。其次,因为所述缺失多型性是一不关于任何特定肿瘤的DNA或肿瘤的种类的生殖细胞多型性,所以个体的多型性状态的评估不需要肿瘤特异性DNA的分析。在病患的初期表现时间上用于此缺失多型性筛选的能力通过治疗手段提供了用于防止TKI抗药性出现的潜能,比如在初期表现时间上或在对于一或多个TKI治疗药物产生抗性的首次迹象上给予类BH3药物。事实上,在实质肿瘤情况(比如EGFR NSCLC)中,所述缺失多型性是一生殖细胞多型性,所述生殖细胞多型性与任何特定肿瘤的DNA是不相关的;或者所述生殖细胞多型性中的肿瘤类型是特别有利的,其中用于肿瘤特异性组织的第二次活组织检查通常需要一种侵入性手术,这样的过程可能带来的风险,包括感染。治疗预测TKI反应性之前,在肿瘤中,这些诊断测量可以与BIM RNA水平的测量一起进行(A.Faber等人,“于未曾治疗癌症中BIM表现预测对激酶抑制剂的反应性”,CancerDiscov.1:352-365(2011));然而,曝露于TKI后,如上所述的缺失多型性的发现强调生物标志物的重要性,所述生物标志物还可以预测BIM的功能性异构体的诱导。
结果如上所述,在BIM功能上说明所述缺失多型性的作用中,于CML 及EGFR驱动的NSCLC中还描述一新颖的剪接机制,通过这样的剪接机制所述多型性有助于抗药性。这表明,在这两个癌症中,BIM功能的药理恢复可以克服此特定形式的TKI抗药性。在人类疾病内基因的剪接样式中,这些结果也支持改变的逐渐公认作用(L.Cartegni等人,“倾听沉默及了解无义:外显子突变影响剪接”,Nat.Rev.Genet.3:285-298(2002);N.López-Bigas 等人,“剪接突变是遗传性疾病的最常见原因吗?”,FEBS Lett.579: 1900-1903(2005),这两者皆通过引用将其包括在内)且提供一继承生殖细胞突变的新例子,所述继承生殖细胞突变对于标靶癌症治疗有助于抵抗。尽管所述缺失多型性的存在与临床TKI抗药性及较短的PFS是密切相关的,其他的基因因子,后天及继承两者,在任何个体病患中将可能决定TKI疗法的最终反应。EGFR非依赖型抗药性的几个其他机制已被描述,包含上调肝细胞生长因子相关性讯息传递(S.Yano等人,“肝细胞生长因子诱发具有表皮生长因子受体活化性突变的肺腺癌的吉非替尼抗药性”,Cancer Res.68: 9479-9487(2008),并通过引用将其包括在内),活化类B细胞(NF-κB)相关性讯息传递的核因子κ轻链强化子(T.G.Bivona等人,“在突变的EGFR上肺癌的FAS及NF-κB信号调节依赖性”,Nature 471:523-526(2011),并通过引用将其包括在内)以及v-Ki-ras2 Kirsten大鼠肉瘤病毒癌基因同源物(KRAS) 突变(M.Takeda等人,“在具有非小细胞肺癌的EGFR-突变阳性患者中对表皮生长因子受体酪氨酸激酶抑制剂的再一次(De Novo)抗药性”,J.Thorac. Oncol.5:399-400(2010),并通过引用将其包括在内)。
临床TKIs抗性通常被归类为原发性或次发性,其中后者定义为发生于对TKI疗法经历一初步反应的个体,然后发展成抗药性。通常认为,次发抗药性是通过后天体细胞突变而介导的,所述后天体细胞突变在TKI疗法的选择性压力下出现,然而抗药性的先天机制,包含生殖细胞多型性,其更可能呈现原发抗药性及缺乏任何前期反应。这种推理方法是基于这样的假设:赋予抗性的生殖细胞多型性引起绝对而非相对TKIs抗性。然而,述结果表明,通过创造具有所述缺失多型性的CML及EGFR NSCLC细胞两者,结果表明:所述BIM多型性引起相对的TKI抗药性。在BIM蛋白浓度中,此与对小变化敏感的癌细胞是一致的(Kuroda等人(2006);A Egle等人,“Bim是一Myc 诱导的小鼠B细胞白血病的抑制基因”,Proc.Natl.Acad.Sci.USA 101: 6164-6169(2004),并通过引用将其包括在内)。这意味着,携带缺失多型性的细胞对于TKIs没有完全的抗药性,且在某些情况下,可能会出现一些反应。
这些结果还表明,其他的多型性可以解释癌症生物学的其他方面之间的异质性。还有可能是于特定人群中这些多型性可能存在或可能发生在更高的频率,特别是已知于本质上同系交配的人群。许多这类人群是已知的,并且,在某些情况下,这些人群中已被证明具有增加特定类型的癌症的风险。特别是,如下所述,本文中所述的具有烷基化己醣醇衍生物的疗法在具有生殖细胞突变或体细胞突变的病患中是有用的。
在一家族被称为具有促细胞凋亡活性及抗细胞凋亡活性两者的BCL-2 家族中,所述BH3区域是许多蛋白之间所共享的区域。这些蛋白除了BIM 以外包含BAK、BAX、BIK、BID及HRK。所述BH3区域是保留于促细胞凋亡BCL-2家族蛋白及抗细胞凋亡BCL-2家族蛋白两者中。所述促细胞凋亡蛋白的BH3区域提供一双重功能。它是其细胞死亡活性及调节具有抗细胞凋亡蛋白的异源二聚合作用的必要条件。
天然人类BIMEL的序列,BIM的主要形式,其如下:
MAKQPSDVSSECDREGRQLQPAERPPQLRPGAPTSLQTEPQGNPEGNHGG EGDSCPHGSPQGPLAPPASPGPFATRSPLFIFMRRSSLLSRSSSGYFSFDTDRS PAPMSCDKSTQTPSPPCQAFNHYLSAMASMRQAEPADMRPEIWIAQELRRI GDEFN AYYARRVFLN NYQAAEDHPRMVILRLLRYIVRLVWRMH(SEQ ID NO:1)。
此蛋白中的BH3区域是残基148-162(15个胺基酸),其具有序列 IAQELRRIGDEFNAY(SEQ ID NO:2)。于BIMEL中类似于BH3部分的序列被发现于其他蛋白中,包含:在BIK中的LACIGDEMD(SEQ ID NO:3)、在 HRK中的LKALGDELD(SEQ ID NO:4)、在BAK中的LAIIGDDIN(SEQ ID NO:5)、在BID中的LAQVGDSMD(SEQ ID NO:6)、在BAX中的 LKRIGDELD(SEQ ID NO:7)、在BNIP3中的LKKNSDWIW(SEQ ID NO:8)、在BAD中的LRRMSDEFE(SEQ ID NO:9)、在BCL-2中的LRQAGDDFS (SEQ ID NO:10),以及在BCL-XL中的LREAGDEFE(SEQ ID NO:11)。在通常中这些序列具有多个胺基酸,包含一初级白胺酸(L),相应于BIMEL中所述BH3区域的第五个胺基酸;以及一天冬胺酸(D),相应于BIMEL中所述 BH3区域的第十个胺基酸。其他相同或适当地取代的胺基酸发生于这些序列中。这些同源性被描述于M.Yasuda等人,“腺病毒E1B-19K/BCL-2相互作用的蛋白BNIP3包含一BH3区域及一粒线体靶向序列”,J.Biol.Chem.273:12415-12421(1998),并通过引用将其包括在内。
如上所述,许多BH3类似物已经被发现,包含ABT-737。BH3类似物被描述于G.Lessene等人,“用于癌症疗法的BCL-2家族拮抗物,”Nature Rev. Drug Discovery 7:989-1000(2007),并通过引用将其包括在内。
在哺乳动物细胞中,通过促活及促凋亡蛋白(特别是蛋白的BCL-2家族的成员)之间的交互作用来控制细胞凋亡的发生或非发生。在哺乳动物细胞中,五个促活蛋白,BCL-2、BCL-X
所述五个促活蛋白和BAK及BAX所有共享四个序列同源性的区域(称为BCL-2同源区1(BCL-2homology 1,BH1)、BH2、BH3及BH4)一样。它们还具有一羧基端膜锚定序列及一相似三级结构。然而,还存在其他编排细胞凋亡的蛋白。这些额外的具有促细胞凋亡功能的蛋白为仅有BH-3蛋白(BH-3 only proteins),指明,因为它们缺乏BH1、BH2及BH4区域。在哺乳动物中,八个单一BH3区段蛋白是已知的,亦即BIM、BID、PUMA、NOXA、BAD、 BMF、HRK及BIK;在对于胁迫信号的反应中,这些蛋白是通过转录或后转译加工而上调的。
促细胞凋亡蛋白的BH3区域是具有抗凋亡(促活)家族成员的交互作用的主要媒介物。比如,已经被提出,一促活蛋白,BCL-X
单一BH3区段蛋白及促活蛋白之间有选择性交互作用。当BAD及NOXA 具有互补结合剖面的时候,BIM及PUMA连接至所有的五个促活蛋白。具有改变选择性型态的突变BH3序列已经被发现,如下面进一步讨论的。
潜在的BH3类似物包含:
(1)胜肽;
(2)修饰胜肽;
(3)三砒啶基拟胜肽类;
(4)对苯酰胺基拟胜肽类;
(5)苯甲酰脲基拟胜肽类;
(6)欧巴妥拉(obatoclax);
(7)TW37;
(8)(-)棉子酚;
(9)棉子酚衍生物;
(10)异恶唑啉衍生物;
(11)A-385358;
(12)ABT-737;
(13)ABT-263;以及
(14)TM-1206。
适用于BH3类似物的设计的一般原则包含下列:(1)结合作用发生于一坐落于Bcl-X
下面详细描述这些BH3类似物。
修饰胜肽包含钉住的BID BH3螺旋,其被描述于L.D.Walensky等人“钉住(stapled)的BID BH3螺旋直接结合并活化BAX”,Mol.Cell 24:199-210 (2006)及L.D.Walensky等人,“通过一碳氢化物钉住的BH3螺旋活化体内细胞凋亡”,Science 305:1466-1470(2004),这两者皆通过引用将其包括在内。这些钉住的螺旋是通过在两个位置上S-戊烯基丙氨酸衍生物至所述BH3螺旋中的取代、以及在一相邻的位置上用于甲硫胺酸的正白胺酸的取代而产生的。然后,一碳氢化物交联通过钌催化的烯烃置换而生成,留下一具有一个双键的架桥结构。
另一用于修饰胜肽BH3类似物的选择方案包含基于螺旋形的胜肽的折迭体,其描述于J.D.Sadowsky等人,“BH3区域/Bcl-x
三砒啶基拟胜肽类被描述于J.M.Davis等人,“作为a-螺旋类似物的2,3 ˊ;6ˊ,3″-三砒啶支架的合成”,Org.Letters 7:5404-5408(2005),并通过引用将其包括在内。
对苯酰胺基拟胜肽类被描述于H.Yin&A.D.Hamilton,“Bak胜肽标靶 Bcl-xL蛋白的螺旋形区域的作为类似物的对苯酰胺基衍生物”,Bioorg.Med. Chem.Lett.14:1375-1379(2004),并通过引用将其包括在内。
苯甲酰脲基BH3类似物透过Lessene等人被描述于美国专利申请公开第 2008/0153802号,并通过引用将其包括在内。
欧巴妥拉是2-(2-((3,5-二甲基-1H-吡咯-2-基)甲烯基)-3-甲氧基-2H-吡咯 -5-基)-1H-吲哚,且具有显示于下述式(II)的结构。
TW37是N-[(2-叔-丁基-苯磺酰基)-苯基]-2,3,4-三羟基-5-(2-异丙基-苄基)-苯甲酰胺,且具有显示于下述式(III)的结构,且被描述于G.P.Wang等人,“抗凋亡Bcl-2蛋白的有效小分子抑制剂的基于结构的设计”,J.Med.Chem. 50:3163-3166(2006)且被描述于M.A.Verhaegen等人,“一新颖BH3类似物公开了一p53及活性氧类所控制的黑色素瘤细胞死亡的丝裂原活化蛋白激酶依赖性机制”,Cancer Res.66:11348-11359(2006),这两者皆通过引用将其包括在内。
另外的BH3类似物包含但不限于TW37的类似物及衍生物,包含下面: (1)TW37的类似物,其中以低级烷基取代一或多个苯环的氢;以及(2)TW37 的类似物,其中以低级烷基取代一或多个于三羟苯基部分的羟基团中的氢。
ABT-263是(R)-4-(4-((4’-氯基-4,4-二甲基-3,4,5,6-四氢-[1,1’-联苯]-2-基)甲基)哌嗪-1-基)-N-((4-((4-吗啉基-1-(苯硫基)丁-2-基)胺基)-3-((三氟代甲基) 磺酰基)苯基)磺酰基)苯甲酰胺,且具有显示于下述式(IV)的结构。ABT-263 的活性被描述于C.Tse等人,“ABT-263:一有效的及口服的生物可利用Bcl-2 家族抑制剂”,Cancer Res.68:3421-3428(2008)及被描述于A.R.Shoemaker 等人,“在一组小细胞肺癌异种移植模式中Bcl-2家族抑制剂ABT-263的活性”,Clin.Cancer Res.14:3268-3277(2008)。
另外的BH3类似物包含但不限于ABT-263的类似物或衍生物,包含下面:(1)ABT-263的类似物,其中以氟、溴或碘取代键结至苯环的氯;(2) ABT-263的类似物,其中以低级烷基取代一或多个苯环的氢;(3)ABT-263的类似物,其中以低级烷基取代一或多个哌嗪基部分的氢;以及(4)ABT-263的类似物,其中以包含一或两个低级烷基团的另一部分取代二甲胺基部分,所述低级烷基团是被连接至二乙胺基部分的胺基。
一具有部分结构同源性(具有ABT-737)的化合物是A-385358。A-385358 是[(R)-4-(3-二甲胺基-1-苯基磺酰基甲基-丙基胺基)-N-[4-(4,4-二甲基-哌啶-1- 基)-苄酰基]-3-硝基-苯磺酰胺,且具有如下面分子式(V)所示的结构。A-385358 的活性被描述于A.R.Shoemaker等人,“一Bcl-XL的小分子抑制剂增强体外及体内细胞毒杀药物的活性”,Cancer Res.66:8731-8739(2006),并通过引用将其包括在内。
另外的BH3类似物包含但不限于A-385358的类似物或衍生物,包含下面:(1)A-385358的类似物,其中以低级烷基取代一或多个苯环的氢;以及 (2)A-385358的类似物,其中以包含一或两个低级烷基团的另一部分取代二甲胺基部分,所述低级烷基团是被连接至二乙胺基部分的胺基。
棉子酚的(-)对映异构物已被证明具有BH3类似的活性;此对映异构物的 BH3类似的活性被描述于J.P.Qiu等人,“通过棉子酚棉子酚对映异构物的细胞杀伤的不同途径”,Exp.Biol.Med.227:398-401(2002),并通过引用将其包括在内。
在BH3类似的活性方面,棉子酚衍生物及类似物的活性被描述于G.Z. Tang等人,“抗细胞凋亡Bcl-2蛋白的作为抑制剂的酰基苯三酚 (Acylpyrogallols)”,J.Med.Chem.51:717-720(2008),并通过引用将其包括在内。一个特别显著的棉子酚衍生物是阿珀棉酚(apogossypolone);阿珀棉酚的BH3类似的活性被描述于A.A.Arnold等人,“阿珀棉酚的临床前研究:在囊泡状小型有裂隙细胞淋巴瘤模式中,一个新的Bcl-2、Bcl-X-L及Mcl-1蛋白的非肽Pan小分子抑制剂”,Mol.Cancer 7:20-30(2008),并通过引用将其包括在内。
TM-1206具有显示于下述式(VI)的结构。TM-1206的BH3类似的活性被描述于G.Z.Tang等人(2007)。
另外的BH3类似物包含但不限于TM-1206的类似物或衍生物,包含下面:(1)TM-1206的类似物,其中以低级烷基取代一或多个苯环的氢;以及(2) TM-1206的类似物,其中以低级烷基取代一或多个于三羟苯基部分的羟基团中的氢。
其他的BH3类似物于本领域中为已知的。
在具有上述生殖细胞缺失多型性的病患中,一系列的提供选择性治疗方式及作为烷化剂的药剂可以治疗抗TKI恶性肿瘤。当问题的严重程度被考虑,这些用以治疗抗药性恶性肿瘤的选择性药剂的能力是显著的。如上所述,在东亚血统的人(约15%的情况下)中,上述生殖细胞缺失多型性被认为是起因于TKIs抗性,比如格列卫(Gleevec)。这意味着,在所述类别中,出约552,000 病患,每年发生于携带有生殖细胞缺失多型性的病患中有至少约78,000肺癌病例,从而对比如为格列卫的TKIs产生抗性。此外,据估计,每100,000人口至少有1例的耐药性慢性骨髓细胞性白血病(CML),或每年约2500例;这是一个非常保守的估计,在承载生殖细胞缺失多型性的人群中耐药性的CML 的病例数很可能更高。
这些药剂,进一步如下所述,其也可以用在其他抗TKI的恶性肿瘤的治疗,其中一或多个突变,生殖细胞突变或影响特定细胞或组织形式的体细胞突变的任一个,防止所述BIM蛋白或另一蛋白(在其中磷酸化对于TKI启动细胞凋亡是必须的)的磷酸化。
可成功地使用于具有生殖细胞缺失多型性的病患的一类治疗试剂为半乳糖醇、取代的半乳糖醇(galacitols)、卫矛醇及取代的卫矛醇,包含卫康醇、二乙酰二脱水卫矛醇、二溴卫矛醇及其衍生物及类似物。
这些半乳糖醇、取代的半乳糖醇、卫矛醇及取代的卫矛醇为烷化剂或烷化剂的前驱物,如下面进一步讨论的。
卫康醇的结构如下述式(VII)所示。
此外,在本发明的范围之内具有卫康醇的衍生物,比如其具有一或两个以低级烷基取代的卫康醇的两个羟基团的氢、其具有一或多个以低级烷基取代的连接于两个环氧环的氢,或者其具有存在于卫康醇的甲基团,所述甲基团是被连接于相同的碳,所述碳带有以C
二乙酰二脱水卫矛醇的结构如下述式(VIII)所示。
此外,在本发明的范围之内具有二乙酰二脱水卫矛醇的衍生物,比如其具有一或两个以C
二溴卫矛醇的结构如下述式(IX)所示。二溴卫矛醇可以通过于高温下以氢溴酸与卫矛醇的反应来生成,随后结晶化所述二溴卫矛醇。一些二溴卫矛醇的特性被描述于N.E.Mischler等人,“二溴卫矛醇”,Cancer Treat.Rev.6: 191-204(1979),并通过引用将其包括在内。特别是,二溴卫矛醇,作为一α, ω-二溴化的己醣醇,二溴卫矛醇分配许多生化及生物性质的相似药物,比如二溴甘露醇及甘露醇马里兰。二溴卫矛醇至二环氧化卫康醇的活化发生于体内,且卫康醇可以代表一主要的活性形式的药物;这意味着,二溴半乳糖醇具有许多前驱物的性质。通过口服途径吸收的二溴卫矛醇是迅速的且相当完整。在黑色素瘤、乳房淋巴瘤(何杰金氏(Hodgkins)及非何杰金氏两者)、结肠直肠癌、急性淋巴性白血病中,二溴卫矛醇具有已知的活性;并且,二溴卫矛醇已被证明能够降低中枢神经系统白血病、非小细胞肺癌、子宫颈癌、膀胱癌及转移性血管外皮细胞瘤的发病率。
此外,在本发明的范围之内具有二溴卫矛醇的衍生物,比如其具有一或多个以低级烷基取代的羟基团的氢,或其具有一或两个以另一卤素基(比如氯、氟或碘)取代的溴基团。
如上所述,且正如下面通常所提到的,应用于本发明的方法或组合物的烷基化己醣醇衍生物、BH3类似物及其他治疗活性剂的衍生物及类似物可以以一或多个基团任意地取代,所述基团不会显著影响所述衍生物或类似物的药理活性。这些基团于本领域中通常是已知的。下面提供一些常用的基团(其可以作为任意取代基)的定义;然而,只要满足用于任意取代基的化学及药理的需求,从这些定义的任何基团的省略不能被认为意味着这样的一基团不能被使用作为一任意取代基。
如本文所使用的术语“烷基”指的是一1-20个碳原子的未分枝的、分枝的或环状的饱和烃基残基或其组合,其可以被任意地取代;所述烷基残基仅含有未取代的C及H。通常,所述未分枝的或分枝的饱和烃基残基为1至6 碳原子,其被称为“低级烷基”。当所述烷基残基是环状的,且包含一个环,可以理解的是,所述烃基残基包含至少三个碳原子,其为最小数形成一环。如本文所使用的术语“烯基”指的是一未分枝的、分枝的或环状的烃基残基具有一或多个碳碳双键。如本文所使用的术语“炔基”指的是一未分枝的、分枝的或环状的烃基残基,其具有一或多个碳碳三键;所述残基还可以包含一或多个双键。关于“烯基”或“炔基”的使用,多个双键的存在不能产生芳香环。如本文所使用的术语“羟基烷基”、“羟基烯基”及“羟基炔基”分别指的是含有一或多个羟基团(作为取代基)的烷基团、烯基基团或炔基基团;如下文所述,可以任意地包含进一步的取代基。如本文所使用的术语“芳基”指的是一单环部分或稠合双环部分,其具有众所周知芳香性的特性;例子包含苯基及萘基,其可以被任意地取代。如本文所使用的术语“羟基芳基”指的是一芳基,其包含一或多个作为取代基的羟基团;正如下面进一步所提到的,可以任意地包含进一步的取代基。如本文所使用的术语“杂芳基”指的是具有芳香性的特性的单环系统或稠合双环系统,且包含一或多个选自于氧、硫及氮的杂原子。在5元环以及6元环中,杂原子的内含物允许芳香性。典型的杂芳香系统包含单环的C
如本文所使用的术语“随意取代的”表明任意取代的所提到的所述特定基团(group)或基团(groups)可以不具有非氢取代基,或所述特定基团或基团可以具有一或多个与生成的分子的化学及药理活性一致的非氢取代基。如果不是另有规定,这样的可能存在的取代基的总数是等于存在于描述未取代形式的基团的氢原子的总数;少于这样的取代基的最大数可能存在。其中一任意取代基经由一双键而被连接,比如一羰基氧(C=O),在所述任意取代基连接至所述碳原子上,所述基团占用两个可用价数,所以根据可用价数的数量减少可被包含的取代基的总数。如本文所使用的术语“取代的”,无论是使用作为部分的“随意取代的”,或以其他方式使用,当用来修改一特定基团、部分或自由基,意味着一或多个氢原子是各自彼此独立地以相同或不同的取代基 (含单数或复数)来取代。
在特定基团、部分或自由基中对于取代的饱和碳原子有用的取代基团包含但不限于—Z
同样地,在特定基团、部分或自由基中对于取代的未饱和碳原子有用的取代基团包含但不限于—Za、卤基、—O-、—OZb、—SZb、—S-、—NZcZc、三卤甲基、—CF3、—CN、—OCN、—SCN、—NO、—NO2、—N3、—S(O)2Zb、—S(O2)O-、—S(O2)OZb、—OS(O2)OZb、—OS(O2)O-、—P(O)(O-)2、— P(O)(OZb)(O-)、—P(O)(OZb)(OZb)、—C(O)Zb、—C(S)Zb、—C(NZb)Zb、—C(O)O-、—C(O)OZb、—C(S)OZb、—C(O)NZcZc、—C(NZb)NZcZc、— OC(O)Zb、—OC(S)Zb、—OC(O)O-、—OC(O)OZb、—OC(S)OZb、—NZbC(O)OZb、—NZbC(S)OZb、—NZbC(O)NZcZc、—NZbC(NZb)Zb,以及—NZbC(NZb)NZcZc,其中Za、Zb及Zc是如上所定义的。
同样地,在特定基团、部分或自由基中对于取代的未饱和碳原子有用的取代基团包含但不限于—Z
同样地,在杂烷基及环杂烷基团中对于取代氮原子有用的取代基团包含但不限于—Z
本文所述的化合物可以包含一或多个旋光中心和/或双键,因此,可能存在立体异构物,比如双键异构物(亦即比如为E及Z的几何异构物)、对映异构物或非对映立体异构物。在不同旋光纯度的程度或E及Z的百分比中,本发明包含每个隔离型立体异构形式(比如所述对映异构地纯异构物,所述E 及Z异构物,以及用于立体异构物的其他选择方案)以及立体异构物的混合物,包含外消旋混合物、非对映立体异构物的混合物,以及E及Z异构物的混合物。因此,本文所描述的化学结构包含所示化合物的所有可能的对映异构物及立体异构物,所述化合物包含立体异构的纯形式(比如纯几何异构地、纯对映异构地或纯非对映立体异构地)以及对映异构的及立体异构的混合物。对映异构的及立体异构的混合物可以被分解于它们组成的对映异构物或使用本领域技术人员众所周知的分离技术或旋光合成技术的立体异构物。在不同程度的旋光纯度中,本发明包含每个隔离型立体异构形式以及立体异构物的混合物,包含外消旋混合物。它还包含各种非对映立体异构物。其他结构或许会描绘一特定异构物,但其仅仅是为了方便,且非意在将本发明限制于描述的烯烃异构体。当所述化学名称没有指定化合物的异构形式,它表示为可能的异构形式或这些异构形式的化合物的混合物的任何一个。
所述化合物也可能存在几个互变异构形式,本文描绘的一个互变异构物仅是为了方便,且还需理解包含其他所示形式的互变异构物。因此,本文所描述的化学结构包含所有可能互变异构形式的说明的化合物。如本文所使用的术语"互变异构物"指的是异构物,所述异构物非常轻松地改变成另一种,以致它们可以同时处于平衡状态;所述平衡状态可以强烈地倾向于所述互变异构物的一种,这取决于对稳定性的考虑。比如,酮和烯醇是两个互变异构形式的一种化合物。
如本文所使用的术语“溶剂化物”意指一通过溶合作用形成的化合物(具有溶质的分子或离子的溶剂分子的结合)或一由溶质离子或分子组成的聚集物(亦即本发明化合物具有一或多个溶剂分子)。当水为溶剂时,相应的溶剂化物为“水合物”。水合物的例子包含但不限于半水合物、一水合物、二水合物、三水合物、六水合物及其他含水物种。本领域中的普通技术人员应当理解的是,本化合物的药学上可接受盐类和/或前驱物也可能存在一溶剂化物形式。所述溶剂化物通常是经过水合作用(部分制备本化合物或以本发明无水化合物通过自然吸收水分)而形成的。
如本文所使用的术语“酯”意指本化合物的任何酯类,其中是以--COOR 官能基取代所述分子的任何--COOH官能基,其中所述酯类的R部分为任何的形成一稳定的酯类部分的含碳基团,包含烷基、烯基、炔基、环烷基、环烷基烷基、芳基、芳基烷基、杂环基、杂环烷基及其取代的衍生物,但本发明并不局限于此。本化合物的可水解酯是羧基以可水解酯基团的形式呈现的化合物。亦即,这些酯类为药学上可接受的,且于体内可以水解成羧酸。
除了如上所述的取代基以外,烷基团、烯基基团及炔基基团可以被交换地或额外地被C
“杂烷基”、“杂烯基”及“杂炔基”等等被类似的定义到相应的烃基(烷基、烯基及炔基)基团,但是所述‘杂基’术语指的是包含骨干残基内1-3个氧、硫或氮的杂原子或其组合的基团;因此,通过一特定杂原子取代相应烷基团、烯基基团或炔基基团的至少一碳原子而分别形成杂烷基基团、杂烯基基团或杂炔基基团。由于化学稳定性的缘故,其还可以理解的是,除非另有规定,这样的基团不包括两个以上相邻的杂原子,但有下列情形除外:如在硝基团或磺酰基团中,一氧基团是存在于N或S上。
当如本文所使用的术语“烷基”包含环烷基团及环烷基烷基团的时候,所述术语“环烷基”在此可用于描述一经由环碳原子连接的碳环非芳香基团,且“环烷基烷基”可用于描述一通过烷基连接基连接于所述分子的碳环非芳香基团。
同样地,“杂环基”可用于描述一非芳香环基环,其包含至少一个作为环成员的杂原子(通常选自于氮、氧及硫),且经由一环原子连接于所述分子,其可以是C(碳链结型)或N(氮链结型);且“杂环烷基”可用于描述这样的一个透过一连接基连接于另一分子的基团。所述杂环基可以是充分饱和的或部分饱和的,但非芳香族。可用于环烷基团、环烷基烷基、杂环基及杂环烷基团的尺寸及取代基是相同于那些上述烷基团。所述杂环基团通常包含1、2 或3个作为环成员的杂分子,所述杂原子是选自于氮、氧及硫;在杂环系统中所述N或S可以以常见于这些原子的基团来取代。如本文所使用的这些术语还包括含有一个双键或两个的环,只要连接的环不是芳香族。所述取代的环烷基团及杂环基团还包含稠合至一芳环或杂芳环的环烷环或杂环,提供所述基团的连接的点是至所述环烷环或杂环,而非至所述芳香/杂芳香环。
如本文所使用的“酰基”包含含有烷基、烯基、炔基、芳基或芳基烷基自由基的基团,所述烷基、烯基、炔基、芳基或芳基烷基自由基是连接于一羰基碳原子的两个可用价位置的一个上,且杂酰基指的是所述相应的基团,其中至少一碳(除了羰基碳)已通过一选自于氮、氧及硫的杂原子而取代。
酰基团及杂酰基团通过羰基碳原子的开价而结合于任何基团或分子至它们所连接的。通常,它们为C
同样地,“芳基烷基”及“杂芳基烷基”指的是芳香环系统及杂芳香环系统,所述芳香环系统及杂芳香环系统是通过一连接基团键结至其接合点,所述连接基团比如是伸烷基,包含取代或未取代的、饱和或未饱和的、环状或非环状的连接基。通常所述连接基为C
其中一芳基烷基团或杂芳基烷基团被描述为随意取代基的,所述取代基可以在烷基或杂烷基部分,或者在所述基团的芳基或杂芳基部分。随意地存在于烷基或杂烷基部分的所述取代基与以上通常烷基团所描述的那些是一样的;随意地存在于芳基或杂芳基部分的所述取代基与以上通常芳基所描述的那些是一样的。
在此使用的“芳基烷基”基团为烃基基团(如果它们是未取代的),且在环及伸烷基或相似连接基中其是通过碳原子的总数而被描述的。因此苄基团是一C7-芳基烷基,且苯基乙基是一C8-芳基烷基。
如上所述的“杂芳基烷基”指的是一含有芳基的部分,所述芳基是通过连接基团而被连接,且不同于“芳基烷基”之处在于,至少一芳基部分的环原子或于所述连接基团中的一原子是一选自于氮、氧及硫的杂原子。在所述环与连接基相结合中,依据原子总数,所述杂芳基烷基团被描述于本文中,且它们包含通过杂烷基连接基所连接的芳基团;通过烃基连接基(比如伸烷基) 所连接的杂芳基团;以及通过杂烷基连接基所连接的杂芳基团。如此,比如为C7-杂芳基烷基将包含吡啶甲基、苯氧基及N-吡咯基甲氧基。
在此使用的“伸烷基"指的是一二价烃基基团;因为其为二价,所以其可以一起连接两个其他的基团。通常其指的是—(CH
一般地,任一包含于取代基的烷基团、烯基团、炔基团、酰基团或芳基团或芳基烷基团可自身随意地通过附加的取代基来取代。如果所述取代基不是另有描述,这些取代基的性质与关于主要的取代基它们自己所描述的那些是类似的。
在此使用的“胺基”指的是—NH
如本文所使用的术语“碳环化合物”、“碳环基”或“碳环”指的是一仅包含于所述环中的碳原子的循环环(cyclic ring),而所述术语“杂环”或“杂环的”指的是一包含杂原子的环。所述碳环基可以是充分饱和的或部分饱和的,但非芳香族。比如,所述碳环基包含环烷基。所述碳环及杂环结构包含具有单环、双环或多环系统的化合物;且这样的系统可以混合芳香环、杂环及碳环。混合环系统是根据连接于所述化合物的支架的环而描述的。
如本文所使用的术语“杂原子”指的是任何非碳或氢的原子,比如氮、氧或硫。当它为链或环的骨干或骨架的一部分,一杂原子必须至少为二价,且通常是选自于氮、氧、磷及硫。
如本文所使用的术语“烷酰基”指的是一共价连接于一羰基团(C=O)的烷基团。所述术语“低级烷酰基”指的是一烷酰基团,其中所述烷酰基团的烷基部分为C
如本文所使用的术语“烷氧基”指的是一共价连接于一氧原子的烷基团;所述烷基团可以被认为是取代一羟基团的氢原子。所述术语“低级烷氧基”指的是一烷氧基团,其中所述烷氧基团的烷基部分为C
如本文所使用的术语“磺酸基”指的是一磺酸(—SO
如本文所使用的术语“胺磺酰基”指的是一具有结构—S(O
如本文所使用的术语“羧基”指的是结构—C(O
如本文所使用的术语“胺甲酰基”指的是结构—C(O
如本文所使用的术语“单烷基胺烷基”及“二烷基胺烷基”指的是结构—Alk
如本文所使用的术语“烷基磺酰基”指的是结构—S(O)
如本文所使用的术语“烷氧基羰基”指的是一含有烷基团的酯取代基,其中所述羰基碳是连接于所述分子的点。一例子为乙氧基羰基,其是 CH
其他的取代基的组合于本领域中为已知的,且比如透过Jung等人被描述于美国专利第8,344,162号,并通过引用将其包括在内。比如,所述术语“硫代羰基”及含有“硫代羰基”的取代基的组合包含一羰基团,其中在所述基团内一双键的硫取代正常双键的氧。所述术语“烷叉基”及类似术语指的是烷基团、烯基基团、炔基基团或环烷基团,根据规定,其具有两个从单一碳原子移除的氢原子,以致所述基团被双键结至所述结构的残基。
卫康醇及其他取代的己醣醇具有许多使用于治疗抗TKI非小细胞肺癌 (NSCLC)及慢性骨髓性白血病(CML)的优势。这些药剂可以抑制癌症干细胞 (cancer stem cells,CSC)的生长,且对于O
如下文所述,在TKIs抗性的病患中,卫康醇及其他取代的己醣醇可以被使用来治疗恶性肿瘤。
本发明的一方面是提供一于患有具有一赋予TKIs抗性的生殖细胞缺失多型性的恶性肿瘤的目标对象中的恶性肿瘤的治疗方法,其包含:给予一治疗试剂的治疗有效量至目标对象以治疗所述恶性肿瘤的步骤,所述治疗试剂是选自于由卫康醇、卫康醇的衍生物或类似物、二乙酰二脱水卫矛醇、二乙酰二脱水卫矛醇的衍生物或类似物、二溴卫矛醇,以及二溴卫矛醇的衍生物或类似物所组成的群组。
如上文所述,所述恶性肿瘤可以是慢性骨髓性白血病(CML)或非小细胞肺癌(NSCLC),但是,如下文所述,所述治疗试剂可以使用来治疗其他的恶性肿瘤,包含三阴性乳癌。
通常,一特定目标对象是否患有一具有赋予TKIs抗性的生殖细胞缺失多型性的恶性肿瘤的确认是通过执行DNA双末端标记(DNA-paired end tag, DNA-PET)分析来进行的。PET序列的连通性允许检测缺失。比如,所述片段可以被选择为特定大小的全部。在基因图谱分析后,因此所述PET序列被预期始终是一彼此之间的特定距离。从这个距离差异表明一PET序列之间的结构变异。比如,在定序的基因组中的缺失将读取,在作为参考基因组的参考基因组中远大于预期的图谱将具有一未存在于定序基因组的DNA的片段。这可以被用来检测缺失的存在,比如上述2903bp或另一具有赋予TKIs抗性作用的缺失的生殖细胞缺失多型性。通常,所述生殖细胞DNA缺失多型性引发一得到缺乏一BH3区域的BIM蛋白的异构体的表现的剪接变异,从而抑制所述细胞凋亡的诱发。
本发明的另一方面是提供一种结合筛选及治疗的方法,其结合:生殖细胞缺失多型性的筛选,以及如果所述生殖细胞缺失多型性被发现存在时于一目标对象中TKIs抗性的恶性肿瘤的治疗。
一般地,此方法包含下面步骤:
(1)于一具有恶性肿瘤的目标对象中筛选所述生殖细胞缺失多型性;以及
(2)如果所述生殖细胞缺失多型性被发现存在于所述具有恶性肿瘤的目标对象中,给予一治疗试剂的治疗有效量至目标对象以治疗所述恶性肿瘤,所述治疗试剂是选自于由卫康醇、卫康醇的衍生物或类似物、二乙酰二脱水卫矛醇、二乙酰二脱水卫矛醇的衍生物或类似物、二溴卫矛醇,以及二溴卫矛醇的衍生物或类似物所组成的群组。
如上文所述,所述恶性肿瘤可以是慢性骨髓性白血病(CML)、非小细胞肺癌(NSCLC)或三阴性乳癌。然而,可以通过类似的方法治疗其他的恶性肿瘤(其中酪氨酸激酶活性与异常或不受控制的细胞增殖相关)。
这些方法可以进一步包含:于一患有一生殖细胞缺失多型性存在的恶性肿瘤的目标对象中给予(1)一BH3类似物;或(2)一BH3类似物及一抗酪氨酸激酶治疗试剂两者的治疗有效量的步骤。
合适的BH3类似物为以上所提及的。一特别优选的BH3类似物为 ABT-737。
合适的TKI治疗试剂包含伊马替尼、伯舒替尼、尼罗替尼及达沙替尼,所有的这些都靶向所述Bcr-Abl受体酪氨酸激酶。一般地,优选为伊马替尼。另外的TKI治疗试剂包含厄洛替尼、阿法替尼及达可替尼,其是主动朝向原生型或突变的EGFR的EGFR抑制剂(可逆型或非可逆型);这些药剂专门靶向所述EGFR酪氨酸激酶。
可以依据下面用于提高一或多个这些己醣醇衍生物的投药的治疗效果的原则来实现用以治疗一抗TKI肿瘤的卫康醇、卫康醇的衍生物或类似物、二乙酰二脱水卫矛醇、卫康醇的衍生物或类似物、二溴卫矛醇或二溴卫矛醇的衍生物或类似物(在本文中通常称为“烷基化己醣醇衍生物”)的投药。
因此,本发明的一方面是提供一恶性肿瘤的治疗方法,其中所述恶性肿瘤的特征是对至少一酪氨酸激酶抑制剂(tyrosine kinase inhibitor,TKI)有抗性,原因为:(1)一基因中的至少一突变,其编码一蛋白,所述蛋白是一至少一TKI的标靶;或(2)于一原生型或突变状态中的至少一额外的基因的存在,其编码一给予至少一TKI的治疗效果的抗性的产物,所述方法包含:给予一烷基化己醣醇衍生物的治疗有效量。通常,所述烷基化己醣醇衍生物是卫康醇、卫康醇的衍生物或类似物、二乙酰二脱水卫矛醇、二乙酰二脱水卫矛醇的衍生物或类似物、二溴卫矛醇或二溴卫矛醇的衍生物或类似物。
在一选择方案中,所述编码给予至少一TKI的治疗效果的抗性的产物的于原生型或突变状态中的至少一额外的基因为AHI-1。
如下文所述,这些方法可以进一步包含:(1)给予一BH3类似物的治疗有效量至目标对象的步骤;(2)给予一TKI的治疗有效量至目标对象的步骤; (3)给予一JAK2抑制剂的治疗有效量至目标对象的步骤;(4)给予一STAT5 抑制剂的治疗有效量至目标对象的步骤;(5)给予一Src激酶抑制剂的治疗有效量至目标对象的步骤;或(6)给予一激酶抑制剂的组合的治疗有效量至目标对象以治疗所述恶性肿瘤的步骤;其中所述激酶抑制剂的组合是一选自于由下列所组成的群组的组合:(a)一JAK2抑制剂及一STAT5抑制剂;(b) 一JAK2抑制剂及一Src抑制剂;(c)一STAT5抑制剂及一Src抑制剂;以及 (d)一JAK2抑制剂、一STAT5抑制剂及一Src抑制剂,至目标对象。此外,当所述方法包含:给予一JAK2抑制剂、一STAT5抑制剂、一Src抑制剂或两个或多个的JAK2抑制剂、一STAT5抑制剂及一Src抑制剂的治疗有效量至目标对象的步骤,所述方法可以进一步包含:给予一BH3类似物或一TKI 的治疗有效量至目标对象的步骤。下面描述用于这些方法的合适的BH3类似物、TKIs、JAK2抑制剂、STAT5抑制剂及Src抑制剂。
本发明的另一方面是提供一种在用于治疗抗TKI肿瘤的烷基化己醣醇衍生物的治疗应用中的改良方法,其是通过改变给予化合物的时间、使用控制所述化合物代谢率的剂量改质药剂、正常组织保护剂及其他变质剂来进行的。一般的例子包含:注入时间表的变化(比如,单次静脉注射与持续性注入相比)、使用治疗抗体以增加白血球总数,而用以提高免疫反应或用于防止骨髓抑制药剂所引起的贫血,或使用援救剂(比如用于5-FU的甲酰四氢叶酸或用于顺铂治疗的硫代硫酸盐)。用于治疗一抗TKI肿瘤的烷基化己醣醇衍生物的具体发明例子包含:持续性静脉注射数小时至数天;双周投药;剂量大于5 mg/m
本发明的另一方面是提供一种在用于治疗抗TKI肿瘤的烷基化己醣醇衍生物的治疗应用中的改良方法,其是通过改变给予化合物的途径来进行的。一般的例子包含:从口服途径改变至静脉给药(反之亦然);或使用特定途径,比如皮下、肌肉内、动脉内、腹膜内、病灶内、淋巴内、瘤内、脊髓内、静脉内、头颅内。用于治疗一抗TKI肿瘤的烷基化己醣醇衍生物的具体发明例子包含:日常投药;每周投药三周、每周投药二周、双周投药;双周投药三周(1-2周休息周期);间歇性增加剂量投药;一周每日投药,接着多周每周一次投药;用剂达到40mg/m
本发明的再一方面是提供一种在用于治疗抗TKI肿瘤的烷基化己醣醇衍生物的治疗应用中的改良方法,其是通过改变给予化合物的诊断/恶化上的疾病阶段来进行的。一般的例子包含:使用用于非可切除局部疾病、预防性使用而防止转移性扩散或抑制疾病恶化或者转移至更多恶性肿瘤阶段的化学疗法。用于治疗一抗TKI肿瘤的烷基化己醣醇衍生物的具体发明例子包含:使用具有血管生成抑制剂(比如癌思停、VEGF抑制剂)的烷基化己醣醇衍生物,以防止或限制转移性扩散,特别是在中枢神经系统、使用用于新诊断疾病的烷基化己醣醇衍生物、使用用于复发性疾病的烷基化己醣醇衍生物,以及使用用于抗药性或难治性疾病的烷基化己醣醇衍生物。
本发明的再一方面是提供一种在用于治疗抗TKI肿瘤的烷基化己醣醇衍生物的治疗应用中的改良方法,其是通过改变从化合物的使用最好忍受或受益的病患类型来进行的。一般的例子包含:使用用于老年患者的小儿剂量、用于肥胖病患的变动剂量;利用共病疾病情况,比如糖尿病、肝硬化或其他可以独特地开发一化合物的功能的情况。用于治疗一抗TKI肿瘤的烷基化己醣醇衍生物的具体发明例子包含:具有高水平的代谢酵素、组蛋白去乙酰酶、蛋白激酶、鸟胺酸脱羧酶的疾病情况的病患;具有低水平的代谢酵素、组蛋白去乙酰酶、蛋白激酶、鸟胺酸脱羧酶的疾病情况的病患;具有低或高易感性至血小板减少症、嗜中性白血球减少症的病患;无法忍受GI毒性的病患; jun的过表现或低表现、GPCR’s及信号传递蛋白、VEGF、摄护腺特殊基因、蛋白激酶或端粒酶。
本发明的再一方面是提供一种在用于治疗抗TKI肿瘤的烷基化己醣醇衍生物的治疗应用中的改良方法,其是通过更精确识别病患对于忍受、代谢及利用化合物的能力来进行的。一般的例子包含:肿瘤或正常组织的活组织检查样品(比如中枢神经系统的神经胶细胞或其他细胞),其还可以被采取及分析,针对基因标靶以明确地制定或监视特定药物的使用;独特肿瘤基因表现形态或单核苷酸多型性(single nucleotide polymorphisms,SNPs)的分析的研究,以提高功效或避免特定药物敏感性正常组织毒性。用于治疗一抗TKI肿瘤的烷基化己醣醇衍生物的具体发明例子包含:用以确认患者的特定基因型的诊断工具、技术、套组及试验;基因/蛋白表现芯片及分析;单核苷酸多型性(SNPs)评估;用于组蛋白去乙酰酶、鸟胺酸脱羧酶、GPCR’s、蛋白激酶、端粒酶或jun的SNPs;以及代谢酵素及代谢物的识别及量测。
本发明的再一方面是提供一种在用于治疗抗TKI肿瘤的烷基化己醣醇衍生物的治疗应用中的改良方法,其是通过化学治疗试剂的病患之前或之后使用的专业制备来进行的。一般的例子包含:代谢酵素的诱发或抑制、敏感性正常组织或器官系统的特定防护。用于治疗一抗TKI肿瘤的烷基化己醣醇衍生物的具体发明例子包含:使用秋水仙素或类似物;使用利尿药或促尿酸药 (比如丙磺舒);使用尿酸酶;非口服使用烟碱酰胺;持续释放形式的烟碱酰胺;使用聚ADP核糖聚合酶的抑制剂;使用咖啡因;甲酰四氢叶酸援救;感染控制;抗高血压药。
本发明的再一方面是提供一种在用于治疗抗TKI肿瘤的烷基化己醣醇衍生物的治疗应用中的改良方法,其是通过使用额外的药物或工序以防止或减少潜在副作用或毒性来进行的。一般的例子包含:使用止吐辅助剂、抗恶心辅助剂、用以限制或防止嗜中性白血球减少症、贫血、血小板减少症的血液辅助剂、维生素、抗忧郁剂、用于性功能障碍的治疗剂,以及其他辅助技术。用于治疗一抗TKI肿瘤的烷基化己醣醇衍生物的具体发明例子包含:使用秋水仙素或类似物;使用利尿药或促尿酸药(比如丙磺舒);使用尿酸酶;非口服使用烟碱酰胺;持续释放形式的烟碱酰胺;使用聚ADP核糖聚合酶的抑制剂;使用咖啡因;甲酰四氢叶酸援救;使用持续释放异嘌呤醇;非口服使用异嘌呤醇;骨髓移植促进剂、血液、血小板注入、优保律(Neupogen)、G-CSF; GM-CSF;疼痛控制;抗发炎药物;流体;皮质类固醇;胰岛素控制药物;解热剂;抗恶心治疗剂;止泻治疗剂;N-乙酰半胱胺酸;抗组织胺。
本发明的再一方面是提供一种在用于治疗抗TKI肿瘤的烷基化己醣醇衍生物的治疗应用中的改良方法,其是通过给药后使用监控药物水平而企图最大化病患的药物血中水平、监视毒性代谢物产生、监视于药物–药物交互作用方面可能有益或有害的辅助药物来进行的。一般的例子包含:药物血浆蛋白结合率的监控,以及其他药物动力学变量或药效学变量的监控。用于治疗抗TKI肿瘤的烷基化己醣醇衍生物的具体发明例子包含:药物血中水平的多次测定;于血液或尿液中代谢物的多次测定。
本发明的再一方面是提供一种在用于治疗抗TKI肿瘤的烷基化己醣醇衍生物的治疗应用中的改良方法,其是通过利用独特并用药来进行的,所述独特并用药于功效或副作用管理中可以提供一超过加成或协力的改良。用于治疗一抗TKI肿瘤的烷基化己醣醇衍生物的具体发明例子包含:与位置异构酶抑制剂结合;使用伪核苷;使用伪核苷酸;使用胸苷合成酶抑制剂;使用信号传递抑制剂;使用顺铂或铂类似物;使用烷化剂,比如亚硝基尿(BCNU、格立得植入剂(Gliadel wafers)、CCNU、尼莫司汀(nimustine)(ACNU)、苯达莫司汀(Treanda));使用烷化剂,其在与卫康醇或另一烷基化己醣醇衍生物不同地方上破坏DNA(TMZ、BCNU、CCNU及其他的烷化剂,所有破坏DNA在鸟嘌呤的O
本发明的再一方面是提供一种在用于治疗抗TKI肿瘤的烷基化己醣醇衍生物的治疗应用中的改良方法,其是通过利用烷基化己醣醇衍生物作为化学致敏剂(在当于功效中观察到一超过加成或协力的改良(单独使用但结合其他治疗)时,观察到无可量测活性的情况下)来进行的。用于治疗一抗TKI肿瘤的烷基化己醣醇衍生物的具体发明例子包含:作为与位置异构酶抑制剂结合的化学致敏剂;作为与伪核苷结合的化学致敏剂;作为与伪核苷酸结合的化学致敏剂;作为与胸苷合成酶抑制剂结合的化学致敏剂;作为与信号传递抑制剂结合的化学致敏剂;作为与顺铂或铂类似物结合的化学致敏剂;作为与烷化剂(比如BCNU、格立得植入剂、CCNU、苯达莫司汀(Treanda)或替莫唑胺(Temodar))结合的化学致敏剂;作为与抗微管蛋白剂结合的化学致敏剂;作为与抗代谢物结合的化学致敏剂;作为与小蘗碱结合的化学致敏剂;作为与芹菜素结合的化学致敏剂;作为与氨萘非特结合的化学致敏剂;作为与秋水仙素或其类似物结合的化学致敏剂;作为与染料木黄酮结合的化学致敏剂;作为与伊妥普赛结合的化学致敏剂;作为与阿拉伯糖基胞嘧啶结合的化学致敏剂;作为与喜树碱结合的化学致敏剂;作为与长春花生物碱类结合的化学致敏剂;作为与位置异构酶抑制剂结合的化学致敏剂;作为与5-氟代尿嘧啶结合的化学致敏剂;作为与姜黄素结合的化学致敏剂;作为与NF-κB抑制剂结合的化学致敏剂;作为与迷迭香酸结合的化学致敏剂;作为与丙脒腙结合的化学致敏剂;或作为与粉防己碱结合的化学致敏剂。
本发明的再一方面是提供一种在用于治疗抗TKI肿瘤的烷基化己醣醇衍生物的治疗应用中的改良方法,其是通过利用烷基化己醣醇衍生物作为化学增效剂(在当于功效中观察到一超过加成或协力的改良(单独使用但结合其他治疗的独特药物)时,观察到最小治疗活性的情况下)来进行的。用于治疗一抗TKI肿瘤的烷基化己醣醇衍生物的具体发明例子包含:作为与位置异构酶抑制剂结合的化学增效剂;作为与伪核苷结合的化学增效剂;作为与胸苷合成酶抑制剂结合的化学增效剂;作为与信号传递抑制剂结合的化学增效剂;作为与顺铂或铂类似物结合的化学增效剂;作为与烷化剂(比如BCNU、BCNU 片、格立得或苯达莫司汀(Treanda))结合的化学增效剂;作为与抗微管蛋白剂结合的化学增效剂;作为与抗代谢物结合的化学增效剂;作为与小蘗碱结合的化学增效剂;作为与芹菜素结合的化学增效剂;作为与氨萘非特结合的化学增效剂;作为与秋水仙素或其类似物结合的化学增效剂;作为与染料木黄酮结合的化学增效剂;作为与伊妥普赛结合的化学增效剂;作为与阿拉伯糖基胞嘧啶结合的化学增效剂;作为与喜树碱结合的化学增效剂;作为与长春花生物碱类结合的化学增效剂;作为与位置异构酶抑制剂结合的化学增效剂;作为与5-氟代尿嘧啶结合的化学增效剂;作为与姜黄素结合的化学增效剂;作为与NF-κB抑制剂结合的化学增效剂;作为与迷迭香酸结合的化学增效剂;作为与丙脒腙结合的化学增效剂;作为与粉防己碱结合的化学增效剂。
本发明的再一方面是提供一种在用于治疗抗TKI肿瘤的烷基化己醣醇衍生物的治疗应用中的改良方法,其是通过考虑最大受益至以化合物治疗的病患的药物、治疗及诊断来进行的。一般的例子包含:疼痛控制、营养支持、止吐、抗恶心疗法、抗贫血疗法、抗发炎。用于治疗一抗TKI肿瘤的烷基化己醣醇衍生物的具体发明例子包含:使用与疼痛控制相关的疗法;使用营养支持;使用止吐剂;使用抗恶心疗法;使用抗贫血疗法;使用抗发炎药物;使用解热剂;使用免疫促进剂。
本发明的再一方面是提供一种在用于治疗抗TKI肿瘤的烷基化己醣醇衍生物的治疗应用中的改良方法,其是通过通过使用互补疗法或用以提高有效性或减少副作用的方法的应用来进行的。用于治疗一抗TKI肿瘤的烷基化己醣醇衍生物的具体发明例子包含:催眠;针灸;身心疗法;通过合成或提取产生的草本药物包含NF-κB抑制剂(比如小白菊内酯、姜黄素、迷迭香酸);天然的抗发炎药物(包含大黄酸、小白菊内酯);免疫促进物(比如于紫锥花中发现的);抗微生物药(比如小蘗碱);类黄酮、异黄酮及黄酮(比如芹菜素、染料木黄酮、染料木苷、6″-O-丙二酰基染料木苷、6″-O-乙酰基染料木苷、大豆黄酮、大豆苷、6″-O-丙二酰基大豆苷、6″-O-乙酰基染料木苷、黄豆黄素、黄豆黄苷、6″-O-丙二酰基黄豆黄苷及6-O-乙酰基黄豆黄苷);应用人体运动学。
本发明的再一方面是提供一种在用于治疗抗TKI肿瘤的烷基化己醣醇衍生物的治疗应用中的改良方法,其是通过改变药物原料来进行的。一般的例子包含:盐类形成、同质晶体结构、纯异构物。用于治疗一抗TKI肿瘤的烷基化己醣醇衍生物的具体发明例子包含:盐类形成;同质晶体结构;纯异构物;提高纯度;低残留溶剂及重金属。
本发明的再一方面是提供一种在用于治疗抗TKI肿瘤的烷基化己醣醇衍生物的治疗应用中的改良方法,其是通过改变使用于溶解及释放/存在用于投药的化合物的稀释剂来进行的。一般的例子包含:氢化蓖麻油聚氧乙烯、用于难溶于水的化合物的环糊精。用于治疗抗TKI肿瘤的烷基化己醣醇衍生物的具体发明例子包含:乳化液的使用、二甲亚砜(dimethyl sulfoxide,DMSO)、甲基甲酰胺(N-methylformamide,NMF)、二甲基甲酰胺(dimethylformamide, DMF)、二甲基乙酰胺(dimethylacetamide,DMA)、乙醇、苯甲醇、用于注射的含有葡萄糖的水、聚氧乙烯蓖麻油、环糊精、PEG。
本发明的再一方面是提供一种在用于治疗抗TKI肿瘤的烷基化己醣醇衍生物的治疗应用中的改良方法,其是通过改变溶解用于投药或用于进一步稀释的化合物所使用或所需要的溶剂来进行的。一般的例子包含:乙醇、二甲基乙酰胺(DMA)。用于治疗一抗TKI肿瘤的烷基化己醣醇衍生物的具体发明例子包含:乳化液的使用、二甲亚砜(DMSO)、N-甲基甲酰胺(NMF)、二甲基甲酰胺(DMF)、二甲基乙酰胺(DMA)、乙醇、苯甲醇、用于注射的含有葡萄糖的水、聚氧乙烯蓖麻油、PEG或盐系统。
本发明的再一方面是提供一种在用于治疗抗TKI肿瘤的烷基化己醣醇衍生物的治疗应用中的改良方法,其是通过改变稳定及存在用于适当投药的化学化合物所需的材料/赋形剂、缓冲剂或防腐剂来进行的。一般的例子包含:甘露醇、白蛋白、EDTA、亚硫酸氢钠、苯甲醇。用于治疗一抗TKI肿瘤的烷基化己醣醇衍生物的具体发明例子包含:使用甘露醇;使用白蛋白;使用 EDTA;使用亚硫酸氢钠;使用苯甲醇;使用碳酸盐缓冲液;使用磷酸盐缓冲液;使用聚乙二醇(polyethylene glycol,PEG);使用维生素A;使用维生素 D;使用维生素E;使用酯酵素抑制剂;使用细胞色素P450抑制剂;使用多重抗药性(multi-drugresistance,MDR)抑制剂;使用有机树脂;使用洗涤剂;使用紫苏醇或其类似物;或使用通道形成受体的活化剂。
本发明的再一方面是提供一种在用于治疗抗TKI肿瘤的烷基化己醣醇衍生物的治疗应用中的改良方法,其是通过改变取决于投药的途径、影响期间、血浆水平要求、曝露于副作用正常组织及代谢酵素的化合物的潜在剂型来进行的。一般的例子包含:片剂、胶囊、外用凝胶、药膏、贴剂、栓塞剂。烷基化己醣醇衍生物的使用的具体发明例子包含:使用片剂;胶囊;外用凝胶;外用药膏;贴剂;栓塞剂;冻干剂量填充物;使用速释制剂;使用缓释制剂;使用控释制剂;或使用液体胶囊。
本发明的再一方面是提供一种在用于治疗抗TKI肿瘤的烷基化己醣醇衍生物的治疗应用中的改良方法,其是通过改变剂型、容器/密闭系统、混合精度,以及剂量配制及呈现来进行的。一般的例子包含:避免光照的棕色瓶、具有专用涂层的瓶塞。用于治疗一抗TKI肿瘤的烷基化己醣醇衍生物的具体发明例子包含:避免光照的棕色瓶的使用;用以增加储存寿命稳定性的具有专用涂层的瓶塞。
本发明的再一方面是提供一种在用于治疗抗TKI肿瘤的烷基化己醣醇衍生物的治疗应用中的改良方法,其是通过使用传递系统以改善药物产物的潜在属性(比如便利、作用期间、减少毒性)来进行的。一般的例子包含:纳米晶体、生物溶蚀型聚合物、脂质体、持续释放注射凝胶、微球。用于治疗一抗TKI肿瘤的烷基化己醣醇衍生物的具体发明例子包含:使用口服剂型;使用纳米晶体;使用纳米颗粒;使用共溶剂;使用浆体;使用糖浆、使用生物溶蚀型聚合物;使用脂质体;使用持续释放注射凝胶;使用微球;或使用具有表皮生长因子受体结合胜肽的靶向组合物。
本发明的再一方面是提供一种在用于治疗抗TKI肿瘤的烷基化己醣醇衍生物的治疗应用中的改良方法,其是通过改变具有共价、离子或氢键结部分的初始烷基化己醣醇衍生物以修改功效、毒性、药物动力学、代谢或投药的途径来进行的。一般的例子包含:聚合物系统(比如聚乙二醇)、聚乳酸、聚甘醇酸、胺基酸、胜肽或多价连接符。用于治疗一抗TKI肿瘤的烷基化己醣醇衍生物的具体发明例子包含:使用聚合物系统(比如聚乙二醇);使用聚乳酸;使用聚甘醇酸;使用胺基酸;使用胜肽;使用多价连接符;使用免疫球蛋白;使用环糊精聚合物;使用修饰运铁蛋白;使用疏水性聚合物或疏水- 亲水性聚合物;使用具有一磷甲酸钠偏酯的共轭物;使用具有加入一带电交联剂的细胞粘合剂的共轭物;或使用具有通过一连接剂的β-葡萄醛酸的共轭物。
本发明的再一方面是提供一种在用于治疗抗TKI肿瘤的烷基化己醣醇衍生物的治疗应用中的改良方法,其是通过改变具有额外化学官能性的分子的初始结构来进行的,所述额外化学官能性可以改变功效、降低毒性、提高药物性能、兼容一个特定的给药途径,或改变所述治疗试剂的代谢。一般的例子包含:侧链的变动以增加或减少亲脂性;额外的化学官能性以改变反应活性、电子亲和力及结合能力;盐类型。用于治疗一抗TKI肿瘤的烷基化己醣醇衍生物的具体发明例子包含:侧链的变动以增加或减少亲脂性;额外的化学官能性以改变反应活性、电子亲和力或结合能力;或盐类型。
本发明的再一方面是提供一种在用于治疗抗TKI肿瘤的烷基化己醣醇衍生物的治疗应用中的改良方法,其是通过改变分子以致于在于裂解一部分分子导入体内而显示优选活性分子后以活性分子的变异体得到改进药物性能来进行的。一般的例子包含:酵素敏感酯类、二聚体、希夫碱(Schiff bases)。用于治疗一抗TKI肿瘤的烷基化己醣醇衍生物的具体发明例子包含:使用酵素敏感酯类;使用二聚体;使用希夫碱;使用吡哆醛复合物;使用咖啡因复合物;使用一氧化氮释放前驱物;使用具有纤维原细胞激活蛋白α-可分裂寡肽的前驱物;使用与一乙酰化剂或一胺甲酰化剂反应的产物的前驱物;使用己酸盐共轭物的前驱物;使用聚合物-剂的共轭物的前驱物;或受到氧化还原活化的前驱物的使用。
本发明的再一方面是提供一种在用于治疗抗TKI肿瘤的烷基化己醣醇衍生物的治疗应用中的改良方法,其是通过使用额外的化合物、生物剂(当在适当的类型中投药,一独特的和有益的效果可以被实现)来进行的。一般的例子包含:多重抗药性的抑制剂、特异的抗药性抑制剂、特异的选择性酵素的抑制剂、信号传递抑制剂、修复抑制剂。用于治疗一抗TKI肿瘤的烷基化己醣醇衍生物的具体发明例子包含:使用多重抗药性的抑制剂;使用特异的抗药性抑制剂;使用特异的选择性酵素的抑制剂;使用信号传递抑制剂;使用修复抑制剂;或使用具有非重迭副作用的位置异构酶抑制剂。
本发明的再一方面是提供一种在用于治疗抗TKI肿瘤的烷基化己醣醇衍生物的治疗应用中的改良方法,其是通过结合使用烷基化己醣醇衍生物与具有生物反应修饰物的敏化剂/增效剂来进行的。一般的例子包含:结合使用具有生物反应修饰物、细胞激素、治疗抗体、治疗抗体、反义治疗、基因治疗的敏化剂/增效剂。用于治疗一抗TKI肿瘤的烷基化己醣醇衍生物的具体发明例子包含:结合使用具有生物反应修饰物的敏化剂/增效剂;结合使用具有细胞激素的敏化剂/增效剂;结合使用具有治疗抗体的敏化剂/增效剂;结合使用具有治疗抗体的敏化剂/增效剂;结合使用具有反义治疗(比如癌思停、贺癌平、利妥昔及爱必妥)的敏化剂/增效剂;结合使用具有基因治疗的敏化剂/ 增效剂;结合使用具有核醣酶的敏化剂/增效剂;结合使用具有RNA干扰的敏化剂/增效剂。
本发明的再一方面是提供一种在用于治疗抗TKI肿瘤的烷基化己醣醇衍生物的治疗应用中的改良方法,其是通过利用烷基化己醣醇衍生物的选择性使用来克服生物疗法的有效使用的发展中或完全的抗性来进行的。一般的例子包含:生物反应修饰物的作用抗性的肿瘤、细胞激素、治疗抗体、治疗抗体、反义治疗、基因治疗。用于治疗一抗TKI肿瘤的烷基化己醣醇衍生物的具体发明例子包含:用来抵抗耐生物反应改良剂影响的肿瘤;用来抵抗耐细胞激素影响的肿瘤;用来抵抗耐淋巴激素影响的肿瘤;用来抵抗耐治疗抗体影响的肿瘤;用来抵抗耐反义治疗影响的肿瘤;用来抵抗耐如癌思停、利妥昔、贺癌平、爱必妥这样的治疗影响的肿瘤;用来抵抗耐基因治疗影响的肿瘤;用来抵抗耐核酶影响的肿瘤;或用来抵抗耐RNA干扰影响的肿瘤。
本发明的再一方面是提供一种在用于治疗抗TKI肿瘤的烷基化己醣醇衍生物的治疗应用中的改良方法,其是通过利用烷基化己醣醇衍生物与电离辐射、光疗法、热疗法或射频产生疗法的结合使用来进行的。一般的例子包含:低氧敏化剂、辐射敏化剂/保护剂、光敏化剂、辐射修复抑制剂。用于治疗一抗TKI肿瘤的烷基化己醣醇衍生物的具体发明例子包含:与电离辐射结合使用;与低氧敏化剂结合使用;与辐射敏化剂/保护剂结合使用;与光敏化剂结合使用;与辐射修复抑制剂结合使用;与硫醇耗尽结合使用;与血管标靶剂结合使用;与放射株结合使用;与放射性核种结合使用;与放射性标帜抗体结合使用;或与近接疗法结合使用。在这样的放射线疗法或起到通过结合放射线疗法与烷基化己醣醇衍生物的投药的协力效应的能力的功效中的改良是显著的。
本发明的再一方面是提供一种在用于治疗抗TKI肿瘤的烷基化己醣醇衍生物的治疗应用中的改良方法,其是通过通过确定各种新颖作用机制、用于更深地理解及精确的更优选利用分子的效用的化合物的生物靶而优化其效用来进行的。用于治疗一抗TKI肿瘤的烷基化己醣醇衍生物的具体发明例子包含:使用聚ADP核糖聚合酶的抑制剂;使用影响血管的药剂;使用促进血管扩张的药剂;使用致癌基因靶向药物;使用信号传递抑制剂;使用引起EGFR 抑制作用的药剂;使用引起蛋白激酶C抑制的药剂;使用引起磷脂酶C递减调节的药剂;使用引起jun递减调节的药剂;使用调节组织蛋白基因的表现的药剂;使用调节VEGF的表现的药剂;使用调节鸟胺酸脱羧酶的表现的药剂;使用调节jun D的表现的药剂;使用调节v-jun的表现的药剂;使用调节 GPCRs的表现的药剂;使用调节蛋白激酶A的表现的药剂;使用调节端粒酶的表现的药剂;使用调节摄护腺特殊基因的表现的药剂;使用调节蛋白激酶 (除了蛋白激酶A)的表现的药剂;使用调节组蛋白去乙酰酶的表现的药剂。
本发明的再一方面是提供一种在用于治疗抗TKI肿瘤的烷基化己醣醇衍生物的治疗应用中的改良方法,其是通过更精确识别及显露化合物至这些选择的细胞群(在所述细胞群可以最大地利用化合物的效果的情况下)来进行的。用于治疗一抗TKI肿瘤的烷基化己醣醇衍生物的具体发明例子包含:用来抵抗辐射敏感细胞、用来抵抗辐射抗性细胞或用来抵抗能量耗尽细胞。
本发明的再一方面是提供一种在用于治疗抗TKI肿瘤的烷基化己醣醇衍生物的治疗应用中的改良方法,其是通过使用用以提高烷基化己醣醇衍生物的活性的药剂来进行的。一般的例子包含:使用烟碱酰胺、咖啡因、粉防己碱或小蘗碱。用于治疗一抗TKI肿瘤的烷基化己醣醇衍生物的具体发明例子包含:使用烟碱酰胺;使用咖啡因;使用粉防己碱;或使用小蘗碱。
于一烷基化己醣醇衍生物的治疗应用中的这些改良还可以被使用在:(1) 一AHI1关联恶性肿瘤的治疗,比如一以AHI1的突变或失调为特征的恶性肿瘤;以及(2)三阴性乳癌的治疗。
因此,本发明的另一方面是提供一种用以使用于治疗一抗TKI肿瘤的烷基化己醣醇衍生物的投药的功效增加和/或副作用减少的方法,所述方法包含下面步骤:
(1)确认烷基化己醣醇衍生物的投药的功效和/或副作用发生相关的至少一因子或参数,所述烷基化己醣醇衍生物是用于治疗一抗TKI肿瘤;以及
(2)修改所述因子或参数,用以使所述烷基化己醣醇衍生物的投药的功效增加和/或副作用减少,所述烷基化己醣醇衍生物是用于治疗所述抗TKI肿瘤。
通常,所述因子或参数是选自于由下列所组成的群组:
(1)剂量调整;
(2)投药的途径;
(3)投药的时间表;
(4)使用适应症;
(5)病程阶段的选择;
(6)其他适应症;
(7)选择病患;
(8)病患/疾病表现型;
(9)病患/疾病基因型;
(10)前/后治疗准备;
(11)毒性管理;
(12)药物动力学/药效学监测;
(13)并用药;
(14)化学敏化作用;
(15)化学增效作用;
(16)后治疗病患管理;
(17)选择性医疗/治疗支持;
(18)原料药产品改良;
(19)稀释剂系统;
(20)溶剂系统;
(21)赋形剂;
(22)剂型;
(23)剂量套组及包装;
(24)药物传递系统;
(25)药物共轭形式;
(26)化合物类似物;
(27)前驱物;
(28)多重药物系统;
(29)生物治疗加强作用;
(30)抗生物治疗调控;
(31)放射线疗法增强;
(32)新颖作用机制;
(33)选择性靶细胞群疗法;以及
(34)使用一增加其活性的药剂。
如上文所述,于一烷基化己醣醇衍生物的治疗应用中的这些改良还可以被使用在:(1)一AHI1关联恶性肿瘤的治疗,比如一以AHI1的突变或失调为特征的恶性肿瘤;以及(2)三阴性乳癌的治疗。下面提供于AHI1关联恶性肿瘤的治疗中及于三阴性乳癌的治疗中关于使用烷基化己醣醇衍生物的更多细节。
如上文所述,所述烷基化己醣醇衍生物通常是一半乳糖醇、一取代的半乳糖醇、一卫矛醇或一取代的卫矛醇,比如卫康醇、二乙酰二脱水卫矛醇、二溴卫矛醇及其衍生物及类似物,但本发明并不局限于此。这些化合物是烷化剂或烷化剂的前驱物。
这些烷基化己醣醇衍生物包含但不限于:(1)卫康醇;(2)卫康醇的衍生物,比如其具有以低级烷基取代的羟基团的氢、其具有以低级烷基取代的连接到环氧环的氢,或者其具有连接于相同的碳的甲基团,所述碳带有以低级烷基取代的羟基团或带有比如以卤素基取代的羟基团;(3)二乙酰二脱水卫矛醇;(4)二乙酰二脱水卫矛醇的衍生物,比如其具有以低级烷基取代部份乙酰基的甲基团、其具有以低级烷基取代的连接于环氧环的氢,或者其具有连接于相同的碳的甲基团,所述碳带有以低级烷基取代的乙酰基团或带有比如以卤素基取代的乙酰基团;(5)二溴卫矛醇;以及(6)二溴卫矛醇的衍生物,比如其具有一或多个以低级烷基取代的羟基团的氢,或者具有一或两个以另一卤素基(比如氯或氟)取代的溴基团。
当所述改良是通过剂量调整来进行的,所述剂量调整可以是但不限于至少一选自于由下列所组成的群组的剂量调整:
(a)持续性静脉注射数小时至数天;
(b)双周投药;
(c)剂量大于5mg/m
(d)基于患者的耐受性由1mg/m
(e)使用用以调节代谢的咖啡因;
(f)使用用以调节代谢的异烟碱酰;
(g)剂量投药的选定及间歇性增加;
(h)通过单次从mg/m
(i)口服剂量低于30mg/m
(j)口服剂量超过130mg/m
(k)口服剂量达到40mg/m
(l)用剂在一持续期的较低水平(比如21天);
(m)用剂在一较高的水平;
(n)用剂以超过21天的最低/恢复周期;
(o)使用一作为单一细胞毒杀剂的烷基化己醣醇衍生物;
(p)速释用剂;
(q)持续释放用剂;以及
(r)控释用剂。
速释用剂的使用透过Flanner等人被描述于美国专利第8,299,052号,并通过引用将其包括在内。持续释放用剂的使用透过Vergnault等人被描述于美国专利第8,303,986号,并通过引用将其包括在内。控释用剂的使用Dzierba 等人透过被描述于美国专利第8,304,577号,并通过引用将其包括在内。可以通过使用生物分解性聚合物完成控释用剂,所述生物分解性聚合物比如为聚乳酸、聚ε-己内酯、聚羟基丁酸、聚原酸酯、聚缩醛、聚二氢吡喃、聚氰丙烯酸酯及水胶的交联或双极性嵌段共聚合物,但本发明并不局限于此。
当所述改良是通过投药的途径来进行的,所述投药的途径可以是但不限于至少一选自于由下列所组成的群组的投药的途径:
(a)局部投药;
(b)口服投药;
(c)持续释放口服传递;
(d)脊髓注射;
(e)动脉注射;
(f)持续性注入;
(g)间歇性注入;
(h)静脉给药,比如静脉给药30分钟;
(i)通过一个较长的注入投药;
(j)通过静脉推注给药;以及
(k)腹膜内投药。
当所述改良是通过投药的时间表来进行的,所述投药的时间表可以是但不限于至少一选自于由下列所组成的群组的投药的时间表:
(a)日常投药;
(b)每周投药;
(c)每周投药三周;
(d)双周投药;
(e)双周投药三周(1-2周休息周期);
(f)间歇性增加剂量投药;以及
(g)日常投药一周至多周。
当所述改良是通过病程阶段的选择来进行的,所述病程阶段的选择可以是但不限于至少一选自于由下列所组成的群组的病程阶段的选择:
(a)使用于一用于抗TKI肿瘤的适当的病程阶段;
(b)新诊断疾病的用途;
(c)复发性疾病的用途;
(d)抵抗性或难治性疾病的用途;
(e)一AHI1关联恶性肿瘤的治疗的用途;
(f)三阴性乳癌的治疗的用途;以及
(g)转移性疾病的治疗的用途。
当所述改良是通过选择病患来进行的,所述选择病患可以是但不限于一通过一基准所实现的选择病患,所述基准是选自于由下列所组成的群组:
(a)选择具有以一代谢酵素的高水平为特征的疾病情况的病患,所述代谢酵素是选自于由组蛋白去乙酰酶及鸟胺酸脱羧酶所组成的群组;
(b)选择具有一低或高易感性的条件的病患,所述条件选自于由血小板减少症及嗜中性白血球减少症所组成的群组;
(c)选择无法忍受GI毒性的病患;
(d)选择以一基因的过表现或低表现为特征的病患,所述基因是选自于由 c-Jun、一GPCR、一信号传递蛋白、VEGF、一摄护腺特殊基因及一蛋白激酶所组成的群组;
(e)选择基于BIM共缺失的病患;以及
(f)选择基于AHI1的突变或失调存在的病患。
所述细胞原致癌基因c-Jun编码一蛋白,其与c-Fos结合,而形成AP-1 早期反应转录因子。这个原致癌基因在转录中扮演一个关键的角色,且与大量影响转录及基因表现的蛋白相互作用。其还涉及于形成许多组织之一部分的细胞的增殖及细胞凋亡,包含子宫内膜的细胞及腺上皮细胞。G蛋白偶合受体(G-protein coupled receptors,GPCRs)是重要的信号转导受体。所述G蛋白偶合受体的超级家族包含大量的受体。这些受体是以含有七个疏水区域的胺基酸序列为特征的完整的膜蛋白,预测表示所述蛋白的横跨膜的生成区域。他们被发现于广泛的生物体中,且涉及到由于它们与异三聚体G蛋白的交互作用而使信号传输至细胞内部。它们对不同种类的药剂产生反应,所述药剂包含脂肪类似物、胺基酸衍生物、小分子物(比如肾上腺素及多巴胺)及各种感觉刺激物。许多已知的GPCR的属性被概述于S.Watson及S.Arkinstall,“G蛋白连结受体纪实丛书”(Academic Press,London,1994),并通过引用将其包括在内。GPCR受体包含但不限于乙酰胆碱受体、β-肾上腺素能受体、β3-肾上腺素能受体、血清素(5-羟色胺)受体、多巴胺受体、腺苷受体、第二型血管收缩素受体、舒缓素受体、抑钙素受体、抑钙素基因相关受体、大麻素受体、胆囊收缩素受体、趋化激素受体、细胞激素受体、胃泌激素受体、内皮素受体、γ-胺基丁酸(γ-aminobutyric acid、GABA)受体、甘丙胺酸素受体、升糖素受体、麸胺酸受体、黄体激素受体、绒毛膜激性腺素受体、促滤泡激素受体、促甲状腺激素受体、激性腺素释放激素受体、白三烯素受体、神经胜肽Y受体、脑内啡受体、副甲状腺激素受体、血小板活化因子受体、类前列腺素(前列腺素)受体、生长抑制素受体、促甲状腺素释放激素受体、血管升压素及催产素受体。
AHI1(Abelson-Helper Integration Site-1)是一个新的致癌基因,其已经被发现在一些白血病细胞株中有调控异常的情形,包含CML(慢性骨髓细胞性白血病)。AHI1增强体内BCR-ABL1的作用,且诱发许多激酶,包含JAK2、 STAT5及Src家族激酶,最终介导胸腺核苷激酶抑制剂(thymidine kinase inhibitors,TKIs)的反应性或抗性。许多研究已表明于原生造血细胞中AHI1 过度表现:赋予一体外生长优势至这样的细胞、可诱发体内白血病,且增强 BCR-ABL1的作用。从具有CML的病患的细胞的研究还表明AHI1通过调解 TKI抗药性还有助于BCR-ABL1诱发恶性转换。此外,ABL1蛋白的激酶区域内的突变也有助于伊马替尼及其他TKIs的抗药性;未经常发现的突变是一涉及单一胺基酸取代的点突变,T315I(T.B.Balci等人,“于慢性骨髓性白血病患者中AHI1基因表现水平及BCR-ABL1 T315I突变”,Hematology 16: 357-360(2011),并通过引用将其包括在内)。所述AHI1基因座于Abelson前 B细胞淋巴瘤中被初步认定为一常见的辅助前病毒整合位点,且被证明是紧密连接于c-myb原致癌基因。在窝藏AHI1基因座内所插入的前病毒的Abelson鼠白血病病毒诱导前B淋巴瘤中,发现因为c-myb表现没有显著的改变,这表明,此基因座包含至少一其他的涉及到肿瘤形成的失调的基因。在此基因座上的此基因为AHI1基因。在反转录方向中基因的3’末端上发现前病毒插入,且大多数的前病毒插入被设置于基因的最后外显子的周围或下游;另一个插入被发现于基因的内含子22中。此外,另一个以往确定的前病毒插入位点,Mis-2,被发现绘制于AHI1基因的内含子16中。所述AHI1 cDNA 编码一1047胺基酸残基的蛋白。所述预测AHI1蛋白是一模蛋白,其含有一个SH3基序(motif)及七个WD-40重复序列。在哺乳动物中所述AHI1基因是高度保守的,且编码5及4.2kb的两个主要RNA物种及其他几个、较短的、剪接变异体。所述AHI1基因被表现于小鼠胚胎中及被表现于几个小鼠及大鼠的器官中;尤其是,在大脑和睾丸中表现发生于高水平。在AHI1内窝藏插入性突变的肿瘤细胞中,截短的病毒稠合转录体已经被确定,包含一些具有SH3区域的缺失的剪接变异体。在正常细胞内信号传递中,AHI1呈现几个讯息分子的特征,且被认为是扮演一个重要的角色。由于在窝藏v-abl缺失性反转录病毒或c-myc转基因的肿瘤中,或者在呈现Nf1的缺失中确认到 AHI1插入位点,所以AHI1也可能涉及肿瘤发展,可能与其他致癌基因(比如v-abl及c-myc)或肿瘤抑制基因(Nf1)合作。全长人类AHI1基因是由29个外显子组成的(亦即外显子1至33,外显子24、28、29及32除外);所述终止密码子坐落于外显子33中。然而,在外显子24中,有一缺乏所述SH3区域的截短异构体,并是由24个外显子(只有外显子1至24)所组成,且包含一框内终止密码子。有一由32个外显子(外显子1至33,外显子24除外)组成的第二截短异构体,然而在外显子28中有一终止密码子。一些来源于前病毒插入的3’末端插入性突变可以改变标准的剪接机制,且可以强制病毒或内含子序列内的选择性剪接接近它们,从而删除通常会编码AHI1蛋白的羧基端区域的外显子。基因证据表明,在一些不同的细胞系中AHI1基因的前病毒插入突变有助于肿瘤形成。所述AHI1基因座被确定为一常见的前病毒整合位点,最初是在Abelson(v-abl诱导)前B淋巴瘤中,后来是在MMTV
此外,所述AHI1蛋白与所述BCR-ABL融合蛋白及其他的涉及到造血及造血的调节的蛋白交互作用。此交互作用被描述于L.L.Zhou等人“AHI-1 与BCR-ABL交互作用而调节BCR-ABL转化活性及伊马替尼对CML干/前驱细胞的反应”,J.Exp.Med.11:2657-2671(2005),并通过引用将其包括在内。明确地说,通过已经获得一BCR-ABL融合基因的CML干细胞的罕见群体来启动或增殖慢性骨髓细胞性白血病(chronic myelocytic leukemia,CML),其编码一嵌合致癌蛋白(BCR-ABL),所述嵌合致癌蛋白显示本质地提高驱动 CML发病机制的酪氨酸激酶活性。这涉及到,在其他效果之中,细胞增殖及细胞凋亡控制的去调节通过对多个讯息传递途径上产生影响,包含Ras、磷脂酰肌醇-3激酶(phosphatidylinositol3-kinase,PI3K)、JAK-STAT及NF-κB途径。如上文所述,甲磺酸伊马替尼,一BCR-ABL酪氨酸激酶的抑制剂,经常使用于治疗CML,其受到所述酪氨酸激酶抑制剂抗性的发展,而导致复发或治疗失败。尽管其他酪氨酸激酶抑制剂(比如达沙替尼及尼罗替尼)在抗药性已发展或可能发展到伊马替尼的案例中可以是有用的,仍然有需要通过不依赖于酪氨酸激酶抑制剂及所述BCR-ABL酪氨酸激酶之间的特定交互作用的机制来防止抗药性的发展。
最近的研究已经表明,于慢性期患者中CML干/前驱细胞是很少反应至 IM及其他酪氨酸激酶抑制剂,且是一用于甲磺酸伊马替尼抗药性的发展的关键标靶群体。这样的CML干/前驱细胞具有基因不稳定性的性质,且经常引起在体外(in vitro)甲磺酸伊马替尼抵抗性突变体。
如上所述,在作为候选协同致癌基因的v-abl诱导小鼠前B细胞淋巴瘤中,所述Ahi-1基因(在小鼠中)或其保留性人类同源物(AHI-1)是一通过前病毒插入性突变所确定的新颖基因。所述小鼠Ahi-1基因编码一具有SH3区域、多个SH3结合位置及WD40-重复区域的唯一性蛋白,其全部被认为是重要的蛋白-蛋白交互作用的媒介物,这表明,正常的Ahi-1蛋白(在小鼠中)或人类同源AHI-1蛋白具有异常信号活性,且它的去调节可以影响特定细胞讯息传递途径。所述保留性人类同源物AHI-1在其氨基末端区域中具有一额外的卷曲螺旋区域。在高度保留于小鼠及人类之间的方式中,所述Ahi-1基因(在小鼠中)或所述AHI-1基因(在人类中)的表现已经被证明调控于多个造血的阶段。一般地,这些基因,在小鼠及人类两者中,于大多原生造血细胞中它的最高水平上被表现,然后像细胞开始分化一样迅速下调。一般地,AHI-1表现的去调节已经出现在许多人类白血病细胞株中,特别是在一CML细胞株 (K562)中及在费城染色体阳性(Ph
发现独自于原生造血细胞中所述小鼠基因Ahi-1的过度表现,以赋予体外增生的优势,且诱发体内致命的白血病;这些效应是通过BCR-ABL增强的。在中BCR-ABL-转导原始人脐带血及从CML患者获取的原生白血病细胞,通过小干扰核糖核酸(small interferingRNA,siRNA)的同源人类基因AHI-1的稳定抑制减少了其体外(in vitro)生长自主性,因此将有望使得这样的细胞减少白血病的生成。一用于siRNA的选择方案为siRNA分子,其被描述于A. Ringrose等人,“在Sezary症候群、人类皮肤T细胞淋巴瘤的白血病变异体中AHI-1的致癌作用的证据”,Leukemia 20:1593-1601(2006),并通过引用将其包括在内。编码这些siRNAs的寡核苷酸是 5’-GATCCCCGTGATGATCCCGACACTATTTCAAGAGAATAGTGTCGGGATCATCACTTTTTA-3’(SEQ ID NO:12)及5’-AGCTTAAAAAGTGATGATCCCGACACTATTCTCTTGAAATAGTGTCGG GATCATCACGGG-3’(SEQ ID NO:13)。此外,Ahi-1的过度表现(在小鼠中) 诱发异常分化(包含系群切换)。因此,独自于IL-3-依赖性造血细胞内小鼠中的Ahi-1的过度表现导致体外及体内强的转化活性,且此为具有BCR-ABL作用的添加剂。Ahi-1的过度表现在小鼠造血干/前驱细胞上赋予一生长优势,且增强BCR-ABL的作用。所述过度表现是通过mRNA转录体的Q-RT-PCR 分析而监视的。此外,尽管表现的水平是少于以Ahi-1转导的细胞,在以BCR-ABL转导的细胞单独与Ahi-1本身没有传递中,已经观察到提高的Ahi-1 表现。
此外,以在一定程度上Ahi-1表现的抑制恢复生长控制且减少生长因子依赖性增殖、形成群落的大小,以及从单一细胞形成表现株的能力,调查于 K562细胞中人类同源物AHI-1、一源自于病患的细胞株的抑制或过度表现的影响,所述病患具有CML且以AHI-1的高度增加表现为特征。相比之下,与控制细胞相比,AHI-1的过度表现会导致群落形成能力的急剧增加;由于 AHI-1的抑制,于细胞中AHI-1基因或AHI-1蛋白的恢复表现受到生长缺陷逆转的siRNA干扰。在AHI-1中,类似的结果被视为在体内表现变化。
同样地,在体内,在原生BCR-ABL-转导人类CB细胞及初级CML干/ 前驱细胞中,AHI-1表现的抑制减少了其生长自主性。通过使用慢病毒RNA 干扰来完成所述抑制。结果表明,在CML中,AHI-1可以在骨髓细胞的过度生成中起到作用。
此外,在从具有对甲磺酸伊马替尼疗法的后来的临床反应(反应者及无反应者两者)的患者的前治疗lin
在一BCR-ABL-转导BaF3细胞株中调查Ahi-1的协同效应,所述 BCR-ABL-转导BaF3细胞株其中的p210
此外,它也被证明,于BCR-ABL-诱导细胞中Ahi-1的共表现遭受了 BCR-ABL的酪氨酸磷酸化,且提高了JAK2及STAT5的活化。明确地说,即使于多西环素的存在中,在已由Ahi-1及BCR-ABL两者所共转导的细胞中,没有显著地抑制p210
此外,在通过免疫共沉淀的CML细胞中,于AHI-1及ABL之间检测一物理性交互作用。在使用抗磷酸酪氨酸抗体的K562细胞中,酪胺酸磷酸化的p210
同时具有JAK2的BCR-ABL及AHI-1的交互作用调解BCR-ABL+细胞的甲磺酸伊马替尼敏感性/抗性。在实验中,以不同剂量的甲磺酸伊马替尼治疗BCR-ABL-转导BaF3细胞及以Ahi-1所共转导的细胞,在IL-3的存在或缺乏中的对于甲磺酸伊马替尼治疗的群落形成细胞的输出反应中,BCR-ABL- 转导细胞表现出显著减少。然而,以BCR-ABL及Ahi-1两者所共传导的BaF3 细胞对于甲磺酸伊马替尼没有反应,且在IL-3的存在中产生一样多的群落形成细胞,还有通过相同的细胞所产生的不能以甲磺酸伊马替尼治疗。尽管这些细胞与在IL-3的存在中的细胞相比对于甲磺酸伊马替尼治疗是更敏感的,但在IL-3的缺乏中的群落形成细胞生成方面,共转导细胞对于甲磺酸伊马替尼也显示了更大的抗药性。这些结果表明,当在这些细胞中活化IL-3信号, Ahi-1能够在BCR-ABL
与这些观察结果一致看法在lin
一个重要的观察结果是,于BCR-ABL–诱导细胞中Ahi-1的共表现可以援救BCR-ABL的递减调节所抑制的生长因子非依赖性细胞生长。有趣的是,此具有Ahi-1的引入的重建的GF非依赖性似乎是通过BCR-ABL的持续磷酸化来调节的,而不是它的持续表现,如同这些作用在体外诱导抑制BCR-ABL 表现的共转导细胞中被观察到。这些结果表明,Ahi-1(在小鼠中)或AHI-1(在人类中)及BCR-ABL之间的物理性交互作用可以稳定一蛋白-蛋白交互作用复合物,所述蛋白-蛋白交互作用复合物能持续活化BCR-ABL酪氨酸激酶活性且进一步改变解除细胞增殖及细胞凋亡控制的特定下游BCR-ABL讯息传递途径。在具有IL-3的GF刺激器的存在中的Ahi-1的具有共传递的BCR-ABL –诱导细胞中,这进一步通过于此交互作用复合物中做为关联蛋白的JAK2 的识别以及JAK2–STAT5途径的活性增强的观察结果来支持。它是已知的, BCR-ABL信号密切模仿细胞激素受体的讯息传递途径,且其IL-3/GM-CSF 受体活化及所述BCR-ABL致癌蛋白两者可以诱发许多蛋白的酪氨酸磷酸化连锁反应,包含JAK2及STAT5,是常见的基质。有趣的是,BCR-ABL-表现细胞与IL-3刺激器所诱发的细胞或具有强迫IL-3过度表现的细胞有许多相似之处。其已经表明,BCR-ABL可以与常见的IL-3/GM-CSF受体的β链交互作用,而本质地活化JAK2。特别是,STAT5的磷酸化增加,其以往被认为是一BCR-ABL致癌蛋白的立即功能,现在已经被证明在初级CML CD34
因此,本发明的另一方面是提供一于患有具有一赋予胸腺核苷激酶抑制剂(TKIs)抗性的生殖细胞缺失多型性的恶性肿瘤的目标对象中的恶性肿瘤的治疗方法,其包含给予:(1)一治疗试剂的治疗有效量至目标对象以治疗所述恶性肿瘤;以及(2)一JAK2抑制剂的治疗有效量至目标对象以治疗所述恶性肿瘤的步骤,所述治疗试剂是选自于由卫康醇、卫康醇的衍生物或类似物、二乙酰二脱水卫矛醇、二乙酰二脱水卫矛醇的衍生物或类似物、二溴卫矛醇及二溴卫矛醇的衍生物或类似物所组成的群组。
所述JAK2抑制剂可以是但不限于(E)-N-苄基-2-氰基-3-(3,4-二羟苯基)- 丙烯酰胺(AG490)、鲁索利替尼(ruxolitinib)、托法替尼(tofacitinib)、托法替尼柠檬酸盐、N-叔-丁基-3-(5-甲基-2-(4-(2-(吡咯啶-1-基)乙氧基)苯胺基)嘧啶-4- 基胺)苯磺酰胺(TG-101348)、(S)-5-氯基-N2-(1-(5-氟代嘧啶-2-基)乙基)-N4-(5- 甲基-1H-吡唑-3-基)嘧啶-2,4-二胺(AZD1480)、N-(-氰甲基-)-4-(2-(4-吗啉基苯胺基)嘧啶-4-基)苯甲酰胺(CYT387)、巴利西替尼(baricitinib)、(S,E)-3-(6-溴吡啶-2-基)-2-氰基-N-(1-苯基乙基)-丙烯酰胺(WP1066)、S-鲁索利替尼、N-叔- 丁基-3-(5-甲基-2-(4-(4-甲基哌嗪-1-基)苯胺基)嘧啶-4-基胺)苯磺酰胺 (TG101209)、N-[3-(4-甲基-1-哌嗪基)苯基]-8-[4-(甲磺酰基)苯基]-[1,2,4]三唑 [1,5-a]吡啶-2-胺(CEP33779)、8-(3,5-二氟代-4-(吗啉甲基)苯基)-2-(1-(哌啶-4- 基)-1H-吡唑-4-基)喹喔啉(NVP-BSK805)、(S)-5-氟代-2-(1-(4-氟代苯基)乙胺基)-6-(5-甲基-1H-吡唑-3-基胺)烟碱甲腈(AZ 960)、3-(4-氯基-2-氟代苄基)-2- 甲基-N-(3-甲基-1H-吡唑-5-基)-8-(吗啉甲基)咪唑并[1,2-b]哒嗪-6-胺(LY2784544)、1-环丙基-3-(3-(5-(吗啉甲基)-1H-苯并[d]咪唑-2-基)-1H-吡唑-4- 基)尿素(AT9283)、帕克利替尼(pacritinib,SB1518)、(S)-N-(4-(2-((4-吗啉苯基)胺基)嘧啶-4-基)苯基)吡咯啶-2-羧酰胺(XL019),以及N-叔-丁基-3-(5-甲基 -2-(4-(2-(吡咯啶-1-基)乙氧基)苯胺基)嘧啶-4-基胺)苯磺酰胺(TG101348)。其他的JAK2抑f制剂于本领域中为已知的,且透过Sayeski等人被描述于美国专利第8,367,078号,并通过引用将其包括在内,包含2-甲基-1-苯基-4-吡啶 -2-基-2-(2-吡啶-2-基乙基)丁-1-酮、3-[5-[(4-氧基-4-苯基-丁-2-亚基)胺基]戊基亚胺基]-1-苯基-丁-1-酮、2-(二乙胺基甲基)-4-[4-[3-(二乙胺基甲基)-4-羟基- 苯基]己-3-烯-3-基]苯酚、2-二丁氧基磷酰氧基戊腈、镱(+3)阳离子三羟化物,以及4-[(1S)-6,7-二乙氧基-1,2,3,4-四氢异喹啉-1-基]氰苯。尽管如此,其他的JAK2抑制剂透过Bourke等人被描述于美国专利第8,354,408号,并通过引用将其包括在内,包含7-碘基-N-(4-吗啉苯基)噻咔[3,2-d]嘧啶-2-胺、7-(4-胺苯基)-N-(4-吗啉苯基)噻咔[3,2-d]嘧啶-2-胺、N-(4-(2-(4-吗啉基苯胺基)噻咔 [3,2-d]嘧啶-7-基)苯基)-丙烯酰胺、7-(3-胺苯基)-N-(4-吗啉苯基)噻咔[3,2-d]嘧啶-2-胺、N-(3-(2-(4-吗啉基苯胺基)噻咔[3,2-d]嘧啶-7-基)苯基)-丙烯酰胺、甲基2-(4-吗啉基苯胺基)噻咔[3,2-d]嘧啶-7-羧酸盐、7-(4-胺基-3-甲氧苯基)-N-(4-吗啉苯基)噻咔[3,2-d]嘧啶-2-胺、4-(2-(4-吗啉基苯胺基)噻咔[3,2-d] 嘧啶-7-基)苯磺酰胺、N,N-二甲基-3-(2-(4-吗啉基苯胺基)噻咔[3,2-d]嘧啶-7- 基)苯磺酰胺、1-乙基-3-(2-甲氧基-4-(2-(4-吗啉基苯胺基)噻咔[3,2-d]嘧啶-7- 基)苯基)尿素、N-(4-(2-(4-吗啉基苯胺基)噻咔[3,2-d]嘧啶-7-基)苯基)甲烷磺酰胺、2-甲氧基-4-(2-(4-吗啉基苯胺基)噻咔[3,2-d]嘧啶-7-基)苯酚、2-氰基 -N-(3-(2-(4-吗啉基苯胺基)噻咔[3,2-d]嘧啶-7-基)苯基)乙酰胺、N-(-氰甲基 -)-2-(4-吗啉基苯胺基)噻咔[3,2-d]嘧啶-7-羧酰胺、N-(3-(2-(4-吗啉基苯胺基) 噻咔[3,2-d]嘧啶-7-基)苯基)甲烷磺酰胺、1-乙基-3-(4-(2-(4-吗啉基苯胺基)噻咔[3,2-d]嘧啶-7-基)-2-(三氟代甲氧基)苯基)尿素、N-(3-硝基苯基)-7-苯基噻咔 [3,2-d]嘧啶-2-胺、7-碘基-N-(3-硝基苯基)噻咔[3,2-d]嘧啶-2-胺、N1-(7-(2-乙基苯基)噻咔[3,2-d]嘧啶-2-基)苯-1,3-二胺、叔-丁基-3-(2-(4-吗啉基苯胺基)噻咔[3,2-d]嘧啶-7-基)苯磺酰胺、N1-(7-碘噻咔[3,2-d]嘧啶-2-基)苯-1,3-二胺、 7-(4-胺基-3-(三氟代甲氧基)苯基)-N-(4-吗啉苯基)噻咔[3,2-d]嘧啶-2-胺、7-(2- 乙基苯基)-N-(4-吗啉苯基)噻咔[3,2-d]嘧啶-2-胺、N-(3-(2-(4-吗啉基苯胺基)噻咔[3,2-d]嘧啶-7-基)苯基)乙酰胺、N-(-氰甲基-)-N-(3-(2-(4-吗啉基苯胺基)噻咔 [3,2-d]嘧啶-7-基)苯基)甲烷磺酰胺、N-(-氰甲基-)-N-(4-(2-(4-吗啉基苯胺基) 噻咔[3,2-d]嘧啶-7-基)苯基)甲烷磺酰胺、N-(3-(5-甲基-2-(4-吗啉基苯胺基)-5H-吡咯并[3,2-d]嘧啶-7-基)苯基)甲烷磺酰胺、4-(5-甲基-2-(4-吗啉基苯胺基)-5H-吡咯并[3,2-d]嘧啶-7-基)苯磺酰胺、N-(4-(5-甲基-2-(4-吗啉基苯胺基)-5H-吡咯并[3,2-d]嘧啶-7-基)苯基)甲烷磺酰胺、7-碘基-N-(4-吗啉苯基)-5H-吡咯并[3,2-d]嘧啶-2-胺、7-(2-异丙基苯基)-N-(4-吗啉苯基)噻咔[3,2-d] 嘧啶-2-胺、7-溴-N-(4-吗啉苯基)噻咔[3,2-d]嘧啶-2-胺、N7-(2-异丙基苯基)-N2-(4-吗啉苯基)噻咔[3,2-d]嘧啶-2,7-二胺、N7-(4-异丙基苯基)-N2-(4-吗啉苯基)噻咔[3,2-d]嘧啶-2,7-二胺、7-(5-胺基-2-甲苯基)-N-(4-吗啉苯基)噻咔 [3,2-d]嘧啶-2-胺、N-(-氰甲基-)-4-(2-(4-吗啉基苯胺基)噻咔[3,2-d]嘧啶-7-基) 苯甲酰胺、7-碘基-N-(3-吗啉苯基)噻咔[3,2-d]嘧啶-2-胺、7-(4-胺基-3-硝基苯基)-N-(4-吗啉苯基)噻咔[3,2-d]嘧啶-2-胺、7-(2-甲氧基吡啶-3-基)-N-(4-吗啉苯基)噻咔[3,2-d]嘧啶-2-胺、(3-(7-碘噻咔[3,2-d]嘧啶-2-基胺)苯基)甲醇、N-叔- 丁基-3-(2-(3-吗啉基苯胺基)噻咔[3,2-d]嘧啶-7-基)苯磺酰胺、N-叔-丁基 -3-(2-(3-(羟甲基)苯胺基)噻咔[3,2-d]嘧啶-7-基)苯磺酰胺、N-(4-吗啉苯基)-7-(4-硝基苯基硫基)-5H-吡咯并[3,2-d]嘧啶-2-胺、N-叔-丁基-3-(2-(3,4,5- 三甲氧基苯胺)噻咔[3,2-d]嘧啶-7-基)苯磺酰胺、7-(4-胺基-3-硝基苯基)-N-(3,4-二甲氧苯基)噻咔[3,2-d]嘧啶-2-胺、N-(3,4-二甲氧苯基)-7-(2-甲氧基吡啶-3-基)噻咔[3,2-d]嘧啶-2-胺、N-叔-丁基-3-(2-(3,4-二甲氧基苯胺)噻咔 [3,2-d]嘧啶-7-基)苯磺酰胺、7-(2-胺基嘧啶-5-基)-N-(3,4-二甲氧苯基)噻咔 [3,2-d]嘧啶-2-胺、N-(3,4-二甲氧苯基)-7-(2,6-二甲氧基吡啶-3-基)噻咔[3,2-d] 嘧啶-2-胺;N-(3,4-二甲氧苯基)-7-(2,4-二甲氧基嘧啶-5-基)噻咔[3,2-d]嘧啶-2- 胺、7-碘基-N-(4-(吗啉甲基)苯基)噻咔[3,2-d]嘧啶-2-胺、N-叔-丁基-3-(2-(4-(吗啉甲基)苯胺基)噻咔[3,2-d]嘧啶-7-基)苯磺酰胺、2-氰基-N-(4-甲基-3-(2-(4-吗啉基苯胺基)噻咔[3,2-d]嘧啶-7-基)苯基)乙酰胺、乙基3-(2-(4-吗啉基苯胺基) 噻咔[3,2-d]嘧啶-7-基)苯甲酸盐、7-溴-N-(4-(2-(吡咯啶-1-基)乙氧基)苯基)噻咔[3,2-d]嘧啶-2-胺、N-(3-(2-(4-(2-(吡咯啶-1-基)乙氧基)苯胺基)噻咔[3,2-d]嘧啶 -7-基)苯基)乙酰胺、N-(-氰甲基-)-3-(2-(4-吗啉基苯胺基)噻咔[3,2-d]嘧啶-7-基) 苯甲酰胺、N-叔-丁基-3-(2-(4-吗啉基苯胺基)噻咔[3,2-d]嘧啶-7-基)苯甲酰胺、 N-叔-丁基-3-(2-(4-(1-乙基哌啶-4-基氧基)苯胺基)噻咔[3,2-d]嘧啶-7-基)苯磺酰胺、叔-丁基4-(2-(4-(吗啉甲基)苯胺基)噻咔[3,2-d]嘧啶-7-基)-1H-吡唑-1- 羧酸盐、7-溴-N-(4-((4-乙基哌嗪-1-基)甲基)苯基)噻咔[3,2-d]嘧啶-2-胺、N-叔 -丁基-3-(2-(4-((4-乙基哌嗪-1-基)甲基)苯胺基)噻咔[3,2-d]嘧啶-7-基)苯磺酰胺、N-(4-((4-乙基哌嗪-1-基)甲基)苯基)-7-(1H-吡唑-4-基)噻咔[3,2-d]嘧啶-2- 胺、N-(-氰甲基-)-3-(2-(4-(吗啉甲基)苯胺基)噻咔[3,2-d]嘧啶-7-基)苯甲酰胺、 N-叔-丁基-3-(2-(4-(2-(吡咯啶-1-基)乙氧基)苯胺基)噻咔[3,2-d]嘧啶-7-基)苯磺酰胺、叔-丁基吡咯啶-1-基)乙氧基)苯胺基)噻咔[3,2-d]嘧啶-7-基)苄基胺基甲酸盐、3-(2-(4-(2-(吡咯啶-1-基)乙氧基)苯胺基)噻咔[3,2-d]嘧啶-7-基)苯磺酰胺、7-(3-氯基-4-氟代苯基)-N-(4-(2-(吡咯啶-1-基)乙氧基)苯基)噻咔[3,2-d]嘧啶-2-胺、叔-丁基4-(2-(4-(1-乙基哌啶-4-基氧基)苯胺基)噻咔[3,2-d]嘧啶-7- 基)-1H-吡唑-1-羧酸盐、7-(苯并[d][1,3]二氧基-5-基)-N-(4-(吗啉甲基)苯基)噻咔[3,2-d]嘧啶-2-胺、叔-丁基5-(2-(4-(吗啉甲基)苯胺基)噻咔[3,2-d]嘧啶-7- 基)-1H-吲哚-1-羧酸盐、7-(2-胺基嘧啶-5-基)-N-(4-(吗啉甲基)苯基)噻咔[3,2-d] 嘧啶-2-胺、叔-丁基4-(2-(4-(吗啉甲基)苯胺基)噻咔[3,2-d]嘧啶-7-基)-5,6-二- 氢吡啶-1(2H)-羧酸盐、叔-丁基4-(2-(4-(吗啉甲基)苯胺基)噻咔[3,2-d]嘧啶-7- 基)苄基胺基甲酸盐、N-(3-(2-(4-(吗啉甲基)苯胺基)噻咔[3,2-d]嘧啶-7-基)苯基) 乙酰胺、N-(4-(2-(4-(吗啉甲基)苯胺基)噻咔[3,2-d]嘧啶-7-基)苯基)乙酰胺、 N-(3-(2-(4-(吗啉甲基)苯胺基)噻咔[3,2-d]嘧啶-7-基)苯基)甲烷磺酰胺、7-(4-(4- 甲基哌嗪-1-基)苯基)-N-(4-(吗啉甲基)苯基)噻咔[3,2-d]嘧啶-2-胺、N-(2-甲氧基-4-(2-(4-(吗啉甲基)苯胺基)噻咔[3,2-d]嘧啶-7-基)苯基)乙酰胺、7-溴 -N-(3,4,5-三甲氧苯基)噻咔[3,2-d]嘧啶-2-胺、(3-(2-(3,4,5-三甲氧基苯胺)噻咔[3,2-d]嘧啶-7-基)苯基)甲醇、(4-(2-(3,4,5-三甲氧基苯胺)噻咔[3,2-d]嘧啶-7-基) 苯基)甲醇、(3-(2-(4-吗啉基苯胺基)噻咔[3,2-d]嘧啶-7-基)苯基)甲醇、(4-(2-(4- 吗啉基苯胺基)噻咔[3,2-d]嘧啶-7-基)苯基)甲醇、N-(吡咯啶-1-基)乙氧基)苯胺基)噻咔[3,2-d]嘧啶-7-基)苄基)甲烷磺酰胺、叔-丁基3-(2-(4-(吗啉甲基)苯胺基)噻咔[3,2-d]嘧啶-7-基)苄基胺基甲酸盐、N-(4-(吗啉甲基)苯基)-7-(3-(哌嗪 -1-基)苯基)噻咔[3,2-d]嘧啶-2-胺、7-(6-(2-吗啉乙胺基)吡啶-3-基)-N-(3,4,5-三甲氧苯基)噻咔[3,2-d]嘧啶-2-胺、7-(2-乙基苯基)-N-(4-(吡咯啶-1-基)乙氧基) 苯基)噻咔[3,2-d]嘧啶-2-胺、7-(2-异丙基苯基)-N-(4-(吡咯啶-1-基)乙氧基)苯基 (噻咔[3,2-d]嘧啶-2-胺、7-(4-(胺甲基)苯基)-N-(4-(吗啉甲基)苯基)噻咔[3,2-d] 嘧啶-2-胺、N-(4-(1-乙基哌啶-4-基氧基)苯基)-7-(1H-吡唑-4-基)噻咔[3,2-d]嘧啶-2-胺、N-(2,4-二甲氧苯基)-7-苯基噻咔[3,2-d]嘧啶-2-胺、7-溴-N-(3,4-二甲氧苯基)噻咔[3,2-d]嘧啶-2-胺,或N-(3,4-二甲氧苯基)-7-苯基噻咔[3,2-d]嘧啶 -2-胺。尽管如此,其他JAK2激酶抑制剂透过Li等人被描述于美国专利第8,309,718号,并通过引用将其包括在内,包含4-吡唑基-N-芳基嘧啶-2-胺,以及4-吡唑基-N-杂芳基嘧啶-2-胺。尽管如此,其他JAK2激酶抑制剂透过 Menet等人被描述于美国专利第8,242,274号,并通过引用将其包括在内,包含[1,2,4]三唑[1,5-a]吡啶类。尽管如此,其他JAK2激酶抑制剂透过Rodgers 等人被描述于美国专利第8,158,616号,并通过引用将其包括在内,包含三亚甲亚胺及环丁烷;一优选的化合物为1-(乙磺酰基)-3-[4-(7H-吡咯并[2,3-d] 嘧啶-4-基)-1H-吡唑-1-基]环氮丁烷-3-基}乙睛。尽管如此,其他JAK2激酶抑制剂透过Noronha等人被描述于美国专利第8,138,199号,并通过引用将其包括在内,包含联芳偏嘧啶化合物。尽管如此,其他JAK2激酶抑制剂透过Rodgers等人被描述于美国专利第8,053,433号,并通过引用将其包括在内,包含吡咯并[2,3-b]吡啶-4-基-胺,以及吡咯并[2,3-b]嘧啶-5-基-胺。尽管如此,其他JAK2激酶抑制剂透过Burkholder等人被描述于美国专利第7,897,600 号,并通过引用将其包括在内,包含3-(4-氯基-2-氟代苄基)-2-甲基-N-(5-甲基-1H-吡唑-3-基)-8-(吗啉甲基)咪唑并[1,2-b]哒嗪-6-胺。尽管如此,其他JAK2 激酶抑制剂透过Rodgers等人被描述于美国专利第7,598,257号,并通过引用将其包括在内,包含杂芳基取代的吡咯并[2,3-b]吡啶类,以及杂芳基取代的吡咯并[2,3-b]嘧啶。尽管如此,其他JAK2激酶抑制剂透过Rodgers等人被描述于美国专利第7,355,677号,并通过引用将其包括在内,包含吡咯并[2,3-b] 吡啶-4-基胺类,以及吡咯并[2,3-b]嘧啶-4-基胺类。其他JAK2激酶抑制剂于本领域中为已知的。
所述方法可以进一步包含:给予一BH3类似物的治疗有效量至目标对象的步骤。或者,所述方法可以进一步包含:给予一酪氨酸激酶抑制剂的治疗有效量至目标对象的步骤。
本发明的再一方面是提供一于患有具有一赋予胸腺核苷激酶抑制剂 (TKIs)抗性的生殖细胞缺失多型性的恶性肿瘤的目标对象中的恶性肿瘤的治疗方法,其包含给予:(1)一治疗试剂的治疗有效量至目标对象以治疗所述恶性肿瘤;以及(2)一STAT5抑制剂的治疗有效量至目标对象以治疗所述恶性肿瘤的步骤,所述治疗试剂是选自于由卫康醇、卫康醇的衍生物或类似物、二乙酰二脱水卫矛醇、二乙酰二脱水卫矛醇的衍生物或类似物、二溴卫矛醇及二溴卫矛醇的衍生物或类似物所组成的群组。
STAT5抑制剂包含但不限于N′-((4-氧基-4H-克唍-3-基)甲烯基)烟碱酰肼及派迷清(pimozide)。其他的STAT 5抑制剂透过Frank被公开于美国专利申请公开第2011/0144043号,并通过引用将其包括在内,包含乙胺嘧啶、醋酸胍那苄、阿普洛尔(alprenolol)氢氯化物、硝呋酚酰肼、茄碱α(solanine alpha)、氟洛色丁(fluoxetine)氢氯化物、异环磷酰胺、羟萘酸派文尼(pyrvinium pamoate)、莫雷西嗪(moricizine)氢氯化物、3,3’–氧基双[四氢噻吩,1,1,1’,1’–四氧],2-(1,8-萘啶-2-基)苯酚,以及3-(2-羟苯基)-3-苯基-N,N-二丙基丙酰胺。
所述方法可以进一步包含:给予一BH3类似物的治疗有效量至目标对象的步骤。或者,所述方法可以进一步包含:给予一酪氨酸激酶抑制剂的治疗有效量至目标对象的步骤。
本发明的再一方面是提供一于患有具有一赋予胸腺核苷激酶抑制剂 (TKIs)抗性的生殖细胞缺失多型性的恶性肿瘤的目标对象中的恶性肿瘤的治疗方法,其包含给予:(1)一治疗试剂的治疗有效量至目标对象以治疗所述恶性肿瘤;以及(2)一Src抑制剂的治疗有效量至目标对象以治疗所述恶性肿瘤的步骤,所述治疗试剂是选自于由卫康醇、卫康醇的衍生物或类似物、二乙酰二脱水卫矛醇、二乙酰二脱水卫矛醇的衍生物或类似物、二溴卫矛醇,以及二溴卫矛醇的衍生物或类似物所组成的群组。
Src抑制剂包含达沙替尼、塞卡替尼(saracatinib)、伯舒替尼、N-苄基 -2-(5-(4-(2-吗啉乙氧基)苯基)吡啶-2-基)乙酰胺(KX2-391)、CGP76030,以及 4-甲基-3-(1-甲基-6-(吡啶-3-基)-1H-吡唑并[3,4-d]嘧啶-4-基胺)-N-(3-(三氟代甲基)苯基)苯甲酰胺(NVP-BHG712)。Src激酶抑制剂被描述于M.Missbach 等人,“取代的5,7-二苯基-吡咯并[2,3-d]嘧啶:酪氨酸激酶c-Src的强效抑制剂”,Bioorg.Med.Chem.Lett.10:945-949(2000),并通过引用将其包括在内。其他的Src抑制剂于本领域中为已知的,且透过Desai等人被描述于美国专利第8,389,525号,并通过引用将其包括在内,包含-[[6-(4-溴基苯基)-3-氰基-4-(三氟代甲基)-2-吡啶基]硫基]-苯乙酸、1,4-二氢-2-[[[4-(甲氧基羰基)苯基] 甲基]硫基]-5-甲基-4-氧基-噻咔-[2,3-d]嘧啶-6-羧酸、6-羟基-7-氧基-7H-苯并[e] 呸啶-4-磺酸、7-乙氧基-11H-茚[1,2-b]喹喔啉-11-酮、5-溴-1,3-二氢-3-羟基 -3-[2-氧基-2-(5,6,7,8-四氢-2-萘基)乙基]-2H-吲哚-2-酮、1-(4-氟代苯基)-2-(9H- 噻吨-9-基)-1,3-丁二酮、2-[[(2-氯基-6-氟代苯基)甲基]硫基]-3-(3-吡啶基)-4(3H)-喹唑啉酮、2,7-二硝基-肟-9H-芴-9-酮,以及3-(9H-芴-9-基甲基)酯 -3,4-四氢噻唑二羧酸。Xie等人所申请的美国专利第8,283,441号,通过引用将其包括,其描述胜肽包含连接至Src激酶区域的螺旋或类螺旋结构。 Hangauer,Jr.所申请的美国专利第8,236,799号,通过引用将其包括,其描述 Src激酶抑制剂的联芳化合物。Hangauer,Jr.等人所申请的美国专利第 8,088,768号,并通过引用将其包括在内,其描述萘基Src抑制剂支架、异喹啉基Src抑制剂支架及吲哚基Src抑制剂支架。Byzova等人所申请的美国专利第8,080,252号,通过引用将其包括,其描述作为Src抑制剂的3-(4,5,6,7- 四氢-2-基亚甲基)-2-吲哚酮衍生物,包含2-氧基-3(4,5,6,7-四氢-1-H-吲哚-2- 基-甲烯基)-2,3-二氢-1-H-吲哚-5-磺酸二甲胺,以及2-氧基-3(4,5,6,7-四氢 -1-H-吲哚-2-基-甲烯基)-2,3-二氢-1H-吲哚-5-磺酸胺。Honold等人所申请的美国专利第8,058,283号,并通过引用将其包括在内,其描述作为Src抑制剂的 7H-吡啶并[3,4-d]嘧啶-8-酮类。Bebbington等人所申请的美国专利第7,982,037 号,并通过引用将其包括在内,其描述作为Src激酶抑制剂的吡唑化合物。 Bebbington等人所申请的美国专利第7,951,820号,并通过引用将其包括在内,其描述作为Src激酶抑制剂的三唑化合物。Boschelli等人所申请的美国专利第7,919,625号,并通过引用将其包括在内,其描述作为Src抑制剂的 4-苯胺基-3-甲腈,包含4-[(2,4-二氯-5-甲氧苯基)胺基]-6-甲氧基-7-[3-(4-甲基 -1-哌嗪基)丙氧基]-3-甲腈。Aronov等人所申请的美国专利第7,842,712号,并通过引用将其包括在内,其描述作为Src抑制剂的吲唑啉酮。Fukumoto等人所申请的美国专利第7,842,701号,并通过引用将其包括在内,其描述作为Src抑制剂的吡唑并喹诺酮衍生物,包含3-胺基-2-(2-氯基-5-羟苯基)-7-(3- 吗啉-4-基丙氧基)-2,5-二氢-4H-吡唑并[4,3-c]喹啉-4-酮、3-胺基-2-(2-氯基-5- 羟苯基)-7-(2-吗啉-4-基乙氧基)-2,5-二氢-4H-吡唑并[4,3-c]喹啉-4-酮、3-胺基 -2-(5-羟基-2-甲基苯基)-7-(3-吗啉-4-2,5-二氢-4H-吡唑并[4,3-c]喹啉-4-酮、3- 胺基-2-(5-羟基-2-基丙氧基)甲苯基)-7-(2-吗啉-4-基乙氧基)-2,5-二氢-4H-吡唑并[4,3-c]喹啉-4-酮。Honold等人所申请的美国专利第7,786,113号,并通过引用将其包括在内,其描述作为Src抑制剂的杂环胺基甲酸盐衍生物,包含(2-苯基-1H-吡咯并[2,3-b]吡啶-5-基)-胺甲酸异丙酯、{2-[3-(2-甲氧基-乙氧基)- 苯基]-1H-吡咯并[2,3-b]吡啶-5-基}-胺甲酸异丙酯、(2-苯基-1H-吡咯并[2,3-b] 吡啶-5-基)-胺甲酸2,2-二甲基-丙酯、{2-[4-(2-甲氧基-乙氧基)-苯基]-1H-吡咯并[2,3-b]吡啶-5-基}-胺甲酸乙酯、{2-[4-(2-甲氧基-乙氧基)-苯基]-1H-吡咯并 [2,3-b]吡啶-5-基}-胺甲酸烯丙酯、{2-[3-(2-甲氧基-乙氧基)-苯基]-1H-吡咯并 [2,3-b]吡啶-5-基}-胺甲酸乙酯、{2-[3-(2-甲氧基-乙氧基)-苯基]-1H-吡咯并 [2,3-b]吡啶-5-基}-胺甲酸烯丙酯、(2-苯基-1H-吡咯并[2,3-b]吡啶-5-基)-胺甲酸苄酯、{2-[3-(2-甲氧基-乙氧基)-苯基]-1H-吡咯并[2,3-b]吡啶-5-基}-胺甲酸异丁基酯、{2-[3-(2-甲氧基-乙氧基)-苯基]-1H-吡咯并[2,3-b]吡啶-5-基}-胺甲酸 2-氯基-苄基酯、(2-苯基-1H-吡咯并[2,3-b]吡啶-5-基)-胺甲酸乙酯、(2-苯基-1H- 吡咯并[2,3-b]吡啶-5-基)-胺甲酸2-氯基-苄基酯、(2-苯基-1H-吡咯并[2,3-b] 吡啶-5-基)-胺甲酸烯丙酯、(2-苯基-1H-吡咯并[2,3-b]吡啶-5-基)-胺甲酸异丁基酯、{2-[3-(2-甲氧基-乙氧基)-苯基]-1H-吡咯并[2,3-b]吡啶-5-基}-胺甲酸 2,2-二甲基-丙酯、{2-[3-(2-甲氧基-乙氧基)-苯基]-1H-吡咯并[2,3-b]吡啶-5- 基}-胺甲酸苄酯、{2-[4-(2-甲氧基-乙氧基)-苯基]-1H-吡咯并[2,3-b]吡啶-5- 基}-胺甲酸异丙酯、{2-[4-(2-甲氧基-乙氧基)-苯基]-1H-吡咯并[2,3-b]吡啶-5- 基}-胺甲酸苄酯、{2-[4-(2-甲氧基-乙氧基)-苯基]-1H-吡咯并[2,3-b]吡啶-5- 基}-胺甲酸异丁基酯、[2-(3-乙酰胺基-苯基)-1H-吡咯并[2,3-b]吡啶-5-基]-胺甲酸异丙酯、[2-(4-乙酰胺基-苯基)-1H-吡咯并[2,3-b]吡啶-5-基]-胺甲酸烯丙酯、 [2-(3-乙酰胺基-苯基)-1H-吡咯并[2,3-b]吡啶-5-基]-胺甲酸2-氯基-苄基酯、 [2-(3-乙酰胺基-苯基)-1H-吡咯并[2,3-b]吡啶-5-基]-胺甲酸苄酯、[2-(3-乙酰胺基-苯基)-1H-吡咯并[2,3-b]吡啶-5-基]-胺甲酸2,2-二甲基-丙酯、[2-(4-乙酰胺基-苯基)-1H-吡咯并[2,3-b]吡啶-5-基]-胺甲酸2,2-二甲基-丙酯、[2-(3-乙酰胺基-苯基)-1H-吡咯并[2,3-b]吡啶-5-基]-胺甲酸乙酯、[2-(3-乙酰胺基-苯基)-1H- 吡咯并[2,3-b]吡啶-5-基]-胺甲酸烯丙酯、[2-(3-乙酰胺基-苯基)-1H-吡咯并[2,3-b]吡啶-5-基]-胺甲酸异丁基酯、(2-苯基-3H-咪唑并[4,5-b]吡啶-6-基)-胺甲酸1-甲基-丙烯酯、(2-苯基-1H-咪唑并[4,5-b]吡啶-6-基)-胺甲酸异丙酯、(2- 苯基-3H-咪唑并[4,5-b]吡啶-6-基)-胺甲酸环己酯、(2-苯基-3H-咪唑并[4,5-b] 吡啶-6-基)-胺甲酸异丁基酯、(2-苯基-3H-咪唑并[4,5-b]吡啶-6-基)-胺甲酸烯丙酯、(2-苯基-3H-咪唑并[4,5-b]吡啶-6-基)-胺甲酸叔-丁基酯、(2-苯基-3H-咪唑并[4,5-b]吡啶-6-基)-胺甲酸环戊酯、(2-苯基-3H-咪唑并[4,5-b]吡啶-6-基)- 胺甲酸乙酯、(2-苯基-3H-咪唑并[4,5-b]吡啶-6-基)-胺甲酸仲丁基酯、(2-苯基 -3H-咪唑并[4,5-b]吡啶-6-基)-胺甲酸1-乙基-丙酯、(2-苯基-3H-咪唑并[4,5-b] 吡啶-6-基)-胺甲酸2,2,2-三氟代-1-甲基-乙基酯、(2-苯基-3H-咪唑并[4,5-b]吡啶-6-基)-胺甲酸2,2-二甲基-丙酯、(2-苯基-3H-咪唑并[4,5-b]吡啶-6-基)-胺甲酸1-苯基-乙基酯、(2-苯基-1H-咪唑并[4,5-b]吡啶-6-基)-胺甲酸丙酯、(2-苯基-3H-咪唑并[4,5-b]吡啶-6-基)-胺甲酸1-甲基-丙-2-炔基酯、(2-苯基-3H-咪唑并[4,5-b]吡啶-6-基)-胺甲酸1-甲基-丁-2-炔基酯、(2-苯基-3H-咪唑并[4,5-b] 吡啶-6-基)-胺甲酸环丁基酯、(2-苯基-3H-咪唑并[4,5-b]吡啶-6-基)-胺甲酸 1,3-二甲基-丁基酯、(2-苯基-3H-咪唑并[4,5-b]吡啶-6-基)-胺甲酸1,2-二甲基- 丙酯、{2-[3-(3-甲氧基-丙酰胺基)-苯基]-3H-咪唑并[4,5-b]吡啶-6-基}-胺甲酸异丙酯、(2-苯基-3H-咪唑并[4,5-b]吡啶-6-基)-胺甲酸(E)-1-甲基-丁-2-烯基酯、 (2-苯基-3H-咪唑并[4,5-b]吡啶-6-基)-胺甲酸1,2-二甲基-丙烯酯、{2-[4-(2-二乙胺基-乙氧基)-苯基]-3H-咪唑并[4,5-b]吡啶-6-基}-胺甲酸异丙酯、2-[3-(2- 甲氧基-乙氧基)-苯基]-3H-咪唑并[4,5-b]吡啶-6-基}-胺甲酸异丙酯、{2-[4-(2- 甲氧基-乙氧基)-苯基]-3H-咪唑并[4,5-b]吡啶-6-基}-胺甲酸异丙酯、[2-(3-硝基 -苯基)-3H-咪唑并[4,5-b]吡啶-6-基]-胺甲酸异丙酯、[2-(4-吗啉-4-基-苯基)-3H- 咪唑并[4,5-b]吡啶-6-基]-酰胺酸异丙酯、{2-[4-(4-甲基-哌嗪-1-基)-苯基]-3H- 咪唑并[4,5-b]吡啶-6-基}-胺甲酸异丙酯、{2-[3-(2-羟基-乙基)-苯基]-3H-咪唑并[4,5-b]吡啶-6-基}-胺甲酸异丙酯、[2-(4-硝基-苯基)-3H-咪唑并[4,5-b]吡啶-6-基]-胺甲酸异丙酯、[2-(4-胺磺酰基-苯基)-3H-咪唑并[4,5-b]吡啶-6-基]-胺甲酸异丙酯、[2-(4-甲基磺酰基-苯基)-3H-咪唑并[4,5-b]吡啶-6-基]-胺甲酸异丙酯、[2-(3-胺基-苯基)-3H-咪唑并[4,5-b]吡啶-6-基]-胺甲酸异丙酯、[2-(4-胺基- 苯基)-3H-咪唑并[4,5-b]吡啶-6-基]-胺甲酸异丙酯、[2-(3-乙酰胺基-苯基)-3H- 咪唑并[4,5-b]吡啶-6-基]-胺甲酸异丙酯、[2-(3-甲烷磺酰基胺基-苯基)-3H-咪唑并[4,5-b]吡啶-6-基]-胺甲酸异丙酯、[2-(3-甲基亚磺酰基-苯基)-3H-咪唑并 [4,5-b]吡啶-6-基]-胺甲酸异丙酯、[2-(3-甲磺酰基-苯基)-3H-咪唑并[4,5-b]吡啶 -6-基]-胺甲酸异丙酯、[2-(4-甲烷磺酰基-苯基)-3H-咪唑并[4,5-b]吡啶-6-基]- 胺甲酸异丙酯、[2-(4-甲烷亚磺酰基-苯基)-3H-咪唑并[4,5-b]吡啶-6-基]-胺甲酸异丙酯、[2-(3,4-二氟代-苯基)-3H-咪唑并[4,5-b]吡啶-6-基]-胺甲酸异丙酯、 (2-{4-[双-(2-甲氧基-乙基)-胺基]-3-氟代-苯基}-3H-咪唑并[4,5-b]吡啶-6-基)- 胺甲酸异丙酯、3-(6-异丙氧基羰胺基-3H-咪唑并[4,5-b]吡啶-2-基)-苯甲酸、 {2-[3-(2-甲氧基-I-甲氧基甲基-乙基胺甲酰基)-苯基]-3H-咪唑并[4,5-b]吡啶-6- 基}-胺甲酸异丙酯、{2-[3-(3-甲氧基-丙基胺甲酰基)-苯基]-3H-咪唑并[4,5-b]吡啶-6-基}-胺甲酸异丙酯、(2-噻吩-2-基-3H-咪唑并[4,5-b]吡啶-6-基)-胺甲酸异丙酯、(2-噻吩-3-基-3H-咪唑并[4,5-b]吡啶-6-基)-胺甲酸异丙酯、[2-(2-甲基 -吡啶-4-基)-3H-咪唑并[4,5-b]吡啶-6-基]-胺甲酸异丙酯、[2-(6-甲基-吡啶-3- 基)-3H-咪唑并[4,5-b]吡啶-6-基]-胺甲酸异丙酯、[2-(1H-苯并咪唑基-5-基)-3H- 咪唑并[4,5-b]吡啶-6-基]-胺甲酸异丙酯、[2-(2-氯基-吡啶-4-基)-3H-咪唑并 [4,5-b]吡啶-6-基]-胺甲酸异丙酯,以及{2-[2-(3-甲氧基-丙基胺基)-吡啶-4- 基]-3H-咪唑并[4,5-b]吡啶-6-基}-胺甲酸异丙酯。Boyd等人所申请的美国专利第7,776,878号,并通过引用将其包括在内,其描述作为Src抑制剂的杂环苄基胺基衍生物,包含(1-苯基-乙基)-(2-苯基-3H-咪唑并[4,5-b]吡啶-6-基)-胺、苄基-{2-[3-(2-甲氧基-乙氧基)-苯基]-1H-吡咯并[2,3-b]吡啶-5-基}-胺、{2-[3-(2-甲氧基-乙氧基)-苯基]-1H-吡咯并[2,3-b]吡啶-5-基}-(2-甲基-苄基)-胺、苄基-(2-苯基-1H-吡咯并[2,3-b]吡啶-5-基)-胺、(2-甲基-苄基)-(2-苯基-1H- 吡咯并[2,3-b]吡啶-5-基)-胺、N-[3-(5-苄基胺基-1H-吡咯并[2,3-b]吡啶-2-基)- 苯基]-乙酰胺、N-{3-[5-(2-甲基-苄基胺基)-1H-吡咯并[2,3-b]吡啶-2-基]-苯基}- 乙酰胺、N-[4-(5-苄基胺基-1H-吡咯并[2,3-b]吡啶-2-基)-苯基]-乙酰胺,以及 N-{4-[5-(2-甲基-苄基胺基)-1H-吡咯并[2,3-b]吡啶-2-基]-苯基}-乙酰胺。 Bebbington等人所申请的美国专利第7,691,853号,并通过引用将其包括在内,其描述作为Src激酶抑制剂的吡唑化合物。Engh等人所申请的美国专利第7,655,601号,并通过引用将其包括在内,其公开作为Src激酶抑制剂的 3-苯基二氢嘧啶并[4,5-d]嘧啶酮的酰胺衍生物。Bebbington等人所申请的美国专利第7,625,913号,并通过引用将其包括在内,其描述作为Src激酶抑制剂的吡唑化合物,包含(5-甲基-2H-吡唑-3-基)-(6-苯基-2-苯胺基-嘧啶-4-基)- 胺、(5-环丙基-2H-吡唑-3-基)-(6-苯基-2-苯胺基-嘧啶-4-基)-胺、(5-环丙基-2H- 吡唑-3-基)-[2-(3-甲基苯胺基)-6-苯基-嘧啶-4-基]-胺、[2-(4-氰基甲基苯基胺基)-6-苯基-嘧啶-4-基]-(5-环丙基-2H-吡唑-3-基)-胺、(5-环丙基-2H-吡唑-3- 基)-[6-苯基-2-(吡啶-3-基甲胺基)-嘧啶-4-基]-胺、[2-(3-氯苯基)胺基-6-(3-硝基苯基)-嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺、[2-(3-氯苯基)胺基-6-(3,4,5-三甲氧苯基)-嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺、(5-甲基-2H-吡唑-3-基)-[2-(4- 胺磺酰基苯胺基)-6-(3,4,5-三甲氧苯基)-嘧啶-4-基]-胺、[2-(苯并咪唑-2-基胺 -)-6-乙基-嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺、[2-(4-氯苯基)胺基-6-乙基- 嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺、(5-叔-丁基-2H-吡唑-3-基)-[2-(3-氯苯基) 胺基-6-(3-硝基苯基)-嘧啶-4-基]-胺、[2-(3-氯苯基)胺基-6-(3-硝基苯基)-嘧啶 -4-基]-(5-苯基-2H-吡唑-3-基)-胺、[5-(呋喃-2-基)-2H-吡唑-3-基]-(6-苯基-2-苯胺基-嘧啶-4-基)-胺、[2-(4-氯苯基)胺基-5,6-二甲基-嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺、(5,6-二甲基-2-苯胺基-嘧啶-4-基)-(5-甲基-2H-吡唑-3-基)-胺、[2-(4-氯苯基)胺基-6-甲氧基甲基-嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺、 [2-(苯并咪唑-2-基胺)-6-甲氧基甲基-嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺,以及(6-甲氧基甲基-2-苯胺基-嘧啶-4-基)-(5-甲基-2H-吡唑-3-基)-胺。Lee所申请的美国专利第7,622,472号公开Src抑制剂,包含N-(2-氯基-6-甲苯基)-2-[[6-[4-(2-羟乙基)-1-哌嗪基]-2-甲基-4-嘧啶基]胺基]-5-噻唑羧酰胺。 Honold等人所申请的美国专利第7,618,964号,并通过引用将其包括在内,其描述作为Src激酶抑制剂的苯甲酰胺衍生物,包含2-氯基-N-(2-苯基-1H- 吡咯并[2,3-b]吡啶-5-基)-苯甲酰胺、2-氯基-N-{2-[3-(2-甲氧基-乙氧基)-苯基]-1H-吡咯并[2,3-b]吡啶-5-基}-苯甲酰胺、2-甲氧基-N-(2-苯基-1H-吡咯并 [2,3-b]吡啶-5-基)-苯甲酰胺、2,4-二氯-N-(2-苯基-1H-吡咯并[2,3-b]吡啶-5-基)- 苯甲酰胺、2-氯基-6-甲基-N-(2-苯基-1H-吡咯并[2,3-b]吡啶-5-基)-苯甲酰胺、 N-[2-(3-乙酰胺基-苯基)-1H-吡咯并[2,3-b]吡啶-5-基]-4-甲氧基-苯甲酰胺、2- 甲基-N-(2-苯基-1H-吡咯并[2,3-b]吡啶-5-基)-苯甲酰胺、2-氯基-5-甲氧基 -N-{2-[3-(2-甲氧基-乙氧基)-苯基]-1H-吡咯并[2,3-b]吡啶-5-基}-苯甲酰胺、 2,4-二氯-N-{2-[3-(2-甲氧基-乙氧基)-苯基]-1H-吡咯并[2,3-b]吡啶-5-基}-苯甲酰胺、4-甲氧基-N-{2-[3-(2-甲氧基-乙氧基)-苯基]-1H-吡咯并[2,3-b]吡啶-5- 基}-苯甲酰胺、3,5-二甲氧基-N-{2-[3-(2-甲氧基-乙氧基)-苯基]-1H-吡咯并 [2,3-b]吡啶-5-基-苯甲酰胺、3,5-二甲氧基-N-(2-苯基-1H-吡咯并[2,3-b]吡啶-5- 基)-苯甲酰胺、N-{2-[3-(2-甲氧基-乙氧基)-苯基]-1H-吡咯并[2,3-b]吡啶-5- 基}-2-甲基-苯甲酰胺、2-甲氧基-N-{2-[4-(2-甲氧基-乙氧基)-苯基]-1H-吡咯并 [2,3-b]吡啶-5-基}-苯甲酰胺、N-[2-(3-乙酰胺基-苯基)-1H-吡咯并[2,3-b]吡啶-5-基]-2-氯基-苯甲酰胺、N-[2-(3-乙酰胺基-苯基)-1H-吡咯并[2,3-b]吡啶-5- 基]-2,4-二氯-苯甲酰胺、N-[2-(3-乙酰胺基-苯基)-1H-吡咯并[2,3-b]吡啶-5- 基]-2-甲氧基-苯甲酰胺、N-[2-(3-乙酰胺基-苯基)-1H-吡咯并[2,3-b]吡啶-5- 基]-2-氯基-6-甲基-苯甲酰胺、N-[2-(3-乙酰胺基-苯基)-1H-吡咯并[2,3-b]吡啶-5-基]-2-氯基-5-甲氧基-苯甲酰胺、2-氯基-N-(2-苯基-3H-咪唑并[4,5-b]吡啶-6- 基)-苯甲酰胺、2-氯基-6-甲基-N-(2-苯基-3H-咪唑并[4,5-b]吡啶-6-基)-苯甲酰胺、2-溴-N-(2-苯基-3H-咪唑并[4,5-b]吡啶-6-基)-苯甲酰胺、2-甲基-5-硝基 -N-(2-苯基-3H-咪唑并[4,5-b]吡啶-6-基)-苯甲酰胺、2-氯基-5-硝基-N-(2-苯基 -3H-咪唑并[4,5-b]吡啶-6-基)-苯甲酰胺、N-(2-苯基-3H-咪唑并[4,5-b]吡啶-6- 基)-苯甲酰胺、2-氯基-N-{2-[3-(3-甲氧基-丙酰胺基)-苯基]-3H-咪唑并[4,5-b] 吡啶-6-基}-苯甲酰胺、5-胺基-2-甲基-N-(2-苯基-3H-咪唑并[4,5-b]吡啶-6-基)- 苯甲酰胺、5-胺基-2-氯基-N-(2-苯基-3H-咪唑[4,5-b]吡啶-6-基)-苯甲酰胺、2- 氯基-N-{2-[4-(2-二乙胺基-乙氧基)-苯基]-3H-咪唑并[4,5-b]吡啶-6-基}-苯甲酰胺、2-氯基-N-{2-[4-(2-甲氧基-乙氧基)-苯基]-3H-咪唑并[4,5-b]吡啶-6-基}- 苯甲酰胺、2-氯基-N-{2-[3-(2-甲氧基-乙氧基)-苯基]-3H-咪唑并[4,5-b]吡啶-6- 基}-苯甲酰胺、2-氯基-N-[2-(3-硝基-苯基)-3H-咪唑并[4,5-b]吡啶-6-基]-苯甲酰胺、2-氯基-N-[2-(4-吗啉-4-基-苯基)-3H-咪唑并[4,5-b]吡啶-6-基]-苯甲酰胺、 2-氯基-N-{2-[4-(4-甲基-哌嗪-1-基)-苯基]-3H-咪唑并[4,5-b]吡啶-6-基}-苯甲酰胺、2-氯基-N-{2-[3-(2-羟基-乙基)-苯基]-3H-咪唑并[4,5-b]吡啶-6-基}-苯甲酰胺、3-[6-(2-氯基-苄酰胺基)-3H-咪唑并[4,5-b]吡啶-2-基]-苯甲酸、3-(6-(2- 氯苄酰胺基)-3H-咪唑并[4,5-b]吡啶-2-基)-N-(3-甲氧基-丙基)-苯甲酰胺、 3-(6-(2-氯苄酰胺基)-3H-咪唑并[4,5-b]吡啶-2-基)-N-异丙基-苯甲酰胺、2-氯基 -N-(2-{3-[2-甲氧基-1-甲氧基甲基-乙基胺甲酰基]-苯基}-3H-咪唑并[4,5-b]吡啶-6-基)-苯甲酰胺、2-氯基-N-[2-(3-甲基磺酰基-苯基)-3H-咪唑并[4,5-b]吡啶 -6-基]-苯甲酰胺、2-氯基-N-[2-(4-胺磺酰基-苯基)-3H-咪唑并[4,5-b]吡啶-6- 基]-苯甲酰胺、2-氯基-N-[2-(4-硝基-苯基)-3H-咪唑并[4,5-b]吡啶-6-基]-苯甲酰胺、2-氯基-N-[2-(4-甲基磺酰基-苯基)-3H-咪唑并[4,5-b]吡啶-6-基]-苯甲酰胺、2-氯基-N-[2-(3-甲烷亚磺酰基-苯基)-3H-咪唑并[4,5-b]吡啶--6-基]-苯甲酰胺、 2-氯基-N-[2-(4-甲烷磺酰基-苯基)-3H-咪唑并[4,5-b]吡啶-6-基]-苯甲酰胺、N-[2-(3-胺基-苯基)-3H-咪唑并[4,5-b]吡啶-6-基]-2-氯基-苯甲酰胺、N-[2-(3- 乙酰胺基-苯基)-3H-咪唑并[4,5-b]吡啶-6-基]-2-氯基-苯甲酰胺、N-(2-{4-[双 -(2-甲氧基-乙基)-胺基]-3-氟代-苯基}-3H-咪唑并[4,5-b]吡啶-6-基)-2-氯基-苯甲酰胺、2-氯基-N-(2-噻吩-2-基-3H-咪唑并[4,5-b]吡啶-6-基)-苯甲酰胺、2-氯基-N-(2-噻吩-3-基-3H-咪唑并[4,5-b]吡啶-6-基)-苯甲酰胺、2-氯基-N-[2-(2-甲基-吡啶-4-基)-3H-咪唑并[4,5-b]吡啶-6-基]-苯甲酰胺、2-氯基-N-[2-(6-甲基- 吡啶-3-基)-3H-咪唑并[4,5-b]吡啶-6-基]-苯甲酰胺、N-[2-(1H-苯并咪唑基-5- 基)-3H-咪唑并[4,5-b]吡啶-6-基]-2-氯基-苯甲酰胺、2-氯基-N-[2-(6-吗啉-4-基- 吡啶-3-基)-3H-咪唑并[4,5-b]吡啶-6-基]-苯甲酰胺,以及2-氯基-N-{2-[2-(3- 甲氧基-丙基胺基)-吡啶-4-基]-3H-咪唑并[4,5-b]吡啶-6-基}-苯甲酰胺。Xiao 等人所申请的美国专利第7,583,767号,并通过引用将其包括在内,其描述作为Src抑制剂的取代的吡唑化合物。Honold等人所申请的美国专利第 7,550,589号,并通过引用将其包括在内,其描述作为Src抑制剂的6-(2-烷基 -苯基)-吡啶并[2,3-d]嘧啶,包含2-(4-吗啉-4-基-苯胺基)-6-(2-三氟代甲基-苯基)-吡啶并[2,3-d]嘧啶-7-羧酸(吡咯啶-2-基甲基)-酰胺、2-(3-乙酰胺基-苯胺基)-6-(2-三氟代甲基-苯基)-吡啶并[2,3-d]嘧啶-7-羧酸(吡咯啶-2-基甲基)-酰胺、2-(3-甲烷磺酰基胺基-苯胺基)-6-(2-三氟代甲基-苯基)-吡啶并[2,3-d]嘧啶 -7-羧酸(吡咯啶-2-基甲基)-酰胺、2-(4,4-二氧-3,4-二氢-2H-4λ*6*-苯并[1,4]恶噻英-6-基胺)-6-(2-三氟代甲基-苯基)-吡啶并[2,3-d]嘧啶-7-羧酸(吡咯啶-2-基甲基)-酰胺、2-(4-吗啉-4-基-苯胺基)-6-(2-三氟代甲基-苯基)-吡啶并[2,3-d]嘧啶-7-羧酸(2-甲烷磺酰基胺基-乙基)-酰胺、2-(3-乙酰胺基-苯胺基)-6-(2-三氟代甲基-苯基)-吡啶并[2,3-d]嘧啶-7-羧酸(2-甲烷磺酰基胺基-乙基)-酰胺、2-(3- 甲烷磺酰基胺基-苯胺基)-6-(2-三氟代甲基-苯基)-吡啶并[2,3-d]嘧啶-7-羧酸 (2-甲烷磺酰基胺基-乙基)-酰胺,以及2-(4,4-二氧-3,4-二氢-2H-4λ*6*-苯并[1,4] 恶噻英-6-基胺)-6-(2-三氟代甲基-苯基)-吡啶并[2,3-d]嘧啶-7-羧酸(2-甲烷磺酰基胺基-乙基)-酰胺。Bebbington等人所申请的美国专利第7,531,536号,并通过引用将其包括在内,其描述作为Src激酶抑制剂的吡唑化合物。Engh等人所申请的美国专利第7,494,993号,并通过引用将其包括在内,其公开7- 胺基-3-苯基-二氢嘧啶并[4,5-d]嘧啶酮的酰胺衍生物。Boschelli等人所申请的美国专利第7,479,561号,并通过引用将其包括在内,其描述作为Src抑制剂的4-(2,4-二氯-5-甲氧苯基)胺基-6-甲氧基-7-{[5-取代的-胺基)甲基]-3-呋喃基}-3-甲腈,包含4-[(2,4-二氯-5-甲氧苯基)胺基]-6-甲氧基-7-{5-[(4-甲基哌嗪 -1-基)甲基]-3-呋喃基}-3-甲腈、4-[(2,4-二氯-5-甲氧苯基)胺基]-7-{5-[(二甲胺基)甲基]-3-呋喃基{-3-甲腈、4-[(2,4-二氯-5-甲氧苯基)胺基]-6-甲氧基-7-[5-(吗啉-4-基甲基)]-3-呋喃基]-3-甲腈、4-[(2,4-二氯-5-甲氧苯基)胺基]-6-甲氧基 -7-{5-[(4-苯基哌嗪-1-基)甲基]-3-呋喃基}-3-甲腈、4-[(2,4-二氯-5-甲氧苯基) 胺基]-6-甲氧基-7-(5-{[4-(2,4-二甲氧苯基)哌嗪-1-基]甲基}-3-呋喃基)-3-甲腈、4-[(2,4-二氯-5-甲氧苯基)胺基]-6-甲氧基-7-[5-(吡咯啶-1-基甲基)-3-呋喃基]喹啉-3-羰腈、4-[(2,4-二氯-5-甲氧苯基)胺基]-6-甲氧基-7-[5-(哌啶-1-基甲基)-3-呋喃基]喹啉-3-羰腈、4-[(2,4-二氯-5-甲氧苯基)胺基]-7-{5-[(二乙胺基) 甲基]-3-呋喃基)-6-甲氧基喹啉-3-羰腈、4-[(2,4-二氯-5-甲氧苯基)胺基]-7-{5-[(4-乙基哌嗪-1-基)甲基]-3-呋喃基)-6-甲氧基喹啉-3-羰腈、4-[(2,4- 二氯-5-甲氧苯基)胺基]-6-甲氧基-7-(5-{[4-(1-甲基哌啶-4-基)哌嗪-1-基]甲基}-3-呋喃基)喹啉-3-羰腈、4-[(2,4-二氯-5-甲氧苯基)胺基]-6-甲氧基-7-{5-[(4- 吡咯啶-1-基哌啶-1-基)甲基]-3-呋喃基}喹啉-3-羰腈、7-{5-[(4-丁基哌嗪-1-基) 甲基]-3-呋喃基}-4-[(2,4-二氯-5-甲氧苯基)胺基]-6-甲氧基喹啉-3-羰腈、 4-[(2,4-二氯-5-甲氧苯基)胺基]-6-甲氧基-7-(5-{[4-(2-吗啉-4-基乙基)哌嗪-1- 基]甲基}-3-呋喃基)喹啉-3-羰腈、7-{5-[(4-苄基哌嗪-1-基)甲基]-3-呋喃基}-4-[(2,4-二氯-5-甲氧苯基)胺基]-6-甲氧基喹啉-3-羰腈、4-[(2,4-二氯-5-甲氧苯基)胺基]-6-甲氧基-7-(5-{[4-(2-苯基乙基)哌嗪-1-基]甲基}-3-呋喃基)喹啉-3-羰腈、4-[(2,4-二氯-5-甲氧苯基)胺基]-7-{5-[(二丙基胺基)甲基]-3-呋喃基}-6-甲氧基喹啉-3-羰腈、4-[(2,4-二氯-5-甲氧苯基)胺基]-7-{5-[(1,1-二氧化硫代吗啉-4-基)甲基]-3-呋喃基}-6-甲氧基喹啉-3-羰腈、4-[(2,4-二氯-5-甲氧苯基)胺基]-6-甲氧基-7-{5-[(1-氧化硫代吗啉-4-基)甲基]-3-呋喃基}喹啉-3-碳氮、7-{5-[(4-环己基哌嗪-1-基)甲基]-3-呋喃基}-4-[(2,4-二氯-5-甲氧苯基)胺基]-6-甲氧基喹啉-3-羰腈、4-[(2,4-二氯-5-甲氧苯基)胺基]-6-甲氧基 -7-(5-{[4-(4-甲氧苯基)哌嗪-1-基]甲基}-3-呋喃基)喹啉-3-羰腈、4-[(2,4-二氯 -5-甲氧苯基)胺基]-6-甲氧基-7-{5-[(4-吡啶-4-基哌嗪-1-基)甲基]-3-呋喃基}喹啉-3-羰腈、4-[(2,4-二氯-5-甲氧苯基)胺基]-6-甲氧基-7-(5-{[4-(4-甲苯基)哌嗪 -1-基]甲基}-3-呋喃基)喹啉-3-羰腈、7-(5-{[4-(4-氯苯基)哌嗪-1-基]甲基}-3-呋喃基)-4-[(2,4-二氯-5-甲氧苯基)胺基]-6-甲氧基喹啉-3-羰腈、4-[(2,4-二氯-5- 甲氧苯基)胺基]-7-(5-{[4-(4-羟苯基)哌嗪-1-基]甲基}-3-呋喃基)-6-甲氧基喹啉 -3-羰腈、4-[(2,4-二氯-5-甲氧苯基)胺基]-6-甲氧基-7-[5-({4-[4-(三氟代甲基) 苯基]哌嗪-1-基}甲基)-3-呋喃基]喹啉-3-羰腈、4-[(2,4-二氯-5-甲氧苯基)胺基]-6-甲氧基-7-[5-(硫代吗啉-4-基甲基)-3-呋喃基]喹啉-3-羰腈、4-[(2,4-二氯 -5-甲氧苯基)胺基]-7-(5-{[[2-(二甲胺基)乙基](甲基)胺基]甲基}-3-呋喃基)-6- 甲氧基喹啉-3-羰腈、4-[(2,4-二氯-5-甲氧苯基)胺基]-7-}5-[(4-异丙基哌嗪-1- 基)甲基]-3-呋喃基}-6-甲氧基喹啉-3-羰腈、4-[(2,4-二氯-5-甲氧苯基)胺基]-6- 甲氧基-7-{5-[(4-甲基-1,4-二氮杂环庚烷-1-基)甲基]-3-呋喃基)喹啉-3-羰腈、 4-[(2,4-二氯-5-甲氧苯基)胺基]-6-甲氧基-7-(5-{[4-(2-甲氧基乙基)哌嗪-1-基] 甲基}-3-呋喃基)喹啉-3-羰腈、4-[(2,4-二氯-5-甲氧苯基)胺基]-7-(5-{[4-{2-羟乙基)哌嗪-1-基]甲基}-3-呋喃基)-6-甲氧基喹啉-3-羰腈、4-[(2,4-二氯-5-甲氧苯基)胺基]-7-(5-{[4-(2,6-二甲基苯基)哌嗪-1-基]甲基}-3-呋喃基)-6-甲氧基喹啉-3-羰腈、4-[(2,4-二氯-5-甲氧苯基)胺基]-7-[5-({4-[3-(二乙胺基)丙基]哌嗪 -1-基}甲基)-3-呋喃基]-6-甲氧基喹啉-3-羰腈、4-[(2,4-二氯-5-甲氧苯基)胺基]-6-甲氧基-7-(5-{[4-(吡啶-4-基甲基)哌嗪-1-基]甲基}-3-呋喃基)喹啉-3-羰腈,以及4-[(2,4-二氯-5-甲氧苯基)胺基]-7-(5-{[4-(2,6-二甲基苯基)哌嗪-1-基] 甲基}-3-呋喃基)-6-甲氧基喹啉-3-羰腈。Davies等人所申请的美国专利第 7,473,691号,并通过引用将其包括在内,其描述作为Src抑制剂的吡唑化合物。Boschelli等人所申请的美国专利第7,417,148号,并通过引用将其包括在内,其公开作为Src抑制剂的4-苯胺基-3-甲腈,包含4-[(2,4-二氯-5-甲氧苯基)胺基]-6-甲氧基-7-[3-(4-甲基-1-哌嗪基)丙氧基]-3-甲腈、4-[(2,4-二氯-5-甲氧苯基)胺基]-7-[3-(4-乙基-1-哌嗪基)丙氧基]-6-甲氧基-3-甲腈、4-[(2,4-二氯-5-甲氧苯基)胺基]-6-甲氧基-7-[2-(4-甲基-1-哌嗪基)乙氧基]-3-甲腈、 4-[(2,4-二氯-5-甲氧苯基)胺基]-7-[2-(4-乙基-1-哌嗪基)乙氧基]-6-甲氧基-3-甲腈、4-[(2,4-二氯-5-甲氧苯基)胺基]-6-甲氧基-7-[(1-甲基哌啶-4-基)甲氧基]-3- 甲腈、4-[(2,4-二氯-5-甲氧苯基)胺基]-6-甲氧基-7-[2-(1-甲基哌啶-4-基)乙氧基]-3-甲腈、4-[(2,4-二氯-5-甲氧苯基)胺基]-6-甲氧基-7-[3-(1-甲基哌啶-4-基) 丙氧基]喹啉-3-羰腈、4-[(2,4-二氯-5-甲氧苯基)胺基]-7-[(1-乙基哌啶-4-基)甲氧基]-6-甲氧基喹啉-3-羰腈、4-[(2,4-二氯-5-甲氧苯基)胺基]-6-乙氧基-7-[3-(4- 甲基哌嗪-1-基)丙氧基]喹啉-3-羰腈、4-[(2,4-二氯-5-甲氧苯基)胺基]-6-乙氧基 -7-[(1-甲基哌啶-4-基)甲氧基]喹啉-3-羰腈、4-[(2,4-二氯-5-甲氧苯基)胺基]-6- 乙氧基-7-[3-(4-乙基哌嗪-1-基)丙氧基]喹啉-3-羰腈、4-[(2,4-二氯-5-甲氧苯基) 胺基]-6-乙氧基-7-[3-(1-甲基哌啶-4-基)丙氧基]喹啉-3-羰腈、4-[(2,4-二氯-5- 甲氧苯基)胺基]-6-乙氧基-7-[2-(4-甲基-1-哌嗪基)乙氧基]喹啉-3-羰腈、 4-[(2,4-二氯-5-甲氧苯基)胺基]-6-乙氧基-7-[2-(1-甲基哌啶-4-基)乙氧基]喹啉 -3-羰腈、4-[(2,4-二氯-5-甲氧苯基)胺基]-6-甲氧基-7-[3-(4-丙基-1-哌嗪基)丙氧基]-3-甲腈、4-[(2,4-二氯苯基)胺基]-6-甲氧基-7-[(1甲基哌啶-4-基)甲氧基]-3- 甲腈、6-甲氧基-7-[(1-甲基哌啶-4-基)甲氧基]-4-[(3,4,5-三甲氧苯基)胺基]喹啉 -3-羰腈、4-[(2-氯基-5-甲氧苯基)胺基]-6-甲氧基-7-[(1-甲基哌啶-4-基)甲氧基] 喹啉-3-羰腈、6-甲氧基-4-[(5-甲氧基-2-甲苯基)胺基]-7-[(1-甲基哌啶-4-基)甲氧基]喹啉-3-羰腈、4-[(2,4-二甲苯基)胺基]-6-甲氧基-7-[(1-甲基哌啶-4-基)甲氧基]喹啉-3-羰腈、6-甲氧基-4-[(5-甲氧基-2,4-二甲苯基)胺基]-7-[(1-甲基哌啶 -4-基)甲氧基]喹啉-3-羰腈,以及4-[(2,4-二氯-5-乙氧苯基)胺基]-6-甲氧基 -7-[(1-甲基哌啶-4-基)甲氧基]喹啉-3-羰腈。Davies等人所申请的美国专利第 7,390,815号,并通过引用将其包括在内,其描述作为Src抑制剂的吡唑化合物。Benish等人所申请的美国专利第7,285,556号,并通过引用将其包括在内,其描述作为Src抑制剂的噻咔吡啶化合物,包含2-噻唑甲醛(7-甲基噻咔 [3,2-d]嘧啶-4-基)腙、1H-咪唑-4-甲醛(7-甲基噻咔[3,2-d]嘧啶-4-基)腙、2-氯苯甲醛(7-甲基噻咔[3,2-d]嘧啶-4-基)腙、3-溴苯醛(7-甲基噻咔[3,2-d]嘧啶-4- 基)腙、4-氟代苯甲醛(6,7-二甲基噻咔[3,2-d]嘧啶-4-基)腙、1-(3-吡啶基)乙酮 (7-甲基噻咔[3,2-d]嘧啶-4-基)腙、3-甲基-2-噻吩甲醛(7-甲基噻咔[3,2-d]嘧啶 -4-基)腙、4-丙氧基苯醛(7-甲基噻咔[3,2-d]嘧啶-4-基)腙、3-丙氧基苯醛(7-甲基噻咔[3,2-d]嘧啶-4-基)腙、1H-吲哚-5-甲醛(7-甲基噻咔[3,2-d]嘧啶-4-基)腙、乙醛(7-甲基噻咔[3,2-d]嘧啶-4-基)腙、4-(甲硫基)苯醛(7-甲基噻咔[3,2-d]嘧啶 -4-基)腙、丙醛(7-甲基噻咔[3,2-d]嘧啶-4-基)腙、丁醛(7-甲基噻咔[3,2-d]嘧啶 -4-基)腙、戊醛(7-甲基噻咔[3,2-d]嘧啶-4-基)腙、四氢-3--呋喃甲醛(7-甲基噻咔[3,2-d]嘧啶-4-基)腙、3-环己烯-1-甲醛(7-甲基噻咔[3,2-d]嘧啶-4-基)腙、E-2- 丁烯醛(7-甲基噻咔[3,2-d]嘧啶-4-基)腙、苯乙醛(7-甲基噻咔[3,2-d]嘧啶-4-基) 腙、3-吡啶甲醛(6-溴噻咔[3,2-d]嘧啶-4-基)腙、3-吡啶甲醛(6-(3-噻吩基)噻咔 [3,2-d]嘧啶-4-基)腙、2-噻吩甲醛(吡啶并[3’,2’:4,5]噻咔[3,2-d]嘧啶-4-基)腙、 4-羧基苯醛(吡啶并[3’,2’:4,5]噻咔[3,2-d]嘧啶-4-基)腙、3-羟基-4-甲氧基苯甲醛(吡啶并[3’,2’:4,5]噻咔[3,2-d]嘧啶-4-基)腙、3-羧基苯醛(7-甲基噻咔[3,2-d] 嘧啶-4-基)腙、4-吡啶甲醛(6-苯基噻咔[3,2-d]嘧啶-4-基)腙、3-吡啶甲醛(7-乙基噻咔[3,2-d]嘧啶-4-基)腙、3-吡啶甲醛(7-丙基噻咔[3,2-d]嘧啶-4-基)腙、3- 吡啶甲醛(6-(4-氟代苯基)噻咔[3,2-d]嘧啶-4-基)腙、4-羧基苯醛(6-(4-氟代苯基) 噻咔[3,2-d]嘧啶-4-基)腙、3-吡啶甲醛(6-(4-甲烷硫酰基苯基)噻咔-[3,2-d]嘧啶 -4-基)腙、4-羧基苯醛(6-(4-甲烷硫酰基苯基)噻咔-[3,2-d]嘧啶-4-基)腙、3-吡啶甲醛(6-(3-氯苯基)噻咔[3,2-d]嘧啶-4-基)腙、4-羧基苯醛(6-(3-氯苯基)噻咔 [3,2-d]嘧啶-4-基)腙、3-吡啶甲醛(6-(4-氯苯基)噻咔[3,2-d]嘧啶-4-基)腙、4-羧基苯醛(6-(4-氯苯基)噻咔[3,2-d]嘧啶-4-基)腙、3-吡啶甲醛(6-(2-氯苯基)噻咔 [3,2-d]嘧啶-4-基)腙、4-羧基苯醛(6-(2-氯苯基)噻咔[3,2-d]嘧啶-4-基)腙、2-噻吩甲醛(6-(2-氯苯基)噻咔[3,2-d]嘧啶-4-基)腙、3-羟基-4-甲氧基苯甲醛(6-(2- 氯苯基)噻咔[3,2-d]嘧啶-4-基)腙、3-吡啶甲醛(7-甲基-6-苯基噻咔[3,2-d]嘧啶 -4-基)腙、4-羧基苯醛(7-甲基-6-苯基噻咔[3,2-d]嘧啶-4-基)腙、2-噻吩甲醛(7- 甲基-6-苯基噻咔[3,2-d]嘧啶-4-基)腙、3-羟基-4-甲氧基苯甲醛(7-甲基-6-苯基噻咔[3,2-d]嘧啶-4-基)腙、3-吡啶甲醛(6-碘基-7-甲基噻咔[3,2-d]嘧啶-4-基)腙、 4-羧基苯醛(6-碘基-7-甲基噻咔[3,2-d]嘧啶-4-基)腙、2-噻吩甲醛(6-碘基-7-甲基噻咔[3,2-d]嘧啶-4-基)腙、3-羟基-4-甲氧基苯甲醛(6-碘基-7-甲基噻咔[3,2-d] 嘧啶-4-基)腙、3-吡啶甲醛(7-甲基-6-(3-噻吩基)噻咔[3,2-d]嘧啶-4-基)腙、4- 羧基苯醛(7-甲基-6-(3-噻吩基)噻咔[3,2-d]嘧啶-4-基)腙、2-噻吩甲醛(7-甲基 -6-(3-噻吩基)噻咔[3,2-d]嘧啶-4-基)腙、3-羟基-4-甲氧基苯甲醛(7-甲基-6-(3- 噻吩基)噻咔[3,2-d]嘧啶-4-基)腙、3-吡啶甲醛(6-(2-氯苯基)7-甲基噻咔[3,2-d] 嘧啶-4-基)腙、4-羧基苯醛(6-(2-氯苯基)7-甲基噻咔[3,2-d]嘧啶-4-基)腙、2-噻吩甲醛(6-(2-氯苯基)7-甲基噻咔[3,2-d]嘧啶-4-基)腙、3-吡啶甲醛(6-碘噻咔 [3,2-d]嘧啶-4-基)腙、4-羧基苯醛(6-碘噻咔[3,2-d]嘧啶-4-基)腙、2-噻吩甲醛(6- 碘噻咔[3,2-d]嘧啶-4-基)腙、3-羟基-4-甲氧基苯甲醛(6-碘噻咔[3,2-d]嘧啶-4- 基)腙、3-吡啶甲醛(6-(4-甲氧苯基)噻咔[3,2-d]嘧啶-4-基)腙、4-羧基苯醛(6-(4- 甲氧苯基)噻咔[3,2-d]嘧啶-4-基)腙、2-噻吩甲醛(6-(4-甲氧苯基)噻咔[3,2-d]嘧啶-4-基)腙、3-羟基-4-甲氧基苯甲醛(6-(4-甲氧苯基)噻咔[3,2-d]嘧啶-4-基)腙、3-吡啶甲醛(6-(4-甲氧苯基)-7-甲基噻咔[3,2-d]嘧啶-4-基)腙、4-羧基苯醛(6-(4- 甲氧苯基)-7-甲基噻咔[3,2-d]嘧啶-4-基)腙、2-噻吩甲醛(6-(4-甲氧苯基)-7-甲基噻咔[3,2-d]嘧啶-4-基)腙、3-羟基-4-甲氧基苯甲醛(6-(4-甲氧苯基)-7-甲基噻咔[3,2-d]嘧啶-4-基)腙、3-吡啶甲醛(6-(3-甲氧苯基)-7-甲基噻咔[3,2-d]嘧啶-4- 基)腙、4-羧基苯醛(6-(3-甲氧苯基)-7-甲基噻咔[3,2-d]嘧啶-4-基)腙、2-噻吩甲醛(6-(3-甲氧苯基)-7-甲基噻咔[3,2-d]嘧啶-4-基)腙、3-羟基-4-甲氧基苯甲醛 (6-(3-甲氧苯基)-7-甲基噻咔[3,2-d]嘧啶-4-基)腙、3-吡啶甲醛(6-(2-甲氧苯基)-7-甲基噻咔[3,2-d]嘧啶-4-基)腙、4-羧基苯醛(6-(2-甲氧苯基)-7-甲基噻咔 [3,2-d]嘧啶-4-基)腙、2-噻吩甲醛(6-(2-甲氧苯基)-7-甲基噻咔[3,2-d]嘧啶-4-基) 腙、3-羟基-4-甲氧基苯甲醛(6-(2-甲氧苯基)-7-甲基噻咔[3,2-d]嘧啶-4-基)腙、 3-吡啶甲醛(6-(4-羟甲基苯基)-7-甲基噻咔[3,2-d]嘧啶-4-基)腙、4-羧基苯醛 (6-(4-羟甲基苯基)-7-甲基噻咔[3,2-d]嘧啶-4-基)腙、2-噻吩甲醛(6-(4-羟甲基苯基)-7-甲基噻咔[3,2-d]嘧啶-4-基)腙、3-羟基-4-甲氧基苯甲醛(6-(4-羟甲基苯基)-7-甲基噻咔[3,2-d]嘧啶-4-基)腙、3-吡啶甲醛(6-(3-羟甲基苯基)-7-甲基噻咔[3,2-d]嘧啶-4-基)腙、4-羧基苯醛(6-(3-羟甲基苯基)-7-甲基噻咔[3,2-d]嘧啶 -4-基)腙、2-噻吩甲醛(6-(3-羟甲基苯基)-7-甲基噻咔[3,2-d]嘧啶-4-基)腙、3- 羟基-4-甲氧基苯甲醛(6-(3-羟甲基苯基)-7-甲基噻咔[3,2-d]嘧啶-4-基)腙、3- 吡啶甲醛(7-甲基-6-(反-2-苯乙烯基)-噻咔[3,2-d]嘧啶-4-基)腙、4-羧基苯醛(7- 甲基-6-(反-2-苯乙烯基)-噻咔[3,2-d]嘧啶-4-基)腙、3-羟基-4-甲氧基苯甲醛(7- 甲基-6-(反-2-苯乙烯基)-噻咔[3,2-d]嘧啶-4-基)腙、3-吡啶甲醛(6-(4-羧苯基)-7- 甲基噻咔[3,2-d]嘧啶-4-基)腙、2-噻吩甲醛(6-(4-羧苯基)-7-甲基噻咔[3,2-d]嘧啶-4-基)腙、3-羟基-4-甲氧基苯甲醛(6-(4-羧苯基)-7-甲基噻咔[3,2-d]嘧啶-4- 基)腙、3-吡啶甲醛(6-(1-羟基-1-苯基)甲基-7-甲基噻咔[3,2-d]嘧啶-4-基)腙、 4-羧基苯醛(6-(1-羟基-1-苯基)甲基-7-甲基噻咔[3,2-d]嘧啶-4-基)腙、2-噻吩甲醛(6-(1-羟基-1-苯基)甲基-7-甲基噻咔[3,2-d]嘧啶-4-基)腙、3-羟基-4-甲氧基苯甲醛(6-(1-羟基-1-苯基)甲基-7-甲基噻咔[3,2-d]嘧啶-4-基)腙、3-吡啶甲醛 (6-(1-羟基-1-[3-噻吩基])甲基-7-甲基噻咔[3,2-d]嘧啶-4-基)腙、4-羧基苯醛 (6-(1-羟基-1-[3-噻吩基])甲基-7-甲基噻咔[3,2-d]嘧啶-4-基)腙、2-噻吩甲醛 (6-(1-羟基-1-[3-噻吩基])甲基-7-甲基噻咔[3,2-d]嘧啶-4-基)腙、3-羟基-4-甲氧基苯甲醛(6-(1-羟基-1-[3-噻吩基])甲基-7-甲基噻咔[3,2-d]嘧啶-4-基)腙、3-吡啶甲醛(6-羧基-7-甲基噻咔[3,2-d]嘧啶-4-基)腙、4-羧基苯醛(6-羧基-7-甲基噻咔[3,2-d]嘧啶-4-基)腙、2-噻吩甲醛(6-羧基-7-甲基噻咔[3,2-d]嘧啶-4-基)腙、 3-羟基-4-甲氧基苯甲醛(6-羧基-7-甲基噻咔[3,2-d]嘧啶-4-基)腙、3-吡啶甲醛 (6-(1-环己基-1-羟基)甲基-7-甲基噻咔[3,2-d]嘧啶-4-基)腙、4-羧基苯醛(6-(1- 环己基-1-羟基)甲基-7-甲基噻咔[3,2-d]嘧啶-4-基)腙、2-噻吩甲醛(6-(1-环己基 -1-羟基)甲基-7-甲基噻咔[3,2-d]嘧啶-4-基)腙、3-羟基-4-甲氧基苯甲醛(6-(1- 环己基-1-羟基)甲基-7-甲基噻咔[3,2-d]嘧啶-4-基)腙、3-吡啶甲醛(6-(1-苄基-1- 苯基)甲基-7-甲基噻咔[3,2-d]嘧啶-4-基)腙、2-噻吩甲醛(6-(1-苄基-1-苯基)甲基-7-甲基噻咔[3,2-d]嘧啶-4-基)腙、3-羟基-4-甲氧基苯甲醛(6-(1-苄基-1-苯基) 甲基-7-甲基噻咔[3,2-d]嘧啶-4-基)腙、3-吡啶甲醛(6-羟甲基-7-甲基噻咔[3,2-d] 嘧啶-4-基)腙、4-羧基苯醛(6-羟甲基-7-甲基噻咔[3,2-d]嘧啶-4-基)腙、3-羟基 -4-甲氧基苯甲醛(6-羟甲基-7-甲基噻咔[3,2-d]嘧啶-4-基)腙、3-吡啶甲醛(6-(1- 羟基-1-[3,4,5-三甲氧苯基])甲基-7-甲基噻咔[3,2-d]嘧啶-4-基)腙、4-羧基苯醛(6-(1-羟基-1-[3,4,5-三甲氧苯基])甲基-7-甲基噻咔[3,2-d]嘧啶-4-基)腙、2-噻吩甲醛(6-(1-羟基-1-[3,4,5-三甲氧苯基])甲基-7-甲基噻咔[3,2-d]嘧啶-4-基)腙、3- 羟基-4-甲氧基苯甲醛(6-(1-羟基-1-[3,4,5-三甲氧苯基])甲基-7-甲基噻咔[3,2-d] 嘧啶-4-基)腙、3-吡啶甲醛(6-(1-羟基-1-[3-吡啶基])甲基-7-甲基噻咔[3,2-d]嘧啶-4-基)腙、4-羧基苯醛(6-(1-羟基-1-[3-吡啶基])甲基-7-甲基噻咔[3,2-d]嘧啶 -4-基)腙、2-噻吩甲醛(6-(1-羟基-1-[3-吡啶基])甲基-7-甲基噻咔[3,2-d]嘧啶-4- 基)腙、3-羟基-4-甲氧基苯甲醛(6-(1-羟基-1-[3-吡啶基])甲基-7-甲基噻咔[3,2-d] 嘧啶-4-基)腙、3-吡啶甲醛(6-(1-羟基-1-[2-噻吩基])甲基-7-甲基噻咔[3,2-d]嘧啶-4-基)腙、4-羧基苯醛(6-(1-羟基-1-[2-噻吩基])甲基-7-甲基噻咔[3,2-d]嘧啶 -4-基)腙、2-噻吩甲醛(6-(1-羟基-1-[2-噻吩基])甲基-7-甲基噻咔[3,2-d]嘧啶-4- 基)腙,以及3-羟基-4-甲氧基苯甲醛(6-(1-羟基-1-[2-噻吩基])甲基-7-甲基噻咔 [3,2-d]嘧啶-4-基)腙。Boschelli等人所申请的美国专利第7,276,519号,并通过引用将其包括在内,其描述作为Src抑制剂的噻咔[3,2-b]吡啶-6-羰腈以及噻咔[2,3-b]吡啶-5-羰腈,包含3-溴-7-[(2,4-二氯-5-甲氧苯基)胺基]噻咔[3,2-b] 吡啶-6-羰腈、7-[(2,4-二氯-5-甲氧苯基)胺基]-3-(4-甲酰基苯基)噻咔[3,2-b]吡啶-6-羰腈、7-[(2,4-二氯-5-甲氧苯基)胺基]-3-{4-[(二甲胺基)甲基]苯基}噻咔 [3,2-b]吡啶-6-羰腈、7-[(2,4-二氯-5-甲氧苯基)胺基]-3-{4-[(4-甲基哌嗪-1-基) 甲基]苯基}噻咔[3,2-b]吡啶-6-羰腈,以及7-[(2,4-二氯-5-甲氧苯基)胺基]-3-[4-(吗啉-4-基甲基)苯基]噻咔[3,2-b]吡啶-6-羰腈。Aronov等人所申请的美国专利第7,262,200号,并通过引用将其包括在内,其描述作为Src抑制剂的吲唑啉酮化合物。Arnost等人所申请的美国专利第7,226,920号,并通过引用将其包括在内,其描述作为Src抑制剂的胺基三唑化合物。Honold等人所申请的美国专利第7,189,732号,并通过引用将其包括在内,其描述作为 Src抑制剂的吡啶并[2,3-d]嘧啶二氯-苯基衍生物,包含6-(2,6-二氯-苯基)-2-(4-吗啉-4-基-苯胺基)-吡啶并[2,3-d]嘧啶-7-羧酸(2-甲烷磺酰基胺基-乙基)-酰胺、6-(2,6-二氯-苯基)-2-[4-(2-羟基-乙氧基)-苯胺基]-吡啶并[2,3-d]嘧啶 -7-羧酸(2-甲烷磺酰基胺基-乙基)-酰胺、6-(2,6-二氯-苯基)-2-(3-甲烷磺酰基胺基-苯胺基)-吡啶并[2,3-d]嘧啶-7-羧酸(2-甲烷磺酰基胺基-乙基)-酰胺、6-(2,6- 二氯-苯基)-2-[3-(2-羟基-乙基磺酰基)-苯胺基]-吡啶并[2,3-d]嘧啶-7-羧酸(2- 甲烷磺酰基胺基-乙基)-酰胺、6-(2,6-二氯-苯基)-2-[4-(4-甲基-哌嗪-1-基)-苯胺基]-吡啶并[2,3-d]嘧啶-7-羧酸(2-甲烷磺酰基胺基-乙基)-酰胺,以及6-(2,6-二氯-苯基)-2-(3-甲基磺酰基-苯胺基)-吡啶并[2,3-d]嘧啶-7-羧酸(2-甲烷磺酰基胺基-乙基)-酰胺。Honold等人所申请的美国专利第7,169,781号,通过引用将其包括,其描述作为Src抑制剂的咪唑衍生物,包含2-(2,6-二氯苯基)-4-(3- 溴基苯基)-5-(2-[4-(2-二乙胺基-乙氧基)苯胺基]嘧啶-4-基)-N--H-咪唑、2-(2,6- 二氯苯基)-4-(3-氯苯基)-5-(2-[4-(2-二乙胺基-乙氧基)苯胺基]嘧啶-4- 基)-N--H-咪唑、2-(2,6-二氯苯基)-4-(3-氯苯基)-5-(2-[4-(2-羟基乙氧基)苯基- 胺基]嘧啶-4-基)-N--H-咪唑、2-(2,6-二氯苯基)-4-(3-氯苯基)-5-(2-[4-二甲胺基苯-胺基]嘧啶-4-基)-N--H-咪唑、2-(2,6-二氯苯基)-4-(3-氯苯基)-5-(2-[4-(N-(2-羟乙基)-胺磺酰基)苯胺基]-嘧啶-4-基)-N--H-咪唑、2-(2,6-二氯-4-羟甲基苯基)-4-(3-氯苯基)-5-(2-[4-(2-二乙基胺基乙氧基)-苯胺基]嘧啶-4-基)-N--H-咪唑、2-(2,6-二氯-4-羟甲基苯基)-4-(3-氯苯基)-5-(2-[4-(2-羟基乙氧基)-苯胺基] 嘧啶-4-基)-N--H-咪唑、2-(2,6-二氯-4-[2-羟基乙氧基]苯基)-4-(3-氯苯基)-5-(2-[4-(2-二乙基胺基乙氧基)-苯胺基]嘧啶-4-基)-N--H-咪唑、2-(2,6-二氯 -4-[2-羟基乙氧基]苯基)-4-(3-氯苯基)-5-(2-[4-甲基亚磺酰基-苯胺基]嘧啶-4- 基)-N--H-咪唑、2-(2,6-二氯-4-[2-羟基乙氧基]苯基)-4-(3-氯苯基)-5-(2-[4-(N-(2-羟乙基)-胺磺酰基)苯胺基]嘧啶-4-基)-N--H-咪唑、2-(2,6-二氯苯基)-4-(4-氯苯基)-5-(2-[4-(2-二乙基胺基乙氧基)苯胺基]嘧啶-4-基)-N--H- 咪唑、2-(2,6-二氯苯基)-4-(4-氯苯基)-5-(2-[4-羟苯基-胺]嘧啶-4-基)-N--H-咪唑、2-(2,6-二氯苯基)-4-(4-氯苯基)-5-(2-[4-甲氧苯基-胺]嘧啶-4-基)-N--H-咪唑、2-(2,6-二氯苯基)-4-(4-氯苯基)-5-(2-[4-乙氧苯基-胺]嘧啶-4-基)-N--H-咪唑、2-(2,6-二氯苯基)-4-(3-乙炔苯基)-5-(2-[4-(2-二乙胺基-乙氧基)苯胺基]嘧啶-4-基)-N--H-咪唑、2-(2,6-二氯苯基)-4-(3-乙炔苯基)-5-(2-[4-(2-羟基乙氧基)-苯胺基]嘧啶-4-基)-N--H-咪唑,以及2-(2,6-二氯-4-[2-羟基乙氧基]苯基)-4-(3-乙炔苯基)-5-(2-[4-(2-二乙基胺基乙氧基)-苯胺基]嘧啶-4-基)-N--H- 咪唑。Honold等人所申请的美国专利第7,163,941号,通过引用将其包括,其描述作为Src抑制剂的吡啶并[2,3-d]嘧啶-7-羧酸,包含6-(2-溴-苯基)-2-(4- 吗啉-4-基-苯胺基)-吡啶并[2,3-d]嘧啶-7-羧酸[2-(3H-咪唑-4-基)-乙基]-酰胺、 6-(2-溴-苯基)-2-(4-吗啉-4-基-苯胺基)-吡啶并[2,3-d]嘧啶-7-羧酸(S)-哌啶-3-基酰胺、6-(2-溴-苯基)-2-(3-甲基磺酰基-苯胺基)-吡啶并[2,3-d]嘧啶-7-羧酸(S)- 哌啶-3-基酰胺、6-(2-溴-苯基)-2-(4-吗啉-4-基-苯胺基)-吡啶并[2,3-d]嘧啶-7- 羧酸(2-胺磺酰基-乙基)-酰胺、6-(2-溴-苯基)-2-(3-甲基磺酰基-苯胺基)-吡啶并 [2,3-d]嘧啶-7-羧酸(2-胺磺酰基-乙基)-酰胺、6-(2-溴-苯基)-2-(4-吗啉-4-基-苯胺基)-吡啶并[2,3-d]嘧啶-7-羧酸(2-羟基-乙基)-酰胺、6-(2-溴-苯基)-2-(4-氟代- 苯胺基)-吡啶并[2,3-d]嘧啶-7-羧酸(2-二甲胺基-乙基)-酰胺、6-(2-溴-苯基)-2-(4-氟代-苯胺基)-吡啶并[2,3-d]嘧啶-7-羧酸(2-甲氧基-乙基)-酰胺、6-(2- 溴-苯基)-2-(4-氟代-苯胺基)-吡啶并[2,3-d]嘧啶-7-羧酸(3-二甲胺基-丙基)-酰胺、6-(2-溴-苯基)-2-(4-氟代-苯胺基)-吡啶并[2,3-d]嘧啶-7-羧酸(3-二甲胺基 -2,2-二甲基-丙基)-酰胺、6-(2-溴-苯基)-2-(4-氟代-苯胺基)-吡啶并[2,3-d]嘧啶 -7-羧酸(2-乙酰胺基-乙基)-酰胺、6-(2-溴-苯基)-2-(4-氟代-苯胺基)-吡啶并 [2,3-d]嘧啶-7-羧酸(2-甲胺基-乙基)-酰胺、6-(2-溴-苯基)-2-(4-氟代-苯胺基)- 吡啶并[2,3-d]嘧啶-7-羧酸胺甲酰基甲基-酰胺、6-(2-溴-苯基)-2-(4-氟代-苯胺基)-吡啶并[2,3-d]嘧啶-7-羧酸(2-二甲胺基-1-甲基-乙基)-酰胺、6-(2-溴-苯基)-2-(4-氟代-苯胺基)-吡啶并[2,3-d]嘧啶-7-羧酸甲基胺甲酰基甲基-酰胺、 6-(2-溴-苯基)-2-(4-氟代-苯胺基)-吡啶并[2,3-d]嘧啶-7-羧酸(2-二甲胺基-丙基)-酰胺、6-(2-溴-苯基)-2-(4-氟代-苯胺基)-吡啶并[2,3-d]嘧啶-7-羧酸二甲基胺甲酰基甲基-酰胺、6-(2-溴-苯基)-2-(4-氟代-苯胺基)-吡啶并[2,3-d]嘧啶-7- 羧酸(3-甲胺基-丙基)-酰胺、6-(2-溴-苯基)-2-(4-氟代-苯胺基)-吡啶并[2,3-d]嘧啶-7-羧酸(2-胺磺酰基-乙基)-酰胺、6-(2-溴-苯基)-2-(4-氟代-苯胺基)-吡啶并 [2,3-d]嘧啶-7-羧酸(2-羟基-乙基)-酰胺、6-(2-溴-苯基)-2-(4-氟代-苯胺基)-吡啶并[2,3-d]嘧啶-7-羧酸(3-羟基-丙基)-酰胺、(S)-6-(2-溴-苯基)-2-(4-氟代-苯胺基)-吡啶并[2,3-d]嘧啶-7-羧酸(2,3-二羟基-丙基)-酰胺、(R)-6-(2-溴-苯基)-2-(4- 氟代-苯胺基)-吡啶并[2,3-d]嘧啶-7-羧酸(2,3-二羟基-丙基)-酰胺、6-(2-溴-苯基)-2-(4-氟代-苯胺基)-吡啶并[2,3-d]嘧啶-7-羧酸(2-甲烷亚磺酰基-乙基)-酰胺、6-(2-溴-苯基)-2-(4-氟代-苯胺基)-吡啶并[2,3-d]嘧啶-7-羧酸[2-(2-羟基-乙烷亚磺酰基)-乙基]-酰胺、6-(2-溴-苯基)-2-(4-氟代-苯胺基)-吡啶并[2,3-d]嘧啶 -7-羧酸(2-吗啉-4-基-乙基)-酰胺、6-(2-溴-苯基)-2-(4-氟代-苯胺基)-吡啶并 [2,3-d]嘧啶-7-羧酸[2-(2-氧基-咪唑啉-1-基)-乙基]-酰胺、(R)-6-(2-溴-苯基)-2-(4-氟代-苯胺基)-吡啶并[2,3-d]嘧啶-7-羧酸(吡咯啶-2-基甲基)-酰胺、 6-(2-溴-苯基)-2-(4-氟代-苯胺基)-吡啶并[2,3-d]嘧啶-7-羧酸[3-(2-氧基-吡咯啶 -1-基)-丙基]-酰胺、6-(2-溴-苯基)-2-(4-氟代-苯胺基)-吡啶并[2,3-d]嘧啶-7-羧酸(3-吗啉-4-基-丙基)-酰胺、6-(2-溴-苯基)-2-(4-氟代-苯胺基)-吡啶并[2,3-d] 嘧啶-7-羧酸[2-(1-甲基-吡咯啶-2-基)-乙基]-酰胺、6-(2-溴-苯基)-2-(4-氟代-苯胺基)-吡啶并[2,3-d]嘧啶-7-羧酸[2-(吡啶-2-基胺)-乙基]-酰胺、6-(2-溴-苯基)-2-(4-氟代-苯胺基)-吡啶并[2,3-d]嘧啶-7-羧酸[2-(3H-咪唑-4-基)-乙基]-酰胺、6-(2-溴-苯基)-2-(4-氟代-苯胺基)-吡啶并[2,3-d]嘧啶-7-羧酸(1,5-二甲基 -1H-吡唑-3-基甲基)-酰胺、6-(2-溴-苯基)-2-(4-氟代-苯胺基)-吡啶并[2,3-d]嘧啶-7-羧酸((S)-吡咯啶-2-基甲基)-酰胺、6-(2-溴-苯基)-2-(3-甲基磺酰基-苯胺基)-吡啶并[2,3-d]嘧啶-7-羧酸(2-二甲胺基-乙基)-酰胺、6-(2-溴-苯基)-2-(4-吗啉-4-基-苯胺基)-吡啶并[2,3-d]嘧啶-7-羧酸(2-二甲胺基-乙基)-酰胺、6-(2-溴- 苯基)-2-(4-吗啉-4-基-苯胺基)-吡啶并[2,3-d]嘧啶-7-羧酸(2-甲胺基-乙基)-酰胺、6-(2-溴-苯基)-2-[4-(2-二乙胺基-乙氧基)-苯胺基]-吡啶并[2,3-d]嘧啶-7-羧酸(2-二甲胺基-乙基)-酰胺、6-(2-溴-苯基)-2-(4-吗啉-4-基-苯胺基)-吡啶并 [2,3-d]嘧啶-7-羧酸哌啶-4-基酰胺、6-(2-溴-苯基)-2-(3-甲基磺酰基-苯胺基)- 吡啶并[2,3-d]嘧啶-7-羧酸(2-甲胺基-乙基)-酰胺、(R)-6-(2-溴-苯基)-2-(4-氟代- 苯胺基)-吡啶并[2,3-d]嘧啶-7-羧酸哌啶-3-基酰胺、(S)-6-(2-溴-苯基)-2-(4-氟代-苯胺基)-吡啶并[2,3-d]嘧啶-7-羧酸哌啶-3-基酰胺、6-(2-溴-苯基)-2-(4-氟代-苯胺基)-吡啶并[2,3-d]嘧啶-7-羧酸哌啶-4-基酰胺、1-[6-(2-溴-苯基)-2-(4- 氟代-苯胺基)-吡啶并[2,3-d]嘧啶-7-羰基]半卡肼、6-(2-溴-苯基)-2-(4-氟代-苯胺基)-吡啶并[2,3-d]嘧啶-7-羧酸N’-(2-二甲胺基-乙酰基)-酰肼、6-(2-溴-苯基)-2-(4-氟代-苯胺基)-吡啶并[2,3-d]嘧啶-7-羧酸(1-甲基-哌啶-4-基)-酰胺、 (S)-6-(2-溴-苯基)-2-(4-氟代-苯胺基)-吡啶并[2,3-d]嘧啶-7-羧酸吡咯啶-3-基酰胺、(R)-6-(2-溴-苯基)-2-(4-氟代-苯胺基)-吡啶并[2,3-d]嘧啶-7-羧酸吡咯啶 -3-基酰胺、6-(2-溴-苯基)-2-(4-氟代-苯胺基)-吡啶并[2,3-d]嘧啶-7-羧酸(1-氮- 双环[2.2.2]辛-3-基)-酰胺、6-(2-溴-苯基)-2-(4-氟代-苯胺基)-吡啶并[2,3-d]嘧啶 -7-羧酸(1H-吡唑-3-基)-酰胺、6-(2-溴-苯基)-2-(4-氟代-苯胺基)-吡啶并[2,3-d] 嘧啶-7-羧酸(2-甲基-2H-吡唑-3-基)-酰胺、6-(2-溴-苯基)-2-(4-氟代-苯胺基)- 吡啶并[2,3-d]嘧啶-7-羧酸(4-胺甲酰基-1H-吡唑-3-基)-酰胺、6-(2-溴-苯基)-2-(4-吗啉-4-基-苯胺基)-吡啶并[2,3-d]嘧啶-7-羧酸酰胺、6-(2-溴-苯基)-2-[4-(2-二乙胺基-乙氧基)-苯胺基]-吡啶并[2,3-d]嘧啶-7-羧酸酰胺、6-(2- 溴-苯基)-2-(3-甲基磺酰基-苯胺基)-吡啶并[2,3-d]嘧啶-7-羧酸酰胺、6-(2-溴- 苯基)-2-(4-胺磺酰基-苯胺基)-吡啶并[2,3-d]嘧啶-7-羧酸酰胺、6-(2-溴-苯基)-2-(3-甲基胺磺酰基甲基-苯胺基)-吡啶并[2,3-d]嘧啶-7-羧酸酰胺、6-(2-溴- 苯基)-2-[3-(2-羟基-乙烷磺酰基)-苯胺基]-吡啶并[2,-3-d]嘧啶-7-羧酸酰胺、 6-(2-溴-苯基)-2-(3-甲烷磺酰基-苯胺基)-吡啶并[2,3-d]嘧啶-7-羧酸酰胺、6-(2- 溴-苯基)-2-(3-甲烷磺酰基-苯胺基)-吡啶并[2,3-d]嘧啶-7-羰腈、6-(2-溴-苯基)-2-[4-(2-二乙胺基-乙氧基)-苯胺基]-吡啶并[2,3-d]嘧啶-7-羰腈、6-(2-溴-苯基)-2-[4-(2-羟基-乙氧基)-苯胺基]-吡啶并[2,3-d]嘧啶-7-羰腈、6-(2-溴-苯基)-2-[4-(2-乙胺基-乙氧基)-苯胺基]-吡啶并[2,3-d]嘧啶-7-羰腈、6-(2-溴-苯基)-2-(3-甲烷亚磺酰基-苯胺基)-吡啶并[2,3-d]嘧啶-7-羰腈、6-(2-溴-苯基)-2-[3-(2-羟基-乙烷磺酰基)-苯胺基]-吡啶并[2,3-d]嘧啶-7-羰腈、6-(2-溴-苯基)-2-(4-吗啉-4-基-苯胺基)-吡啶并[2,3-d]嘧啶-7-羧酸(2-甲烷磺酰基胺基-乙基)-酰胺、6-(2-溴-苯基)-2-(3-甲烷磺酰基胺基-苯胺基)-吡啶并[2,3-d]嘧啶-7- 羧酸(2-甲烷磺酰基胺基-乙基)-酰胺、6-(2-溴-苯基)-2-[4-(2-羟基-乙氧基)-苯胺基]-吡啶并[2,3-d]嘧啶-7-羧酸(2-甲烷磺酰基胺基-乙基)-酰胺、6-(2-溴-苯基)-2-[4-(4-甲基-哌嗪-1-基)-苯胺基]-吡啶并[2,3-d]嘧啶-7-羧酸(2-甲烷磺酰基胺基-乙基)-酰胺、6-(2-溴-苯基)-2-(3-甲氧基-苯胺基)-吡啶并[2,3-d]嘧啶-7-羧酸(2-甲烷磺酰基胺基-乙基)-酰胺、2-(3-乙酰胺基-苯胺基)-6-(2-溴-苯基)-吡啶并[2,3-d]嘧啶-7-羧酸(2-甲烷磺酰基胺基-乙基)-酰胺、6-(2-溴-苯基)-2-(4,4-二氧-3,4-二氢-2H-4λ*6*-苯并[1,4]恶噻英-6-基胺)-吡啶并[2,3-d]嘧啶-7-羧酸(2- 甲烷磺酰基胺基-乙基)-酰胺、6-(2-溴-苯基)-2-[3-(2-羟基-乙基磺酰基)-苯胺基]-吡啶并[2,3-d]嘧啶-7-羧酸(2-甲烷磺酰基胺基-乙基)-酰胺、6-(2-溴-苯基)-2-(3-羟甲基-2,3-二氢-苯并[1,4]二恶英-6-基胺)-吡啶并[2,3-d]嘧啶-7-羧酸 (2-甲烷磺酰基-胺基-乙基)-酰胺、6-(2-溴-苯基)-2-(4-氟代-苯胺基)-吡啶并 [2,3-d]嘧啶-7-羧酸(哌啶-2-基甲基)-酰胺、6-(2-溴-苯基)-2-(4-甲烷亚磺酰基- 苯胺基)-吡啶并[2,3-d]嘧啶-7-羧酸(2-羟基-乙基)-酰胺、6-(2-溴-苯基)-2-(3-甲烷亚磺酰基-苯胺基)-吡啶并[2,3-d]嘧啶-7-羧酸(2-羟基-乙基)-酰胺、6-(2-溴-苯基)-2-[3-(2-羟基-乙基胺磺酰基)-苯胺基]-吡啶并[2,3-d]嘧啶-7-羧酸(2-羟基 -乙基)-酰胺、6-(2-溴-苯基)-2-[4-(2-羟基-乙基胺磺酰基)-苯胺基]-吡啶并[2,3-d] 嘧啶-7-羧酸(2-羟基-乙基)-酰胺、6-(2-溴-苯基)-2-(4-甲烷亚磺酰基-苯胺基)- 吡啶并[2,3-d]嘧啶-7-羧酸(2-胺磺酰基-乙基)-酰胺、6-(2-溴-苯基)-2-(3-甲烷亚磺酰基-苯胺基)-吡啶并[2,3-d]嘧啶-7-羧酸(2-胺磺酰基-乙基)-酰胺、6-(2-溴- 苯基)-2-[4-(2-羟基-乙氧基)-苯胺基]-吡啶并[2,3-d]嘧啶-7-羧酸(2-胺磺酰基- 乙基)-酰胺、6-(2-溴-苯基)-2-(3-甲烷磺酰基-苯胺基)-吡啶并[2,3-d]嘧啶-7-羧酸(2-胺磺酰基-乙基)-酰胺、6-(2-溴-苯基)-2-(3-羟甲基-苯胺基)-吡啶并[2,3-d] 嘧啶-7-羧酸(2-羟基-乙基)-酰胺、6-(2-溴-苯基)-2-(3-羟甲基-苯胺基)-吡啶并 [2,3-d]嘧啶-7-羧酸(2-胺磺酰基-乙基)-酰胺、6-(2-溴-苯基)-2-(4,4-二氧-3,4-二氢-2H-4λ*6*-苯并[1,4]恶噻英-6-基胺)-吡啶并[2,3-d]嘧啶-7-羧酸(2-胺磺酰基- 乙基)-酰胺、6-(2-溴-苯基)-2-(4,4-二氧-3,4-二氢-2H-4λ*6*-苯并[1,4]恶噻英-6- 基胺)-吡啶并[2,3-d]嘧啶-7-羧酸(吡咯啶-2-基甲基)-酰胺盐酸盐、6-(2-溴-苯基)-2-(3-甲烷磺酰基胺基-苯胺基)-吡啶并[2,3-d]嘧啶-7-羧酸(吡咯啶-2-基甲基)-酰胺盐酸盐,以及2-(3-乙酰胺基-苯胺基)-6-(2-溴-苯基)-吡啶并[2,3-d]嘧啶-7-羧酸(吡咯啶-2-基甲基)-酰胺盐酸盐。Luk等人所申请的美国专利第 7,129,351号,并通过引用将其包括在内,其描述作为Src抑制剂的嘧啶并化合物,包含(+)-3-(2-溴-苯基)-7-[4-(2-二乙胺基-乙氧基)-苯胺基]-1,4-二甲基 -3,4-二氢-1H-嘧啶并[4,5-d]嘧啶-2-酮,以及(-)-3-(2-溴-苯基)-7-[4-(2-二乙胺基 -乙氧基)-苯胺基]-1,4-二甲基-3,4-二氢-1H-嘧啶并[4,5-d]嘧啶-2-酮。 Bebbington等人所申请的美国专利第7,115,739号,并通过引用将其包括在内,其描述作为Src抑制剂的三唑化合物。Bebbington等人所申请的美国专利第7,098,330号,并通过引用将其包括在内,其描述作为Src抑制剂的吡唑胺基取代的喹诺酮化合物。Cai等人所申请的美国专利第7,091,345号,并通过引用将其包括在内,其描述作为Src抑制剂的胺基-取代的二氢嘧啶并[4,5-d] 嘧啶酮衍生物。Bebbington等人所申请的美国专利第7,087,603号,并通过引用将其包括在内,其描述作为Src抑制剂的吡唑化合物。Bebbington等人所申请的美国专利第7,008,948号,通过引用将其包括,其描述作为Src抑制剂的稠合嘧啶基吡唑化合物。Bebbington等人所申请的美国专利第6,989,385 号,并通过引用将其包括在内,其描述作为Src抑制剂的吡唑化合物,包含{2-[(2-羟乙基)苯胺基]-喹唑啉-4-基}-(5-甲基-2H-吡唑-3--yl)-胺、[2-(甲基苯胺基)-喹唑啉-4-基]-(5-甲基-2H-吡唑-3-基)-胺、(5-甲基-2H-吡唑-3-基)-{2-[N- 甲基-N-(吡啶-3-基甲基)胺基]-喹唑啉-4-基}-胺、(5-甲基-2H-吡唑-3-基)-(2-苯胺基-喹唑啉-4-基)-胺、(2-苄基胺基-喹唑啉-4-基)-(5-甲基-2H-吡唑-3-基)胺、 (2-环己胺基-喹唑啉-4-基)-(5-甲基-2H-吡唑-3-基)-胺、[2-(2,3-二氢苯并[1,4] 二恶英-6-基胺)-喹唑啉-4-基]-(5-甲基-2H-吡唑-3-基)-胺、(2-环己基甲胺基- 喹唑啉-4-基)-(5-甲基-2H-吡唑-3-基)-胺、[2-(1H-吲唑-6-基胺)-喹唑啉-4- 基]-(5-甲基-2H-吡唑-3-基)-胺、(5-甲基-2H-吡唑-3-基)-[2-(吡啶-3-基甲胺基)- 喹唑啉-4-基]-胺、[2-(3-氯苯胺基)-喹唑啉-4-基]-(5-甲基-2H-吡唑-3-基)-胺、 [2-(4-氯苯胺基)-喹唑啉-4-基]-(5-甲基-2H-吡唑-3-基)-胺、[2-(4-氟代苄基胺基)-喹唑啉-4-基]-(5-甲基-2H-吡唑-3-基)-胺、{2-[2-(2-羟乙基)苯胺基]-喹唑啉 -4-基}-(5-甲基-2H-吡唑-3-基)-胺、[2-(4-氰基甲基苯基胺基)-喹唑啉-4-基]-(5- 甲基-2H-吡唑-3-基)-胺、[2-(3-羟甲基苯胺基)-喹唑啉-4-基]-(5-甲基-2H-吡唑 -3-基)-胺、[2-(3-羟基苯胺)-喹唑啉-4-基]-(5-甲基-2H-吡唑-3-基)-胺、(5-环丙基-2H-吡唑-3-基)-(2-苯胺基-喹唑啉-4-基)-胺、(5-环丙基-2H-吡唑-3-基)-[2-(3- 甲基苯胺基)-喹唑啉-4-基]-胺、(5-环丙基-2H-吡唑-3-基)-[2-(6-甲氧基吡啶-3-基胺)-喹唑啉-4-基]-胺、(5-环丙基-2H-吡唑-3-基)-[2-(茚满-5-基胺)-喹唑啉-4- 基]-胺、(5-环丙基-2H-吡唑-3-基)-[2-(1H-吲哚-6-基胺)-喹唑啉-4-基]-胺、[2-(4- 乙酰胺基-3-甲基苯胺基)-喹唑啉-4-基]-(5-环丙基-2H-吡唑-3-基)-胺、[2-(4-氯基-3-甲基苯胺基)-喹唑啉-4-基]-(5-环丙基-2H-吡唑-3-基)-胺、(5-环丙基-2H- 吡唑-3-基)-[2-(4-乙基苯基胺基)-喹唑啉-4-基]-胺、(5-环丙基-2H-吡唑-3- 基)-[2-(4-丙基苯基胺基)-喹唑啉-4-基]-胺、(5-环丙基-2H-吡唑-3-基)-{2-[4-(2- 羟乙基)苯胺基]-喹唑啉-4-基}-胺、(5-环丙基-2H-吡唑-3-基)-(2-苯胺基-喹唑啉 -4-基)-胺、[2-(2-环己基乙胺基)-喹唑啉-4-基]-(5-环丙基-2H-吡唑-3-基)-胺、 [2-(4-羧基甲氧基苯胺)-喹唑啉-4-基]-(5-环丙基-2H-吡唑-3-基)-胺、[2-(4-氰基甲基苯基胺基)-喹唑啉-4-基]-(5-环丙基-2H-吡唑-3-基)-胺、[2-(苯并噻唑-6- 基胺)-喹唑啉-4-基]-(5-环丙基-2H-吡唑-3-基)-胺、(5-环丙基-2H-吡唑-3- 基)-[2-(3,4-二甲基苯胺基)-喹唑啉-4-基]-胺、(5-环丙基-2H-吡唑-3-基)-[2-(2- 苯氧基乙胺基)-喹唑啉-4-基]-胺、(5-环丙基-2H-吡唑-3-基)-[2-(噻吩-2-甲胺基)-喹唑啉-4-基]-胺、[2-(4-羧甲基苯胺基)-喹唑啉-4-基]-(5-环丙基-2H-吡唑-3-基)-胺、(5-环丙基-2H-吡唑-3-基)-[2-(1H-吲唑-5-基胺)-喹唑啉-4-基]-胺、(5-环丙基-2H-吡唑-3-基)-[2-(吡啶-3-基甲胺基)-喹唑啉-4-基]-胺、(5-环丙基-2H- 吡唑-3-基)-[2-(3-甲氧基羰基苯胺基)-喹唑啉-4-基]-胺、[2-(3-羧基苯胺)-喹唑啉-4-基]-(5-环丙基-2H-吡唑-3-基)-胺、(5-环丙基-2H-吡唑-3-基)-[2-(3-乙基苯基胺基)-喹唑啉-4-基]-胺、(5-环丙基-2H-吡唑-3-基)-[2-(2,3-二甲基苯胺基)- 喹唑啉-4-基]-胺、(5-环丙基-2H-吡唑-3-基)-[2-(3,4-二甲氧基苯胺)-喹唑啉-4- 基]-胺、(5-环丙基-2H-吡唑-3-基)-[2-(3-甲氧基苯胺)-喹唑啉-4-基]-胺、(5-甲基-2H-吡唑-3-基)-(2-苯胺基-5,6,7,8-四氢喹唑啉-4-基)-胺、[2-(联苯-3-基胺)- 喹唑啉-4-基]-(5-环丙基-2H-吡唑-3-基)-胺、(5-环丙基-2H-吡唑-3-基)-[2-(3-苯基丙-1-基胺)-喹唑啉-4-基]-胺、[2-(4-乙酰胺基-3-甲基苯胺基)-喹唑啉-4- 基]-(5-甲基-2H-吡唑-3-基)-胺、(5-环丙基-2H-吡唑-3-基)-[2-(茚满-2-基胺)-喹唑啉-4-基]-胺、[2-(3-甲基苯胺基)-喹唑啉-4-基]-(5-甲基-2H-吡唑-3-基)-胺、 [2-(2-氯基-5-甲基苯胺基)-喹唑啉-4-基]-(5-甲基-2H-吡唑-3-基)-胺、(5-环丙基 -2H-吡唑-3-基)-{2-[4-(吗啉-1-基)苯胺基]-喹唑啉-4-基}-胺、[2-(苯并噻唑-6- 基胺)-喹唑啉-4-基]-(5-甲基-2H-吡唑-3-基)-胺、[2-(3,4-二甲基苯胺基)-喹唑啉 -4-基]-(5-甲基-2H-吡唑-3-基)-胺、[2-(3-乙基苯基胺基)-喹唑啉-4-基]-(5-甲基 -2H-吡唑-3-基)-胺、[2-(3-甲氧基苯胺)-喹唑啉-4-基]-(5-甲基-2H-吡唑-3-基)- 胺、[2-(4-乙酰胺基-3-氰基苯胺基)-喹唑啉-4-基]-(5-甲基-2H-吡唑-3-基)-胺、[2-(2-甲氧基联苯基-5-基胺)-喹唑啉-4-基]-(5-甲基-2H-吡唑-3-基)-胺、[2-(4- 乙酰胺基苯胺基)-喹唑啉-4-基]-(5-甲基-2H-吡唑-3-基)-胺、[2-(4-叔-丁氧基羰胺基-苯胺基)-喹唑啉-4-基]-(5-甲基-2H-吡唑-3-基)-胺、[2-(4-氰基苯胺基)-喹唑啉-4-基]-(5-甲基-2H-吡唑-3-基)-胺、(5-甲基-2H-吡唑-3-基)-[2-(6-氧基 -6,10-二氢-4aH-苯并[c]克唍-2-基胺)-喹唑啉-4-基]-胺、[2-(联苯-3-基胺)-喹唑啉-4-基]-(5-甲基-2H-吡唑-3-基)-胺、[2-(4-甲氧基羰基甲基-3-甲基苯胺基)-喹唑啉-4-基]-(5-甲基-2H-吡唑-3-基)-胺、[2-(4-羧基甲基-3-甲基苯胺基)-喹唑啉-4-基]-(5-甲基-2H-吡唑-3-基)-胺、[2-(4-胺基苯基胺基)-喹唑啉-4-基]-(5-甲基 -2H-吡唑-3-基)-胺、[2-(4-溴基苯基胺基)-喹唑啉-4-基]-(5-甲基-2H-吡唑-3- 基)-胺、[2-(4-异丁酰胺基-苯胺基)-喹唑啉-4-基]-(5-甲基-2H-吡唑-3-基)-胺、 (5-乙基-2H-吡唑-3-基)-[2-(5-乙基-2H-吡唑-3-基胺)-喹唑啉-4-基]-胺、(1H-吲唑-3-基)-(2-苯胺基-喹唑啉-4-基)-胺、(1H-吲唑-3-基)-[2-(3-三氟代甲基苯胺基)-喹唑啉-4-基]-胺、(1H-吲唑-3-基)-[2-(4-三氟代甲基苯胺基)-喹唑啉-4-基]- 胺、[2-(金刚烷-2-基胺)-喹唑啉-4-基]-(1H-吲唑-3-基)-胺、(1H-吲唑-3-基)-(2- 甲基-苯基-胺基-喹唑啉-4-基)-胺、[2-(2-氯基-苯基)-胺基-喹唑啉-4-基]-(1H- 吲唑-3-基)-胺、(1H-吲唑-3-基)-[2-(2-三氟代甲基苯胺基)-喹唑啉-4-基]-胺、[2-(4-氰基甲基苯基胺基)-喹唑啉-4-基]-(1H-吲唑-3-基)-胺、[2-(4-氯苯胺基)-5,6,7,8-四氢喹唑啉-4-基]-(5-甲基-2H-吡唑-3-基)-胺、(5-甲基-2H-吡唑-3- 基)-(2-苯胺基-6,7,8,9-四氢-5H-环七嘧啶-4-基)-胺、[2-(苯并咪唑-2-基胺)-7- 苄基-5,6,7,8-四氢-吡啶并[3,4-d]嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺、(7-苄基-2-苯胺基-5,6,7,8-四氢-吡啶并[3,4-d]嘧啶-4-基)-(5-甲基-2H-吡唑-3-基)- 胺、[6-苄基-2-(4-氯苯胺基)-5,6,7,8-四氢-吡啶并[4,3-d]嘧啶-4-基]-(5-甲基-2H- 吡唑-3-基)-胺、[2-(苯并咪唑-2-基胺)-6-苄基-5,6,7,8-四氢-吡啶并[4,3-d]嘧啶 -4-基]-(5-甲基-2H-吡唑-3-基)-胺、(6-苄基-2-苯胺基-5,6,7,8-四氢-吡啶并[4,3-d] 嘧啶-4-基)-(5-甲基-2H-吡唑-3-基)-胺、(5-甲基-2H-吡唑-3-基)-(2-苯胺基 -5,6,7,8-四氢-吡啶并[3,4-d]嘧啶-4-基)-胺、[2-(4-氰基甲基苯基胺基)-喹唑啉 -4-基]-(1H-吡唑并[3,4-b]吡啶-3-基)-胺、[2-(4-氰基苄基胺基)-喹唑啉-4- 基]-(1H-吡唑并[3,4-b]吡啶-3-基)-胺、[2-(4-氰基甲基苯基胺基)-喹唑啉-4- 基]-(4-氟代-1H-吲唑-3-基)-胺、[2-(4-氰基苯胺基)-喹唑啉-4-基]-(1H-吲唑-3- 基)-胺,以及[2-(4-氰基苄基胺基)-喹唑啉-4-基]-(1H-吲唑-3-基)-胺。Boschelli 等人所申请的美国专利第6,987,116号,并通过引用将其包括在内,其描述作为Src抑制剂的噻咔[3,2-b]吡啶-6-羰腈及噻咔[2,3-b]吡啶-5-羰腈。Davies等人所申请的美国专利第6,696,452号,并通过引用将其包括在内,其描述作为Src抑制剂的吡唑化合物。Bebbington等人所申请的美国专利第6,664,247 号,并通过引用将其包括在内,其描述作为Src抑制剂的吡唑化合物,包含 (5-环丙基-2H-吡唑-3-基)-[2-(萘-2-基磺酰基)-6-苯基嘧啶-4-基]-胺、(5-环丙基 -2H-吡唑-3-基)-[2-(3-甲氧基羰基-苯基基磺酰基)-6-苯基嘧啶-4-基]-胺、(5-环丙基-2H-吡唑-3-基)-[2-(萘-2-基磺酰基)-嘧啶-4-基]-胺、(5-环丙基-2H-吡唑-3- 基)-[5,6-二甲基-2-(萘-2-基磺酰基)-嘧啶-4-基]-胺、(5-环丙基-2H-吡唑-3- 基)-[5-甲基-2-(萘-2-基磺酰基)-嘧啶-4-基]-胺、(5-环丙基-2H-吡唑-3-基)-[6- 甲基-2-(萘-2-基磺酰基)-嘧啶-4-基]-胺、(5-环丙基-2H-吡唑-3-基)-[6-(吗啉-4- 基)-2-(萘-2-基磺酰基)-嘧啶-4-基]-胺、(5-环丙基-2H-吡唑-3-基)-[6-(1-甲基哌嗪-4-基)-2-(萘-2yl-磺酰基)-嘧啶-4-基]-胺、[6-(2,6-二甲苯基)-2-(萘-2-基-磺酰基)-嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺、[6-(2-甲苯基)-2-(萘-2-基磺酰基)- 嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺、[2-(4-乙酰胺基-苯基磺酰基)-6-苯基- 嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺、(5-甲基-2H-吡唑-3-基)-[2-(萘-2-基磺酰基)-6-苯基-嘧啶-4-基]-胺、[2-(4-异丁胺-苯基磺酰基)-6-苯基嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺、[6-(4-甲基哌嗪-1-基)-2-甲基磺酰基-嘧啶-4-基]-(5-甲基 -2H-吡唑-3-基)-胺、(5-甲基-2H-吡唑-3-基)-[6-苯基-2-(4-丙酰胺基-苯基磺酰基)-嘧啶-4-基]-胺、[2-(4-环丙烷羰胺-苯基磺酰基)-6-苯基嘧啶-4-基]-(5-甲基 -2H-吡唑-3-基)-胺、(5-甲基-2H-吡唑-3-基)-{6-苯基-2-[4-(丙烷-1-磺酰基胺基)-苯基磺酰基]-嘧啶-4-基}-胺、[2-(4-乙烷磺酰基胺基-苯基磺酰基)-6-苯基- 嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺、[2-(4-乙酰胺基苯基-磺酰基)-6-(2-甲苯基)-嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺、[2-(4-异丁烷羰胺基-苯基-磺酰基)-6-苯基-嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺、[2-(4-乙酰胺基-苯基-磺酰基)-5-甲基-6-苯基-嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺、[2-(4-乙酰胺基-苯基-磺酰基)-6-(4-甲氧苯基)-嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺、[6-(3-乙酰胺基苯基)-2-(4-乙酰胺基-苯基-磺酰基)-嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)- 胺、[2-(4-异丙烷磺酰基胺基-苯基-磺酰基)-6-苯基-嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺、{2-[4-(2-二甲胺基-乙酰胺基)-苯基磺酰基]-6-苯基-嘧啶-4- 基}-(5-甲基-2H-吡唑-3-基)-胺、[2-(3-氯基-苄基磺酰基)-6-吗啉-4-基-嘧啶-4- 基]-(5-甲基-2H-吡唑-3-基)-胺、[2-(3-氯基-苄基磺酰基)-6-(2-甲氧基-乙胺基)- 嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺、[2-苄基磺酰基-6-(4-甲基哌嗪-1-基)- 嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺、[2-苄基磺酰基-6-吗啉-4-基-嘧啶-4- 基]-(5-甲基-2H-吡唑-3-基)-胺、[2-(3-氯基-苄基磺酰基)-6-(4-甲基哌嗪-1-基)- 嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺、[2-(4-甲氧基-苄基磺酰基)-6-(4-甲基哌嗪-1-基)-嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺、[2-(4-乙酰胺基-苯基-磺酰基)-6-叔-丁基-嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺、(5-环丙基-2H-吡唑-3- 基)-[6-苯基-2-(4-丙酰胺基-苯基-磺酰基)-嘧啶-4-基]-胺、[2-(3-氯基-苄基磺酰基)-6-(哌啶-1-基)-嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺、(5-甲基-2H-吡唑-3- 基)-{2-[4-(吗啉磺酰基)-苄基磺酰基]-6-吗啉-4-基-嘧啶-4-基}-胺、{6-(2-甲氧基-乙胺基)-2-[4-(吗啉磺酰基)-苄基磺酰基]-嘧啶-4-基}-(5-甲基-2H-吡唑-3- 基)-胺、{6-(4-甲基哌嗪-1-基)-2-[4-(吗啉磺酰基)-苄基磺酰基]-嘧啶-4-基}-(5- 甲基-2H-吡唑-3-基)-胺、[6-甲氧基甲基-2-(4-丙酰胺基-苯基-磺酰基)-嘧啶-4- 基]-(5-甲基-2H-吡唑-3-基)-胺、[2-(4-甲氧基羰基-苯基-磺酰基)-6-甲氧基甲基 -嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺、[2-(3,5-二甲氧基-苄基磺酰基)-6-吗啉 -4-基-嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺、[2-(3,5-二甲氧基-苄基磺酰基)-6- 吡咯啶-4-基-嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺、(5-甲基-2H-吡唑-3-基)-[6- 吗啉-4-基-2-(萘-2-基-甲基磺酰基)-嘧啶-4-基]-胺、{2-(4-乙酰胺基-苯基-磺酰基)-6-[4-(3-二甲胺基-丙氧基)苯基]-嘧啶-4-基}-(5-甲基-2H-吡唑-3-基)-胺、 [2-(4-乙酰胺基苯基磺酰基)-6-(吗啉-4-基)-嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)- 胺、[6-羟甲基-2-(4-丙酰胺基-苯基-磺酰基)-嘧啶-4-基]-(5-甲基-2H-吡唑-3- 基)-胺、[2-(4-乙酰胺基-苯基-磺酰基)-嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺、 [6-(1-丁氧基羰基)-2-(4-丙酰胺基-苯基-磺酰基)嘧啶-4-基]-(5-甲基-2H-吡唑 -3-基)-胺,以及[6-甲氧基羰基-2-(4-丙酰胺基-苯基-磺酰基)-嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺。Bebbington等人所申请的美国专利第6,660,731号,并通过引用将其包括在内,其描述作为Src抑制剂的吡唑化合物。Bebbington 等人所申请的美国专利第6,653,301号,并通过引用将其包括在内,其描述作为Src抑制剂的吡唑化合物,包含(5-环丙基-2H-吡唑-3-基)-[2-(萘-2-基磺酰基)-6-苯基嘧啶-4-基]-胺、(5-环丙基-2H-吡唑-3-基)-[2-(3-甲氧基羰基-苯基基磺酰基)-6-苯基嘧啶-4-基]-胺、(5-环丙基-2H-吡唑-3-基)-[2-(萘-2-基磺酰基)- 嘧啶-4-基]-胺、(5-环丙基-2H-吡唑-3-基)-[5,6-二甲基-2-(萘-2-基磺酰基)-嘧啶 -4-基]-胺、(5-环丙基-2H-吡唑-3-基)-[5-甲基-2-(萘-2-基磺酰基)-嘧啶-4-基]- 胺、(5-环丙基-2H-吡唑-3-基)-[6-甲基-2-(萘-2-基磺酰基)-嘧啶-4-基]-胺、(5- 环丙基-2H-吡唑-3-基)-[6-(吗啉-4-基)-2-(萘-2-基磺酰基)-嘧啶-4-基]-胺、(5- 环丙基-2H-吡唑-3-基)-[6-(1-甲基哌嗪-4-基)-2-(萘-2-基磺酰基)-嘧啶-4-基]- 胺、[6-(2,6-二甲苯基)-2-(萘-2-基磺酰基)-嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)- 胺、[6-(2-甲苯基)-2-(萘-2-基磺酰基)-嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺、 [2-(4-乙酰胺基-苯基磺酰基)-6-苯基-嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺、 (5-甲基-2H-吡唑-3-基)-[2-(萘-2-基磺酰基)-6-苯基-嘧啶-4-基]-胺、[2-(4-异丁酰胺基-苯基磺酰基)-6-苯基嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺、[6-(4-甲基哌嗪-1-基)-2-甲基磺酰基-嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺、(5-甲基-2H- 吡唑-3-基)-[6-苯基-2-(4-丙酰胺基-苯基磺酰基)-嘧啶-4-基]-胺、[2-(4-环丙烷羰胺-苯基磺酰基)-6-苯基嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺、(5-甲基-2H-吡唑-3-基)-{6-苯基-2-[4-(丙烷-1-磺酰基胺基)-苯基磺酰基]-嘧啶-4-基}-胺、 [2-(4-乙烷磺酰基胺基-苯基磺酰基)-6-苯基-嘧啶-4-基]-(5-甲基-2H-吡唑-3- 基)-胺、[2-(4-乙酰胺基苯基-磺酰基)-6-(2-甲苯基)-嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺、[2-(4-异丁烷羰胺基-苯基-磺酰基)-6-苯基-嘧啶-4-基]-(5-甲基 -2H-吡唑-3-基)-胺、[2-(4-乙酰胺基-苯基-磺酰基)-5-甲基-6-苯基-嘧啶-4- 基]-(5-甲基-2H-吡唑-3-基)-胺、[2-(4-乙酰胺基-苯基-磺酰基)-6-(4-甲氧苯基)- 嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺、[6-(3-乙酰胺基苯基)-2-(4-乙酰胺基- 苯基-磺酰基)-嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺、[2-(4-异丙烷磺酰基胺基 -苯基-磺酰基)-6-苯基-嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺、{2-[4-(2-二甲胺基-乙酰胺基)-苯基磺酰基]-6-苯基-嘧啶-4-基}-(5-甲基-2H-吡唑-3-基)-胺、 [2-(3-氯基-苄基磺酰基)-6-吗啉-4-基-嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺、 [2-(3-氯基-苄基磺酰基)-6-(2-甲氧基-乙胺基)-嘧啶-4-基]-(5-甲基-2H-吡唑-3- 基)-胺、[2-苄基磺酰基-6-(4-甲基哌嗪-1-基)-嘧啶-4-基]-(5-甲基-2H-吡唑-3- 基)-胺、[2-苄基磺酰基-6-吗啉-4-基-嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺、[2-(3-氯基-苄基磺酰基)-6-(4-甲基哌嗪-1-基)-嘧啶-4-基]-(5-甲基-2H-吡唑-3- 基)-胺、[2-(4-甲氧基-苄基磺酰基)-6-(4-甲基哌嗪-1-基)-嘧啶-4-基]-(5-甲基 -2H-吡唑-3-基)-胺、[2-(4-乙酰胺基-苯基-磺酰基)-6-叔-丁基-嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺、(5-环丙基-2H-吡唑-3-基)-[6-苯基-2-(4-丙酰胺基-苯基- 磺酰基)-嘧啶-4-基]-胺、[2-(3-氯基-苄基磺酰基)-6-(哌啶-1-基)-嘧啶-4-基]-(5- 甲基-2H-吡唑-3-基)-胺、(5-甲基-2H-吡唑-3-基)-{2-[4-(吗啉磺酰基)-苄基磺酰基]-6-吗啉-4-基-嘧啶-4-基}-胺、{6-(2-甲氧基-乙胺基)-2-[4-(吗啉磺酰基)-苄基磺酰基]-嘧啶-4-基}-(5-甲基-2H-吡唑-3-基)-胺、{6-(4-甲基哌嗪-1- 基)-2-[4-(吗啉磺酰基)-苄基磺酰基]-嘧啶-4-基}-(5-甲基-2H-吡唑-3-基)-胺、 [6-甲氧基甲基-2-(4-丙酰胺基-苯基-磺酰基)-嘧啶-4-基]-(5-甲基-2H-吡唑-3- 基)-胺、[2-(4-甲氧基羰基-苯基-磺酰基)-6-甲氧基甲基-嘧啶-4-基]-(5-甲基 -2H-吡唑-3-基)-胺、[2-(3,5-二甲氧基-苄基磺酰基)-6-吗啉-4-基-嘧啶-4-基]-(5- 甲基-2H-吡唑-3-基)-胺、[2-(3,5-二甲氧基-苄基磺酰基)-6-吡咯啶-4-基-嘧啶-4- 基]-(5-甲基-2H-吡唑-3-基)-胺、(5-甲基-2H-吡唑-3-基)-[6-吗啉-4-基-2-(萘-2- 基-甲基磺酰基)-嘧啶-4-基]-胺、{2-(4-乙酰胺基-苯基-磺酰基)-6-[4-(3-二甲胺基-丙氧基)苯基]-嘧啶-4-基}-(5-甲基-2H-吡唑-3-基)-胺、[2-(4-乙酰胺基苯基磺酰基)-6-(吗啉-4-基)-嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺、[6-羟甲基-2-(4- 丙酰胺基-苯基-磺酰基)-嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺、[2-(4-乙酰胺基-苯基-磺酰基)-嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺、[6-(1-丁氧基羰基)-2-(4-丙酰胺基-苯基-磺酰基)嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺,以及 [6-甲氧基羰基-2-(4-丙酰胺基-苯基-磺酰基)-嘧啶-4-基]-(5-甲基-2H-吡唑-3- 基)-胺。Bebbington等人所申请的美国专利第6,653,300号,并通过引用将其包括在内,其描述作为Src抑制剂的吡唑化合物,包含(5-甲基-2H-吡唑-3- 基)-[2-(萘-2-基氧基)-喹唑啉-4-基]-胺、(5-甲基-2H-吡唑-3-基)-(2-苯氧基-喹唑啉-4-基)-胺、(5-环丙基-2H-吡唑-3-基)-[2-(5,6,7,8-四氢萘-2-基氧基)-喹唑啉-4- 基]-胺、(5-环丙基-2H-吡唑-3-基)-[2-(3-甲基苯氧基)-喹唑啉-4-基]-胺、[2-(3- 甲氧基苯氧基)-喹唑啉-4-基]-(5-甲基-2H-吡唑-3-基)-胺、[2-(3,4-二甲氧基苯氧基)-喹唑啉-4-基]-(5-甲基-2H-吡唑-3-基)-胺、[2-(苯并[1,3]二氧基-5-基氧基)-喹唑啉-4-基]-(5-甲基-2H-吡唑-3-基)-胺、[2-(3-甲氧基羰基苯氧基)-喹唑啉-4-基]-(5-甲基-2H-吡唑-3-基)-胺、(5-环丙基-2H-吡唑-3-基)-(2-苯氧基甲基- 喹唑啉-4-基)-胺、(2-芐氧甲基-喹唑啉-4-基)-(5-环丙基-2H-吡唑-3-基)-胺、(2- 苄基-喹唑啉-4-基)-(5-环丙基-2H-吡唑-3-基)-胺、(5-环丙基-2H-吡唑-3-基)-(2- 甲基-喹唑啉-4-基)-胺、[2-(4-氯苯氧基甲基)-6,7,8,9-四氢-5H-环七嘧啶-4- 基]-(5-甲基-2H-吡唑-3-基)-胺、[2-(4-氯苯氧基甲基)-5,6,7,8-四氢-喹唑啉-4- 基]-(5-甲基-2H-吡唑-3-基)-胺、[2-(2-氯苯氧基甲基)-6-甲基-嘧啶-4-基]-(5-苯基-2H-吡唑-3-基)-胺、[2-(2-氯苯氧基甲基)-6-甲基-嘧啶-4-基]-[5-(呋喃-2- 基)-2H-吡唑-3-基]-胺、(6-甲基-2-苯氧基甲基-嘧啶-4-基)-(5-苯基-2H-吡唑-3- 基)-胺、[5-(呋喃-2-基)-2H-吡唑-3-基]-(6-甲基-2-苯氧基甲基-嘧啶-4-基)-胺、 [5-(呋喃-2-基)-2H-吡唑-3-基]-(6-甲基-2-苯基磺酰基甲基-嘧啶-4-基)-胺、[6- 甲基-2-(4-甲基-苯基磺酰基甲基)-嘧啶-4-基]-(5-苯基-2H-吡唑-3-基)-胺、 [5-(呋喃-2-基)-2H-吡唑-3-基]-[6-甲基-2-(4-甲基-苯基磺酰基甲基)-嘧啶-4- 基]-胺、[2-(4-氟代-苯氧基甲基)-6-甲基-嘧啶-4-基]-(5-苯基-2H-吡唑-3-基)- 胺、[2-(4-氟代-苯氧基甲基)-6-甲基-嘧啶-4-基]-[5-(呋喃-2-基)-2H-吡唑-3-基]- 胺、(6-乙基-2-苯基磺酰基甲基-嘧啶-4-基)-(5-甲基-2H-吡唑-3-基)-胺、(6-乙基-2-苯氧基甲基-嘧啶-4-基)-(5-甲基-2H-吡唑-3-基)-胺、[6-乙基-2-(4-氟代苯氧甲基)-嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺、[6-乙基-2-(1-甲基-1-苯基-乙基)-嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺、[2-(4-氯苯氧基甲基)-6-甲基-嘧啶 -4-基]-(5-苯基-2H-吡唑-3-基)-胺、[2-(4-氯苯氧基甲基)-6-甲基-嘧啶-4-基]-(5- 甲基-2H-吡唑-3-基)-胺、[2-(4-氯苯氧基甲基)-6-甲氧基甲基-嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺、[2-(4-氯苯氧基甲基)-6-甲基-嘧啶-4-基]-[5-(呋喃-2- 基)-2H-吡唑-3-基]-胺、(5-甲基-2H-吡唑-3-基)-(2-苯基磺酰基甲基-5,6,7,8-四氢-喹唑啉-4-基)-胺、[2-(4-甲基苯基磺酰基甲基)-6,7,8,9-四氢-5H-环七嘧啶-4- 基]-(5-甲基-2H-吡唑-3-基)-胺、[2-(1-甲基-1-苯基-乙基)-6,7,8,9-四氢-5H-环七嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺、[2-(2,6-二氯苄基)-5,6,7,8-四氢-喹唑啉 -4-基]-(5-甲基-2H-吡唑-3-基)-胺、[7-苄基-2-(2,6-二氯苄基)-5,6,7,8-四氢吡啶并[3,4-d]嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺、[6-苄基-2-(4-氯苯氧基甲基)-5,6,7,8-四氢-吡啶并[4,3-d]嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺、[2-(4-氯苯氧基甲基)-5,6,7,8-四氢-吡啶并[4,3-d]嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)- 胺、[2-(2,6-二氯苄基)-6-甲基-嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺、[2-(2,6- 二氯苄基)-5,6-二甲基-嘧啶-4-基]-(5-甲基-2H-吡唑-3-基)-胺、(1H-吲唑-3- 基)-[2-(2-苯基-环丙基)-喹唑啉-4-基]-胺、(7-氟代-1H-吲唑-3-基)-[2-(2-苯基- 环丙基)-喹唑啉-4-基]-胺、(5-氟代-1H-吲唑-3-基)-[2-(2-苯基-环丙基)-喹唑啉 -4-基]-胺、(5-甲基-1H-吡唑-3-基)-[2-(2-苯基-环丙基)-喹唑啉-4-基]-胺、[6- 乙基-2-(1-甲基-1-苯基-乙基)-嘧啶-4-基]-(5-甲基-1H-吡唑-3-基)-胺、[6-甲基-2-(1-甲基-1-苯基-乙基)-嘧啶-4-基]-(5-甲基-1H-吡唑-3-基)-胺、[6-甲基-2-(1- 苯基-环丙基)-嘧啶-4-基]-(5-甲基-1H-吡唑-3-基)-胺,以及[6-乙基-2-(1-甲基 -1-苯基-丙基)-嘧啶-4-基]-(5-甲基-1H-吡唑-3-基)-胺。Davies等人所申请的美国专利第6,638,926号,并通过引用将其包括在内,其描述作为Src抑制剂的吡唑化合物。Knegtel等人所申请的美国专利第6,613,736号,并通过引用将其包括在内,其描述作为Src抑制剂的吡唑化合物。Davies等人所申请的美国专利第6,610,677号,并通过引用将其包括在内,其描述作为Src抑制剂的吡唑化合物。Benish等人所申请的美国专利第6,503,914号,并通过引用将其包括在内,其描述作为Src抑制剂的噻咔嘧啶化合物,包含苯醛(7-甲基噻咔[3,2-d]嘧啶-4-基)腙、4-甲氧基苯甲醛(7-甲基噻咔[3,2-d]嘧啶-4-基)腙、4- 吡啶甲醛(7-甲基噻咔[3,2-d]嘧啶-4-基)腙、3,4-二甲氧基苯甲醛(7-甲基噻咔 [3,2-d]嘧啶-4-基)腙、3,5-二甲氧基苯甲醛(7-甲基噻咔[3,2-d]嘧啶-4-基)腙、3- 氯苯甲醛(7-甲基噻咔[3,2-d]嘧啶-4-基)腙、3,4-二羟基苯甲醛(7-甲基噻咔 [3,2-d]嘧啶-4-基)腙、3-吡啶甲醛(7-甲基噻咔[3,2-d]嘧啶-4-基)腙、2-噻吩甲醛 (7-甲基噻咔[3,2-d]嘧啶-4-基)腙、1H-吡咯-2-甲醛(7-甲基噻咔[3,2-d]嘧啶-4- 基)腙、2--呋喃甲醛(7-甲基噻咔[3,2-d]嘧啶-4-基)腙、3-羟基苯醛(7-甲基噻咔 [3,2-d]嘧啶-4-基)腙、3-噻吩甲醛(7-甲基噻咔[3,2-d]嘧啶-4-基)腙、1H-咪唑-2-甲醛(7-甲基噻咔[3,2-d]嘧啶-4-基)腙、4-乙氧基苯甲醛(7-甲基噻咔[3,2-d]嘧啶 -4-基)腙、4-羟基-3-硝基苯醛(7-甲基噻咔[3,2-d]嘧啶-4-基)腙、3-乙氧基-4-羟基苯醛(7-甲基噻咔[3,2-d]嘧啶-4-基)腙、3-羟基-4-甲氧基苯甲醛(7-甲基噻咔 [3,2-d]嘧啶-4-基)腙、3-氟代苯甲醛(7-甲基噻咔[3,2-d]嘧啶-4-基)腙、4-羟基-3- 甲氧基苯甲醛(7-甲基噻咔[3,2-d]嘧啶-4-基)腙、3-氯基-4-羟基苯醛(7-甲基噻咔[3,2-d]嘧啶-4-基)腙、4-氟代苯甲醛(6-苯基噻咔[3,2-d]嘧啶-4-基)-腙、3-吡啶甲醛(噻咔[3,2-d]嘧啶-4-基)腙、5-甲基-1H-咪唑-4-甲醛(7-甲基噻咔[3,2-d] 嘧啶-4-基)腙、5-甲基-2-噻吩甲醛(7-甲基噻咔[3,2-d]嘧啶-4-基)腙、4-氰基苯醛(7-甲基噻咔[3,2-d]嘧啶-4-基)腙、3-氰基苯醛(7-甲基噻咔[3,2-d]嘧啶-4-基) 腙、3-甲氧基苯甲醛(7-甲基噻咔[3,2-d]嘧啶-4-基)腙、3-乙氧基苯甲醛(7-甲基噻咔[3,2-d]嘧啶-4-基)腙、环丙烷甲醛(7-甲基噻咔[3,2-d]嘧啶-4-基)腙、3-吡啶甲醛(7-溴噻咔[3,2-d]嘧啶-4-基)腙,以及3-吡啶甲醛(6-苯基噻咔[3,2-d]嘧啶-4-基)腙。Budde等人所申请的美国专利第6,100,254号,并通过引用将其包括在内,其描述作为Src抑制剂的基于1,4-苯重氮基-2-酮核的化合物。
所述方法可以进一步包含:给予一BH3类似物的治疗有效量至目标对象的步骤。或者,所述方法可以进一步包含:给予一酪氨酸激酶抑制剂的治疗有效量至目标对象的步骤。
本发明的再一方面是提供一于患有具有一赋予胸腺核苷激酶抑制剂 (TKIs)抗性的生殖细胞缺失多型性的恶性肿瘤的目标对象中的恶性肿瘤的治疗方法,其包含给予:(1)一治疗试剂的治疗有效量至目标对象以治疗所述恶性肿瘤;以及(2)一激酶抑制剂的组合的治疗有效量至目标对象以治疗所述恶性肿瘤的步骤,其中所述治疗试剂是选自于由卫康醇、卫康醇的衍生物或类似物、二乙酰二脱水卫矛醇、二乙酰二脱水卫矛醇的衍生物或类似物、二溴卫矛醇,以及二溴卫矛醇的衍生物或类似物所组成的群组;所述激酶抑制剂的组合是一选自于由下列所组成的群组的组合:(1)一JAK2抑制剂及一 STAT5抑制剂;(2)一JAK2抑制剂及一Src抑制剂;(3)一STAT5抑制剂及一Src抑制剂;以及(4)一JAK2抑制剂、一STAT5抑制剂及一Src抑制剂。
所述方法可以进一步包含:给予一BH3类似物的治疗有效量至目标对象的步骤。或者,所述方法可以进一步包含:给予一酪氨酸激酶抑制剂的治疗有效量至目标对象的步骤。
作为一JAK2抑制剂、一STAT5抑制剂或一Src抑制剂的特异性激酶抑制剂的描述没有排除所述激酶抑制剂被引用或描述朝向另一激酶的抑制活性,比如一或多个JAK2、STAT5、Src或与所述BCR-ABL融合蛋白相关的激酶活性。
当所述改良是通过病患或疾病表现型的分析来进行的,所述病患或疾病表现型的分析可以是但不限于一通过一方法所实现的病患或疾病表现型的分析方法,所述方法是选自于由下列所组成的群组:
(a)使用一诊断工具、诊断技术、诊断套组或诊断试验,以确认患者的特定基因型;
(b)使用一标志物的测量方法,所述标志物是选自于由组蛋白去乙酰酶、鸟胺酸脱羧酶、VEGF、一摄护腺特殊基因的基因产物的蛋白、一jun的基因产物的蛋白、一蛋白激酶、桥粒芯蛋白-3及一凋亡蛋白酶衍生的新抗原决定位所组成的群组;
(c)给予拟似化合物的剂量;以及
(d)低剂量预测试酶的状态。
在由目标对象的样品中,作为肿瘤转移至淋巴结的标志物的蛋白桥粒芯蛋白-3的量测,以及基于桥粒芯蛋白-3的量,适当疗法的选择透过Gutkind 等人被描述于美国专利申请公开第2012/0087892号,并通过引用将其包括在内。
细胞凋亡的作为指示剂的凋亡蛋白酶衍生的新抗原决定位的量测,包含抗肿瘤剂所诱发的细胞凋亡,透过Wells等人被描述于美国专利申请公开第 2012/0028266号,并通过引用将其包括在内。
当所述改良是通过病患或疾病基因型的分析来进行的,所述病患或疾病基因型的分析可以是但不限于一通过一方法所实现的病患或疾病基因型的分析方法,所述方法是选自于由下列所组成的群组:
(a)使用一诊断工具、诊断技术、诊断套组或诊断试验,以确认患者的特定基因型;
(b)使用一基因芯片;
(c)使用基因表现分析;
(d)使用单核苷酸多型性(single nucleotide polymorphism,SNP)分析;以及
(e)测量一代谢物或一代谢酵素的水平。
基因芯片的使用被描述于A.J.Lee&S.Ramaswamy,“在生物发现及病患照顾中的DNA微数组”in Essentials of Genomic and Personalized Medicine (G.S.Ginsburg&H.F.Willard,eds.,Academic Press,Amsterdam,2010),ch.7, pp.73-88,并通过引用将其包括在内。
当所述方法是单核苷酸多型性(SNP)分析的使用时,在一选自于由组蛋白去乙酰酶、鸟胺酸脱羧酶、VEGF、一摄护腺特殊基因、c-Jun及一蛋白激酶所组成的群组的基因上,所述SNP分析可以被实现。SNP分析的使用被描述于S.Levy and Y.-H.Rogers,“用于人类基因变异的发现的DNA定序”in Essentials of Genomic and Personalized Medicine(G.S.Ginsburg&H.F.Willard, eds.,Academic Press,Amsterdam,2010),ch.3,pp.27-37,并通过引用将其包括在内。
尽管如此,可以使用其他的基因丛技术,比如拷贝数目变异分析及DNA 甲基化的分析。拷贝数目变异分析被描述于C.Lee等人,“拷贝数目变异及人体健康”in Essentialsof Genomic and Personalized Medicine(G.S.Ginsburg &H.F.Willard,eds.,AcademicPress,Amsterdam,2010),ch.5,pp.46-59,并通过引用将其包括在内。DNA甲基化分析被描述于S.Cottrell等人,“DNA甲基化分析:提供新见解至人类疾病中”,in Essentials ofGenomic and Personalized Medicine(G.S.Ginsburg&H.F.Willard,eds.,AcademicPress, Amsterdam,2010),ch.6,pp.60-72,并通过引用将其包括在内。
当所述改良是通过前/后治疗准备来进行的,所述前/后治疗准备可以是但不限于一选自于由下列所组成的群组的前/后治疗准备的方法:
(a)使用秋水仙素或其类似物;
(b)使用一促尿酸药;
(c)使用尿酸酶;
(d)非口服使用烟碱酰胺;
(e)使用一持续释放形式的烟碱酰胺;
(f)使用一聚ADP核糖聚合酶的抑制剂;
(g)使用咖啡因;
(h)使用甲酰四氢叶酸援救;
(i)感染控制;以及
(j)使用一抗高血压药剂。
促尿酸药包含但不限于丙磺舒、苯溴马龙及亚磺酰吡唑酮。一特别优选的利尿酸药是丙磺舒。促尿酸药,包含丙磺舒,其也可以具有利尿活性。
聚ADP核糖聚合酶的抑制剂被描述于G.J.Southan&C.Szabó,“聚ADP 核糖抑制剂”,Curr.Med.Chem.10:321-240(2003),并通过引用将其包括在内,且包含烟碱酰胺、3-胺基苯甲酰胺、取代的3,4-二氢异喹啉-1(2H)-酮类及异喹啉-1(2H)-酮类、苯并咪唑、吲哚、酞嗪-1(2H)-酮类、喹唑啉酮类、异吲哚酮类、菲啶酮类以及其他化合物。
甲酰四氢叶酸援救包含亚叶酸(甲酰四氢叶酸)至已给予胺甲基叶酸的病患的投。甲酰四氢叶酸是一种叶酸的简化形式,其绕过二氢叶酸还原酶且恢复造血功能。甲酰四氢叶酸可以静脉注射或口服给药。
在一选择方案中,其中所述前/后治疗是使用一促尿酸药,所述促尿酸药是丙磺舒或其类似物。
当所述改良是通过毒性管理来进行的,所述毒性管理可以是但不限于一选自于由下列所组成的群组的毒性管理的方法:
(a)使用秋水仙素或其类似物;
(b)使用一促尿酸药;
(c)使用尿酸酶;
(d)非口服使用烟碱酰胺;
(e)使用一持续释放形式的烟碱酰胺;
(f)使用一聚ADP核糖聚合酶的抑制剂;
(g)使用咖啡因;
(h)使用甲酰四氢叶酸援救;
(i)使用持续释放异嘌呤醇;
(j)非口服使用异嘌呤醇;
(k)使用骨髓移植;
(l)使用一血液细胞刺激剂;
(m)使用血液或血小板注入;
(n)给予一选自于由惠尔血添
(o)施加一疼痛控制技术:
(p)给予一抗发炎剂;
(q)给予一液体;
(r)给予一皮质类固醇;
(s)给予一胰岛素控制药物;
(t)给予一解热剂;
(u)给予一抗恶心治疗剂;
(v)给予一止泻治疗剂;
(w)给予N-乙酰半胱胺酸;
(x)给予一抗组织胺;以及
(y)给予用于降低胃毒性的药剂。
惠尔血添是一通过用于刺激颗粒球的增殖及分化的重组DNA技术所产生的颗粒球群落刺激因子(granulocytic colony-stimulating factor,G-CSF)类似物,且被使用于治疗嗜中性白血球减少症;G-CSF可以被使用于类似的方法中。GM-CSF是颗粒球-巨噬细胞群落刺激因子,且刺激干细胞以产生颗粒球 (嗜酸性、嗜中性及嗜碱性)及单核细胞;它的投药对于防止或治疗感染是有用的。
抗发炎剂于本领域中是众所周知的且包含皮质类固醇及非类固醇的抗发炎剂(非类固醇抗发炎剂,NSAIDs)。具有抗发炎活性的皮质类固醇包含但不限于氢化可体松(hydrocortisone)、可体松(cortisone)、丙酸倍氯米松、贝皮质醇(betamethasone)、地塞米松(dexamethasone)、普赖松(prednisone)、甲基培尼皮质醇、特安皮质醇(triamcinolone)、丙酮氟洛皮质醇(fluocinolone acetonide) 及氟代可体松(fludrocortisone)。非类固醇的抗发炎剂包含但不限于乙酰水杨酸(阿司匹林)、水杨酸钠、三水杨酸胆碱镁、双柳酸、二氟尼索(diflunisal)、磺胺塞拉金(sulfasalazine)、奥色拉秦(olsalazine)、乙酰胺苯酚、吲哚美辛 (indomethacin)、舒林达酸(sulindac)、托美丁(tolmetin)、双氯氛酸、酮咯酸、布洛芬(ibuprofen)、萘普生(naproxen)、氟白普洛芬(flurbiprofen)、凯妥普洛芬(ketoprofen)、非诺洛芬(fenoprofin)、恶丙嗪、迈菲那密酸(mefenamic acid)、甲氯芬那酸(meclofenamic acid)、匹洛西卡(piroxicam)、美洛西卡(meloxicam)、萘布敉痛(nabumetone)、罗非考昔(rofecoxib)、塞来昔布(celecoxib)、艾特多雷克(etodolac)、尼美苏来(nimesulide)、乙酰氯芬酸(aceclofenac)、阿氯芬酸(alclofenac)、胺普芬(alminoprofen)、氨芬酸(amfenac)、安吡昔康(ampiroxicam)、阿扎丙宗(apazone)、阿拉洛芬(araprofen)、阿扎丙酮(azapropazone)、苄达酸 (bendazac)、苯恶洛芬、苄达明(benzydamine)、柏莫洛芬(bermoprofen)、苄呱立隆(benzpiperylon)、溴芬酸(bromfenac)、布氯酸(bucloxic acid)、布马地宗 (bumadizone)、异丁苯丁酸(butibufen)、卡洛芬(carprofen)、西米考昔 (cimicoxib)、桂美辛(cinmetacin)、辛诺昔康(cinnoxicam)、环氯茚酸(clidanac)、氯非宗(clofezone)、氯尼辛(clonixin)、氯吡酸(clopirac)、达布飞龙(darbufelone)、德拉昔布(deracoxib)、吡罗昔康(droxicam)、依尔替酸(eltenac)、因法来酸 (enfenamic acid)、依匹唑(epirizole)、艾氟洛芬(esflurbiprofen)、乙氧基苯胺、依托芬那酯(etofenamate)、依托考昔(etoricoxib)、联苯乙酸(felbinac)、芬布芬 (fenbufen)、芬氯酸(fenclofenac)、芬克洛酸(fenclozicacid)、芬克洛辛 (fenclozine)、芬度柳(fendosal)、芬替酸(fentiazac)、非普拉宗(feprazone)、菲来拉醇(filenadol)、氟罗布芬(flobufen)、氟尼法林(florifenine)、氟舒胺 (flosulide)、甲磺酸氟必青、氟芬那酸(flufenamic acid)、氟苯柳(flufenisal)、氟尼辛(flunixin)、氟诺洛芬、氟洛芬(fluprofen)、氟丙喹宗(fluproquazone)、呋罗芬酸(furofenac)、异丁芬酸(ibufenac)、艾瑞昔布(imrecoxib)、吲哚布洛芬 (indoprofen)、三苯唑酸(isofezolac)、伊索克酸(isoxepac)、伊索昔康(isoxicam)、利克飞龙(licofelone)、氯布洛芬(lobuprofen)、氯诺昔康(lomoxicam)、氯那唑酸(lonazolac)、洛索洛芬、lumaricoxib、马布洛芬(mabuprofen)、咪洛芬 (miroprofen)、莫非布宗、莫苯唑酸(mofezolac)、吗拉宗(morazone)、奈帕芬胺(nepafanac)、尼氟灭酸(niflumic acid)、硝基芬酸(nitrofenac)、硝基氟吡洛芬(nitroflurbiprofen)、硝基萘普生(nitronaproxen)、奥帕诺辛(orpanoxin)、奥沙西罗、羟吲达酸(oxindanac)、奥皮酸(oxpinac)、羟苯丁唑酮、帕米格雷 (pamicogrel)、帕西他沙(parcetasal)、帕瑞考昔(parecoxib)、帕沙米特(parsalmide)、培鲁比洛芬(pelubiprofen)、培美酸(pemedolac)、苯基丁唑酮、吡拉唑酸(pirazolac)、吡咯布洛芬(pirprofen)、普拉洛芬(pranoprofen)、水杨苷、水杨酰胺、水杨酰水杨酸、沙替格雷(satigrel)、舒多昔康(sudoxicam)、舒洛芬(suprofen)、他美辛(talmetacin)、他尼氟酯(talniflumate)、他唑非隆 (tazofelone)、特丁非隆(tebufelone)、替尼达普(tenidap)、替诺昔康(tenoxicam)、替泊沙林、噻洛芬酸(tiaprofenic acid)、噻拉米特(tiaramide)、替马考昔 (tilmacoxib)、替诺立定(tinoridine)、硫平酸(tiopinac)、硫恶洛芬、托芬那酸(tolfenamic acid)、三氟柳(triflusal)、托呱辛(tropesin)、乌索酸(ursolic acid)、伐地昔布(valdecoxib)、希莫洛芬(ximoprofen)、扎托布洛芬(zaltoprofen)、齐多美辛(zidometacin)及佐美酸(zomepirac),以及盐类、溶剂化物、类似物、同源物、生物电子等排体(bioisosteres)、水解产物、代谢物、前导物及其前驱物。
皮质类固醇的临床应用被描述于B.P.Schimmer及K.L.Parker,“促肾上腺皮质激素;肾上腺皮质类固醇及其合成类似物;肾上腺皮质激素的合成及作用的抑制剂”inGoodman&Gilman’s The Pharmacological Basis of Therapeutics(L.L.Brunton,ed.,11th ed.,McGraw-Hill,New York,2006),ch.59, pp.1587-1612,并通过引用将其包括在内。
抗恶心治疗剂包含但不限于昂丹司琼(ondansetron)、甲氧氯普胺(metoclopramide)、异丙嗪(promethazine)、赛克利嗪(cyclizine)、东莨菪碱(hyoscine)、屈大麻酚(dronabinol)、茶苯海明(dimenhydrinate)、苯海拉明(diphenhydramine)、羟嗪、梅迪辛(medizine)、多拉司琼(dolasetron)、格拉司琼(granisetron)、帕洛诺司琼(palonosetron)、雷莫司琼(ramosetron)、多潘立酮(domperidone)、哈泊度、氯丙嗪(chlorpromazine)、氟奋乃静(fluphenazine)、奋乃静(perphenazine)、丙氯拉嗪(prochlorperazine)、贝皮质醇、地塞米松、劳拉西泮(lorazepam)及硫乙拉嗪(thiethylperazine)。
止泻治疗剂包含但不限于苯乙呱啶、狄芬诺新(difenoxin)、洛呱丁胺、可待因、消旋卡多曲(racecadotril)、奥曲肽及小蘗碱。
N-乙酰半胱胺酸是一抗氧化剂及化痰剂,其还提供生物可近硫。
用于降低胃毒性的药剂包含但不限于铁锈醇(C.Areche等人,“在小鼠和大鼠中铁锈醇的胃保护活性:在胃分泌、内源性前列腺素及非蛋白巯基上的影响”,J.Pharm.Pharmacol.60:245-251(2008)),并通过引用将其包括在内。
当所述改良是通过药物动力学/药效学监测来进行的,所述药物动力学/ 药效学监测可以是但不限于一选自于由下列所组成的群组的方法:
(a)多次测定血浆水平;以及
(b)多次测定于血液或尿液中至少一代谢物。
通常,血浆水平的测定或于血液或尿液中至少一代谢物的测定是通过免疫分析法所完成的。执行免疫分析法的方法于本领域中是众所周知的,且包含放射免疫分析法、酵素连结免疫分析法(enzyme-linked immunosorbent assay, ELISA)、竞争性免疫测定法、使用侧流式试片的免疫分析法,以及其他试验方法。
当所述改良是通过并用药来进行的,所述并用药可以是但不限于一选自于由下列所组成的群组的并用药:
(a)使用伪核苷;
(b)使用伪核苷酸;
(c)使用胸苷合成酶抑制剂;
(d)使用信号传递抑制剂;
(e)使用顺铂或铂类似物;
(f)使用烷化剂;
(g)使用抗微管蛋白剂;
(h)使用抗代谢物;
(i)使用小蘗碱;
(j)使用芹菜素;
(k)使用秋水仙素或其类似物;
(l)使用染料木黄酮;
(m)使用伊妥普赛;
(n)使用阿拉伯糖基胞嘧啶;
(o)使用喜树碱类;
(p)使用长春花生物碱类;
(q)使用位置异构酶抑制剂;
(r)使用5-氟代尿嘧啶;
(s)使用姜黄素;
(t)使用NF-κB抑制剂;
(u)使用迷迭香酸;
(v)使用丙脒腙;
(w)使用甲异靛;
(x)使用伊马替尼;
(y)使用达沙替尼;
(z)使用尼罗替尼;
(aa)使用表观遗传的调节剂;
(ab)使用转录因子抑制剂;
(ac)使用红豆杉醇;
(ad)使用高三尖杉酯碱;
(ae)使用吡哆醛;
(af)使用螺环锗;
(ag)使用咖啡因;
(ah)使用烟碱酰胺;
(ai)使用甲基乙二醛双脒基腙;
(aj)使用Rho激酶抑制剂;
(ak)使用1,2,4-苯并三嗪氧化物;
(al)使用一烷基甘油;
(am)使用一Mer、Ax1或Tyro-3受体激酶的抑制剂;
(an)使用一ATR激酶的抑制剂;
(ao)使用一Fms激酶、Kit激酶、MAP4K4激酶、TrkA激酶或TrkB激酶的调节剂;
(ap)使用河莫昔芬;
(aq)使用一mTOR抑制剂;
(ar)使用一Mnk1a激酶、Mkn1b激酶、Mnk2a激酶或Mnk2b激酶的抑制剂;
(as)使用一M2型丙酮酸激酶的调节剂;
(at)使用一磷酸肌醇3激酶的调节剂;
(au)使用一半胱胺酸蛋白酶抑制剂;
(av)使用苯乙双胍;
(aw)使用辛得比斯病毒基载体;
(ax)使用扮演Smac的类似物及抑制IAPs而促进细胞凋亡的拟胜肽;
(ay)使用一Raf激酶抑制剂;
(az)使用一核转送调节剂;
(ba)使用一酸性神经酰胺酶抑制剂及一胆碱激酶抑制剂;
(bb)使用酪氨酸激酶抑制剂;
(bc)使用抗CS1抗体;
(bd)使用PDK1蛋白激酶的抑制剂;
(be)使用抗鸟苷酸环化酶C(GCC)抗体;
(bf)使用组蛋白去乙酰酶抑制剂;
(bg)使用大麻素;
(bh)使用升糖素类似胜肽(GLP-1)受体促效剂;
(bi)使用Bcl-2或Bcl-xL的抑制剂;
(bj)使用Stat3途径抑制剂;
(bk)使用polo样激酶(Plk1)的抑制剂;
(bl)使用GBPAR1活化剂;
(bm)使用丝氨酸-息宁胺酸蛋白激酶及聚ADP核糖聚合酶(PARP)活性的调节剂;
(bn)使用紫杉烷;
(bo)使用二氢叶酸还原酶的抑制剂;
(bp)使用芳香酶的抑制剂;
(bq)使用苯并咪唑基抗癌剂;
(br)使用一O6-甲基鸟嘌呤-DNA-甲基转移酶(MGMT)抑制剂;
(bs)使用CCR9抑制剂;
(bt)使用酸性鞘磷脂酶抑制剂;
(bu)使用拟胜肽大环类;
(bv)使用胆烷酸胺化物;
(bw)使用取代的氧氮磷环类;
(bx)使用抗TWEAK受体抗体;
(by)使用一ErbB3结合蛋白;
(bz)使用一谷胱甘肽S-转移酶活化抗肿瘤化合物;
(ca)使用取代的磷二酰胺化物;
(cb)使用MEKK蛋白激酶的抑制剂;
(cd)使用COX-2抑制剂;
(ce)使用甲氰咪胍及一半胱胺酸衍生物;
(cf)使用抗IL-6受体抗体;
(cg)使用一抗氧化剂;
(ch)使用一微管蛋白聚合的异恶唑抑制剂;
(ci)使用PARP抑制剂;
(cj)使用极光蛋白激酶抑制剂;
(ck)使用结合至摄护腺特殊性膜抗原的胜肽;
(cl)使用CD19结合剂;
(cm)使用苯重氮基盐;
(cn)使用类铎受体(TLR)促效剂;
(co)使用架桥的双环磺酰胺;
(cp)使用表皮生长因子受体激酶的抑制剂;
(cq)使用一具有肌动蛋白结合活性的T2家族的核醣核酸酶;
(cr)使用萜烯苯酸或其类似物;
(cs)使用一细胞周期素依赖性激酶的抑制剂;
(ct)使用p53及MDM2间交互作用的抑制剂;
(cu)使用所述受体酪氨酸激酶MET的抑制剂;
(cv)使用拉戈唑或拉戈唑类似物;
(cw)使用AKT蛋白激酶的抑制剂;
(cx)使用2’-氟代-5-甲基-β-L-阿拉伯糖呋喃糖基尿苷或L-脱氧胸腺核苷;
(cy)使用HSP90调节剂;
(cz)使用JAK激酶的抑制剂;
(da)使用PDK1蛋白激酶的抑制剂;
(db)使用PDE4抑制剂;
(de)使用原致癌基因c-Met酪氨酸激酶的抑制剂;
(df)使用吲哚胺2,3-二氧合酶的抑制剂;
(dg)使用抑制ATDC(TRIM29)的表现的药剂;
(dh)使用核受体和共活化胜肽的交互作用的拟蛋白抑制剂;
(di)使用XIAP家族蛋白的拮抗物;
(dj)使用肿瘤靶向超抗原;
(dk)使用Pim激酶的抑制剂;
(dl)使用CHK1或CHK2激酶的抑制剂;
(dm)使用血管生成素蛋白4的抑制剂;
(dn)使用Smo拮抗物;
(do)使用烟碱型乙酰胆碱受体拮抗物;
(dp)使用法呢基蛋白转移酶抑制剂;
(dq)使用腺苷A3受体拮抗物;
(dr)使用一癌症疫苗;
(ds)使用一JAK2抑制剂;以及
(dt)使用一Src抑制剂。
这些并用药除了使用一烷基化己醣醇衍生物以外,连同一BH3类似物,如上所述。
此外,进一步如下所述,一烷基化己醣醇衍生物可以与一调节AHI1基因的表现或调节AHI1蛋白的活性的药剂一起使用。
位置异构酶抑制剂包含但不限于伊立替康(irinotecan)、拓扑替康(topotecan)、喜树碱、片螺素D(lamellarin D)、安吖啶(amsacrine)、伊妥普赛、伊妥普赛磷酸盐、替尼泊苷(teniposide)、阿霉素及4-[2-(3,5-二氧-1-哌嗪基)-1- 甲基丙基]哌嗪-2,6-二酮(ICRF-193)。
伪核苷包含但不限于阿糖胞苷、吉西他滨(gemcitabine)及氟达拉滨(fludarabine);其他伪核苷于本领域中为已知的。
伪核苷酸包含但不限于富马酸替诺福韦酯及阿德福韦酯(adefovir dipivoxil);其他伪核苷酸于本领域中为已知的。
胸苷合成酶抑制剂包含但不限于雷替曲塞(raltitrexed)、培美曲塞(pemetrexed)、洛拉曲塞(nolatrexed)、ZD9331、GS7094L、氟代尿嘧啶及BGC 945。
信号传递抑制剂被描述于A.V.Lee等人,“信号传递抑制剂作用的新机制:受体酪氨酸激酶向下调节及信号反式激活的阻断”,Clin.Cancer Res.9: 516s(2003),并通过引用将其整体包括在内。
烷化剂包含但不限于Shionogi 254-S、醛磷胺类似物、六甲蜜胺 (altretamine)、阿那昔酮(anaxirone)、Boehringer Mannheim BBR-2207、苯达莫司汀(bendamustine)、贝拉布昔(bestrabucil)、布朵替坦(budotitane)、Wakunaga CA-102、卡铂普来锭(carboplatin)、卡莫司汀(carmustine)、乙基苯基嘧啶三酮-139(Chinoin-139)、乙基苯基嘧啶三酮-153、氮芥苯丁酸(chlorambucil)、顺铂、环磷酰胺、American Cyanamid CL-286558、Sanofi CY-233、cyplatate、 Degussa D-19-384、Sumimoto DACHP(Myr)
抗微管蛋白剂包含但不限于长春花生物碱类、紫杉烷、鬼臼毒素、软海绵素B(halichondrin B)及拟软海绵素B(homohalichondrin B)。
抗代谢物包含但不限于:胺甲基叶酸、培美曲塞、5-氟代尿嘧啶、卡匹他滨(capecitabine)、阿拉伯糖基胞嘧啶、吉西他滨、6-硫醇嘌呤、喷司他丁 (pentostatin)、阿拉诺新(alanosine)、AG2037(Pfizer)、5-FU-蛋白原 (5-FU-fibrinogen)、老鼠簕属叶酸(acanthifolic acid)、胺基噻二唑、布喹那钠(brequinar sodium)、卡莫氟(carmofur)、Ciba-Geigy CGP-30694、环戊基胞嘧啶、阿拉伯糖基胞嘧啶膦酸酯硬脂酸酯、阿拉伯糖基胞嘧啶共轭物、Lilly DATHF、Merrill-Dow DDFC、去氮鸟嘌呤、双脱氧胞苷、双脱氧鸟苷、didox、 Yoshitomi DMDC、脱氧氟尿苷、Wellcome EHNA、Merck&Co.EX-015、 fazarabine、氟尿苷、磷酸氟达拉滨、N-(2’-呋喃基)-5-氟代尿嘧啶、Daiichi Seiyaku FO-152、异丙基吡咯嗪、Lilly LY-188011、Lilly LY-264618、 methobenzaprim、胺甲基叶酸、WellcomeMZPES、norspermidine、NCI NSC-127716、NCI NSC-264880、NCI NSC-39661、NCI NSC-612567、 Warner-Lambert PALA、吡曲克辛(piritrexim)、普卡霉素(plicamycin)、AsahiChemical PL-AC、Takeda TAC-788、硫鸟嘌呤、噻唑羧胺核苷(tiazofurin)、 ErbamontTIF、三甲曲沙(trimetrexate)、酪氨酸激酶抑制剂、酪氨酸蛋白激酶抑制剂、Taiho UFT及优你生(uricytin)。
小蘗碱具有抗菌活性,且防止和抑制促发炎细胞激素及E选择素的表现,同时增加脂缔素(adiponectin)表现。
芹菜素是一黄酮,其可以颠倒环孢霉素(cyclosporine)的反效应,且具有化学防护活性,单独或与糖衍生。
秋水仙素是一三环生物碱类,其通过连接至所述蛋白微管蛋白发挥其活性。秋水仙素的类似物包含但不限于秋水仙酰胺、N-去乙酰硫秋水仙素、脱羰秋水仙碱(demecolcine)、N-乙酰基碘代秋水仙醇、三甲基秋水仙酸 (trimethylcolchicinie acid、TMCA)甲基醚、N-乙酰基秋水仙醇、TMCA乙基醚、异秋水仙素、异秋水仙酰胺、异TMCA甲基醚、原秋水仙碱(colchiceine)、 TMCA、N-苄酰基TMCA、colchicosamide、秋水仙酰胺苷、秋水仙醇及秋水仙酸(colchinoic acid)(M.H.Zweig及C.F.Chignell,“一些秋水仙素类似物、硫酸长春花碱及具有大鼠脑部微管蛋白的鬼臼毒素的交互作用”,Biochem. Pharmacol.22:2141-2150(1973)及B.Yang等人,“环C修饰秋水仙素类似物的合成及生物评价”,Bioorg.Med.Chem.Lett.20:3831-3833(2010)),这两者皆通过引用将其包括在内。
染料木黄酮是一异黄酮,其化学名为5,7-二羟基-3-(4-羟苯基)克唍-4-酮。染料木黄酮具有许多生物活性,包含PPARs的活化、几个酪氨酸激酶的抑制、位置异构酶的抑制、抗氧化活性、Nrf2抗氧化反应的活化、雌激素受体-β的活化及哺乳动物己糖转运蛋白质GLUT2的抑制。
伊妥普赛是一抗癌症药剂,其主要作为一位置异构酶II抑制剂。伊妥普赛形成一DNA及所述位置异构酶II酵素的三元错合物,防止所述DNA股的再接合,且因此诱发DNA股断裂及促进所述癌细胞的细胞凋亡。
阿拉伯糖基胞嘧啶是一取代具有阿拉伯糖的核糖的核苷类似物。其可以成为DNA的一部分,且也抑制DNA及RNA聚合酶及核苷酸还原酶两者。其在急性骨髓性白血病及急性淋巴性白血病中是特别有用的。
喜树碱类包含喜树碱、高喜树碱、拓扑替康、伊立替康、DB 67、BNP 1350、依喜替康(exatecan)、勒托替康(lurtotecan)、ST 1481及CKD 602。在癌细胞中,这些化合物扮演位置异构酶I抑制剂且阻止DNA合成。
长春花生物碱类包含长春花碱、长春新碱(vincristine)、长春地辛 (vindesine)及长春瑞滨(vinorelbine)。
位置异构酶抑制剂包含位置异构酶I抑制剂以及位置异构酶II抑制剂。位置异构酶I抑制剂包含所述喜树碱类及片螺素D。位置异构酶II抑制剂包含,除了氨萘非特及其衍生物及类似物以外,还包含伊妥普赛、替尼泊苷、阿霉素、道诺霉素(daunorubicin)、双羟蒽醌、安吖啶、玫瑰树碱(ellipticines) 及金精三羧酸。许多植物衍生的自然产生的酚类化合物,比如染料木黄酮、槲皮素及白藜芦醇(resveratrol),表现出朝向位置异构酶I及位置异构酶II的抑制活性。
5-氟代尿嘧啶是一碱基类似物,其作为一胸苷合成酶抑制剂,从而抑制DNA合成。当剥夺足够的胸腺核苷时,通过一称为胸腺嘧啶饥饿的工序快速分裂癌细胞死亡。
姜黄素被认为是具有抗肿瘤、抗发炎性质、抗氧化剂、抗缺血性质、抗关节炎性质及抗淀粉状蛋白性质,且还具有保肝活性。
NF-κB抑制剂包含但不限于硼替佐米(bortezomib)。
迷迭香酸是一自然产生的酚类抗氧化剂,其还具有抗发炎活性。
丙脒腙是一透过S-腺核苷甲硫胺酸脱羧酶的竞争抑制的多胺生物合成的抑制剂。
经由几个可能地新颖作用机制来活化甲异靛。它具有细胞周期特异性效应,包含因AML细胞株在G(O)/G1阻滞以及G2/M因HT-29结肠直肠的细胞株阻滞。其也通过一些机制刺激细胞凋亡,包含在初级AML细胞中p21 与p27的上调节及Bcl-2的递减调节,同时在AML细胞(DKO不敏感于化学疗法)中Bak及Bax的上调节,以及在K562细胞中的新颖凋亡蛋白酶依赖的途径。在粒腺体上甲异靛还具有效果,但没有改变Bcl-2、Bax及Bid蛋白表现。在HL-60骨髓细胞中甲异靛还刺激前凋亡蛋白酶3、8、9及PARP的切割。甲异靛还被定向至多个细胞目标,其可能是协同及互补。比如,它促进人类骨髓胚细胞白血病细胞的分化,伴随着c-myb基因表现的递减调节。在 W256细胞、微管组装、糖原合成酶激酶-3β(GSK-3β)(在5-50nM上)、CDK1/ 细胞周期素B及CDK5/p25(tau微管蛋白磷酸化)中,它还促进DNA及RNA 合成的抑制。此外,甲异靛降低β-连锁蛋白及c-myc(HL-60细胞,但不是在 K562中),通过抑制GSK-3β影响Wnt途径,且下调节β-连锁蛋白及c-myc 蛋白表现。甲异靛还促进CD11b的上调节,促进骨髓分化,且在Jurkat细胞中上调节Ahi-1(引起c-Myb的磷酸化)。此外,甲异靛显示血管生成作用,包含降低VEGF保护、VCAM-1、于HUVEC中的小管制剂及ECV304细胞凋亡。
伊马替尼是一受体酪氨酸激酶酵素ABL的抑制剂,且被使用来治疗慢性骨髓性白血病、胃肠道基质瘤及其他高度增生疾病。
达沙替尼是一BCR/ABL及Src家族酪氨酸激酶的抑制剂,且被使用来治疗慢性骨髓性白血病及急性淋巴性白血病。
尼罗替尼是另一种批准用于治疗慢性骨髓性白血病的酪氨酸激酶抑制剂,其可抑制激酶BCR/ABL、KIT、LCK、EPHA3及许多其他激酶。尼罗替尼的使用透过Aloyz等人被描述于美国专利申请公开号2011/0028422,并通过引用将其包括在内。
表观遗传的调节剂包含多胺基表观遗传的调节剂,比如多胺基表观遗传的调节剂,其被描述于S.K.Sharma等人,“多胺基小分子表观遗传的调节剂”,Med.Chem.Commun.3:14-21(2012),且被描述于L.G.Wang&J.W. Chiao,“透过表观遗传调控的苯乙基异硫氰酸盐的摄护腺癌化学预防活性 (回顾)”,Int.J.Oncol.37:533-539(2010),这两者皆通过引用将其包括在内。
转录因子抑制剂包含1-(4-六苯基)-2-丙烷-1-酮、3-氟代-4-[[2-羟基 -2-(5,5,8,8-四甲基-5,6,7,8,-四氢-2-萘基)乙酰基]胺基]-苯甲酸(BMS 961)、 4-[5-[8-(1-甲基乙基)-4-苯基-2-喹啉基]-1H-吡咯并-2-苯甲酸(ER-50891)、7- 乙烯基-2-(3-氟代-4-羟苯基)-5-苯并恶唑酮(ERB 041),以及其他化合物。转录因子抑制剂被描述于T.Berg,“具有有机小分子的转录因子的抑制”,Curr. Opin.Chem.Biol.12:464-471(2008),并通过引用将其包括在内。
粉防己碱具有化学结构6,6’,7,12-四甲氧基-2,2’-二甲基-1β-berbaman,且其是一具有抗发炎作用、免疫作用及抗过敏作用以及相似于奎尼丁的抗心律不整作用的钙通道阻断剂。其已经从粉防己及亚洲其他药材被隔离。
VEGF抑制剂包含贝伐单抗(癌思停),其针对VEGF而言是一单株抗体;伊曲康唑;及苏拉明(suramin);以及巴马司他(batimastat)及马立马司他 (marimastat),其是基质金属蛋白酶抑制剂,以及大麻素及其衍生物。
癌症疫苗正在开发中。通常,对发生于癌细胞(没有发生于正常细胞)的一蛋白或多个蛋白,癌症疫苗是基于一免疫反应。癌症疫苗包含用于转移性激素难治性摄护腺癌的疫苗、用于肾癌的肿瘤噬菌体、用于肺癌的 CimaVax-EGF、MOBILAN、用于表现癌症(比如乳癌、结肠癌症、膀胱癌及卵巢癌的Her2/neu的Neuvenge、用于乳癌的Stimuvax,以及其他的。癌症疫苗被描述于S.Pejawar-Gaddy及O.Finn,“癌症疫苗:成就及挑战”,Crit.Rev.Oncol.Hematol.67:93-102(2008),并通过引用将其包括在内。
癌症疗法中甲基乙二醛双脒基腙的使用已经被描述于D.D.Von Hoff,“MGBG:教导一老药新用”,Ann.Oncol.5:487-493(1994),并通过引用将其包括在内。
Rho激酶抑制剂的使用,比如(R)-(+)-N-(4-吡啶基)-4-(1-胺乙基)苯甲酰胺、利尿酸、4-[2(2,3,4,5,6-五氟代苯基)丙烯酰基]肉桂酸、(+)-反-4-(1-胺乙基)-1-(4-吡啶基胺甲酰基)环己烷、(+)-10反-N-(1H-吡咯并[2,3-b]吡啶-4- 基)-4-(1-胺乙基)环己烷羧酰胺,以及(R)-(+)-N-(1H-吡咯并[2,3-b]吡啶-4- 基)-4-(1-胺乙基)苯甲酰胺,正如透过Fujii等人于美国专利第6,930,115号中所描述的,并通过引用将其包括在内。
1,2,4-苯并三嗪氧化物的使用,比如:3-羟基-1,2,4-苯并三嗪1,4-二氧化物、3-胺基-7-三氟代甲基-1,2,4-苯并三嗪1-氧化物、3-胺基-7-胺甲酰基-1,2,4- 苯并三嗪1-氧化物、7-乙酰基-3-胺基-1,2,4-苯并三嗪1-氧化肟、3-胺基-6(7) 癸基-1,2,4-苯并三嗪1,4-二氧化物、1,2,4-苯并三嗪二氧化物、7-氯基-3-羟基 -1,2,4-苯并三嗪1,4-二氧化物、7-硝基-3-胺基-1,2,4-苯并三嗪1,4-二氧化物、 3-(3-N,N-二乙胺基丙胺基)-1,2,4-苯并三嗪1,4-二氧化物、7-硝基-3-(2-N,N- 二乙胺基乙胺基)-1,2,4-苯并三嗪1,4-二氧化物、7-烯丙氧基-1,2,4-苯并三嗪 1,4-二氧化物、7-(3-N-乙基乙酰胺基-2-乙酰氧基丙氧基)1,2,4-苯并三嗪1,4- 二氧化物、7-硝基-1,2,4-苯并三嗪1,4-二氧化物.3-丙基-1,2,4-苯并三嗪1,4- 二氧化物,以及3-(1-羟乙基)-1,2,4-苯并三嗪1,4-二氧化物,正如透过Brown 于美国专利第6,277,835号中所描述的,并通过引用将其包括在内。
烷基甘油的使用透过Firshein被描述于美国专利第6,121,245号,并通过引用将其包括在内。
Mer、Ax1或Tyro-3受体酪氨酸激酶的抑制剂的使用透过Graham等人被描述于美国专利申请公开第2012/0230991号,并通过引用将其包括在内。这些抑制剂可以是抗体,包含单株抗体或融合蛋白。
ATR激酶的抑制剂的使用透过Charrier等人被描述于美国专利申请公开第2012/0177748号,通过引用将其包括。这些ATR激酶的抑制剂是取代的吡啶化合物,比如:2-胺基-N-苯基-5-(3-吡啶基)吡啶-3-羧酰胺、5-(4-(甲磺酰基)苯基-3-(5-苯基-1,3,4-恶二唑-2-基)吡啶-2-胺,以及5-(1-乙磺酰基-3,6-二氢 -2H-吡啶-4-基)-3-(5-苯基-1,3,4-恶二唑-2-基)吡啶-2-胺。
调节一或多个Fms激酶、Kit激酶、MAP4K4激酶、TrkA激酶或TrkB 激酶的活性的化合物的使用是透过Ibrahim等人被描述于美国专利申请公开第2012/0165329号,并通过引用将其包括在内。这些化合物包含(6-甲氧基- 吡啶-3-基甲基)[5-(7H-吡咯并[2,3-d]嘧啶-5-基甲基)-嘧啶-2-基]-胺、(5-氟代-2- 甲氧基-吡啶-3-基甲基)-[5-(7H-吡咯并[2,3-d]嘧啶-5-基甲基)-嘧啶-2-y]-胺,以及(5-氟代-6-甲氧基-吡啶-3-基甲基)-[5-(7H-吡咯并[2,3-d]嘧啶-5-基甲基)-嘧啶-2-基]-胺。抑制Trk激酶的化合物,特别是TrkA,其透过Wu等人被描述于美国专利申请公开第2011/0301133号,并通过引用将其包括在内。
河莫昔芬的使用透过Ahmad等人被描述于美国专利申请公开第 2012/0164075号,并通过引用将其包括在内。
一mTOR抑制剂的使用透过Burke等人被描述于美国专利申请公开第 2012/0129881号,并通过引用将其包括在内。合适的mTOR抑制剂包含但不限于40-O-(2-羟乙基)雷帕霉素。这些mTOR抑制剂可以与Raf激酶抑制剂一起使用,正如透过Lane于美国专利申请公开第2011/0301184号中所描述的,并通过引用将其包括在内。Raf激酶抑制剂也透过Ibrahim等人被描述于美国专利申请公开第2010/0286178号,并通过引用将其包括在内;这些化合物包含但不限于丙烷-1-磺酸{2,4-二氟代-3-[5-(2-甲氧基-嘧啶-5-基)-1H-吡咯并[2,3-b]吡啶-3-羰基]-苯基}-酰胺、丙烷-1-磺酸[3-(5-氰基-1H-吡咯并[2,3-b] 吡啶-3-羰基)-2,4-二氟代-苯基]-酰胺、丙烷-1-磺酸[3-(5-氰基-1H-吡咯并[2,3-b] 吡啶-3-羰基)-2-氟代-苯基]-酰胺、N-[3-(5-氰基-1H-吡咯并[2,3-b]吡啶-3-羰基)-2,4-二氟代-苯基]-2,5-二氟代-苯磺酰胺、N-[3-(5-氰基-1H-吡咯并[2,3-b] 吡啶-3-羰基)-2,4-二氟代-苯基]-3-氟代-苯磺酰胺、吡咯啶-1-磺酸[3-(5-氰基 -1H-吡咯并[2,3-b]吡啶-3-羰基)-2,4-二氟代-苯基]-酰胺,以及N,N-二甲胺基- 磺酸[3-(5-氰基-1H-吡咯并[2,3-b]吡啶-3-羰基)-2,4-二氟代-苯基]-酰胺。在恶性肿瘤细胞中,这些mTOR抑制剂还可以与提升pAkt水平的化合物一起使用,正如透过Bhagwat等人于美国专利申请公开第2009/0274698号中所描述的,并通过引用将其包括在内。许多提升pAkt水平的化合物被描述,包含化学治疗试剂、雷帕霉素的类似物及其他药剂。mTOR抑制剂的使用也透过Jin 等人被描述于美国专利第8,268,819号,通过引用将其包括;这些mTOR抑制剂是六氢蝶呤化合物(hexahydrooxazinopterine compounds)。
一Mnk1a激酶、Mnk1b激酶、Mnk2a激酶或Mnk2b激酶的抑制剂的使用透过Austen等人被描述于美国专利申请公开第2012/0128686号,并通过引用将其包括在内。这些化何物包含噻咔嘧啶。另外的这些激酶的一个或多个的噻吩并嘧啶抑制剂透过Heckel等人被描述于美国专利申请公开第 2011/0212103号,且透过Lehmann-Lintz等人被描述于美国专利申请公开第 2011/0212102号,这两者皆通过引用将其包括在内。
一M2型丙酮酸激酶的调节剂的使用透过Salituro等人被描述于美国专利申请公开第2012/0122885号,并通过引用将其包括在内。合适的M2型丙酮酸激酶的调节剂包含但不限于1-(3-氯基-5-(三氟代甲基)吡啶-2-基)-N-(3,5- 二甲苯基)-1H-咪唑-5-磺胺、1-(3-氯基-5-(三氟代甲基)吡啶-2-基)-N-(5-甲氧苯基)-1H-咪唑-5-磺胺,以及N-(4-甲氧苯基)-1-(5-(三氟代甲基)吡啶-2-基)-H- 咪唑-5-磺胺。
一磷酸肌醇3激酶的调节剂的使用透过Ren等人被描述于美国专利申请公开第2012/0122838号,并通过引用将其包括在内。所述磷酸肌醇3激酶的抑制剂也透过Lamb等人被描述于美国专利申请公开第2010/0209420号,并通过引用将其包括在内,且也透过Buhr等人被描述于美国专利申请公开第 2009/0209340号,并通过引用将其包括在内;这些抑制剂包含吡啶并嘧啶酮类。所述磷酸肌醇3激酶的抑制剂也透过Blaquiere等人被描述于美国专利第 8,242,104号,并通过引用将其包括在内;这些抑制剂包含氧氮杂卓。所述磷酸肌醇3激酶的抑制剂也透过Ren等人被描述于美国专利第8,193,182号;这些抑制剂包含异喹啉-1(2H)-酮类。所述磷酸肌醇3激酶的抑制剂也透过 Do等人被描述于美国专利第7,928,428号,并通过引用将其包括在内;这些抑制剂包含苯并哌喃及苯并恶庚英(benzoxepines)。
一半胱胺酸蛋白酶抑制剂的使用透过Cao等人被描述于美国专利申请公开第2012/0114765号,并通过引用将其包括在内。合适的半胱胺酸蛋白酶抑制剂包含但不限于1-[5-(2,4-二氯苯基磺酰基)-4-硝基-2-噻吩基]乙酮、 1-[5-(2,4-二氟代苯基磺酰基)-4-硝基-2-噻吩基]乙酮,以及1-{4-硝基-5-[2-(三氟代甲基)苯基磺酰基]-2-噻吩基}乙酮。
苯乙双胍的使用透过Thompson等人被描述于美国专利申请公开第 2012/0114676号,并通过引用将其包括在内。
辛得比斯基病毒载体的使用透过Meruelo等人被描述于美国专利申请公开第2011/0318430号,并通过引用将其包括在内。这些载体能够结合至表现高亲和性层粘蛋白受体的较高水平的实质肿瘤。
扮演Smac的类似物及抑制IAPs而促进细胞凋亡的拟胜肽的使用透过 Condon等人被描述于美国专利申请公开第2011/0305777号,并通过引用将其包括在内。
核转送调节剂的使用,特别是Crm1的抑制剂,其透过Shacham等人被描述于美国专利申请公开第2011/0275607号,并通过引用将其包括在内。这些Crm1的抑制剂包含但不限于(Z)-3-[3-(3-氯苯基)[1,2,4]-三唑-1-基]-丙烯酸乙基酯、(E)-3-[3-(3-氯苯基)[1,2,4]-三唑-1-基]-丙烯酸乙基酯、(Z)-3-[3-(3-氯苯基)-[1,2,4]-三唑-1-基]-丙烯酸异丙酯、(E)-3-[3-(3-氯苯基)-[1,2,4]-三唑-1- 基]-丙烯酸异丙酯、(Z)-3-[3-(3-氯苯基)-[1,2,4]-三唑-1-基]-丙烯酸t-丁基酯、 (Z)-3-[3-(3-氯苯基)-[1,2,4]-三唑-1-基]-丙烯酸t-丁基酯、(E)-3-[3-(3-氯苯基)-[1,2,4]-三唑-1-基]-N-苯基-丙烯酰胺、(E)-N-(2-氯苯基)-3-[3-(3-氯苯基)-[1,2,4]-三唑-1-基]-丙烯酰胺、(4-{(E)-3-[3-(3-氯苯基)[1,2,4]-三唑-1-基]- 丙烯酰胺基}-苯基-)-胺甲酸t-丁基酯、(E)-3-[3-(3-氯苯基)-[1,2,4]-三唑-1- 基]-N-(4-甲氧苯基)-丙烯酰胺、(E)-3-[3-(3-氯苯基)-[1,2,4]-三唑-1-基]-N-甲基 -N-苯基-丙烯酰胺,以及(E)-N-(4-胺苯基)-3-[3-(3-氯苯基)-[1,2,4]-三唑-1-基]- 丙烯酰胺。
酪氨酸激酶抑制剂的使用透过Zhang等人被描述于美国专利申请公开第 2011/0206661号,其被指向于酪氨酸激酶的三甲氧苯基抑制剂,且被描述于美国专利申请公开第2011/0195066号,其被指向于酪氨酸激酶的喹啉抑制剂,这两者皆通过引用将其包括在内。酪氨酸激酶抑制剂的使用也透过Zhang 等人被描述于美国专利申请公开第2011/053968号,并通过引用将其包括在内,其被指向于酪氨酸激酶的胺吡啶抑制剂。酪氨酸激酶抑制剂的使用也被描述于美国专利申请公开第2010/0291025号,并通过引用将其包括在内,其被指向于酪氨酸激酶的吲唑抑制剂。酪氨酸激酶抑制剂的使用也透过Ren等人被描述于美国专利申请公开第2010/0190749号,并通过引用将其包括在内;这些酪氨酸激酶抑制剂是苯并恶唑化合物;这个种类的化合物还可以抑制mTOR及脂肪激酶(如磷酸肌醇3激酶的调节剂)。酪氨酸激酶抑制剂的使用也透过Lajeunesse等人被描述于美国专利第8,242,270号,并通过引用将其包括在内;这些酪氨酸激酶抑制剂为2-胺基噻唑-5-芳香羧酰胺类。
一酸性神经酰胺酶抑制剂及一胆碱激酶抑制剂的使用透过Ramirez de Molina等人被描述于美国专利申请公开第2011/0256241号,并通过引用将其包括在内。
抗CS1抗体的使用透过Afar被描述于美国专利申请公开第2011/0165154 号,并通过引用将其包括在内。
蛋白激酶CK2抑制剂的使用透过Haddach等人被描述于美国专利申请公开第2011/0152240号,并通过引用将其包括在内。这些蛋白激酶CK2抑制剂包含吡唑并嘧啶。另外的蛋白激酶CK2抑制剂,包含三环化合物,其透过 Haddach等人被描述于美国专利申请公开第2011/0071136号,并通过引用将其包括在内;这些蛋白激酶CK2抑制剂还可以抑制Pim激酶或其他激酶。另外的蛋白激酶CK2抑制剂,包含杂环取代的内酰胺,其也透过Haddach等人被描述于美国专利申请公开第2011/0071115号,并通过引用将其包括在内;这些蛋白激酶CK2抑制剂还可以抑制Pim激酶或其他激酶。
抗鸟苷酸环化酶C(GCC)抗体的使用透过Nam等人被描述于美国专利申请公开第2011/0110936号,并通过引用将其包括在内。
组蛋白去乙酰酶抑制剂的使用透过Thaler等人被描述于美国专利申请公开第2011/0105474号,并通过引用将其包括在内。这些组蛋白去乙酰酶抑制剂包含但不限于(E)-N-羟基-3-{4-[(E)-3-(4-甲基-哌嗪-1-基)-3-氧基-丙烯基]- 苯基}-丙烯酰胺、(E)-N-羟基-3-{3-[(E)-3-(4-甲基-哌嗪-1-基)-3-氧基-丙烯基]- 苯基}-丙烯酰胺、(E)-N-羟基-3-{3-[(E)-3-氧基-3-(4-苯基-哌嗪-1-基)-丙烯基]- 苯基}-丙烯酰胺、(E)-3-[3-((E)-3-[1,4’]二哌啶基-1’-基-3-氧基-丙烯基)-苯基]-N-羟基-丙烯酰胺、(E)-N-羟基-3-{3-[(E)-3-氧基-3-(顺-3,4,5-三甲基-哌嗪 -1-基)-丙烯基]-苯基}-丙烯酰胺、(E)-3-{3-[(E)-3-((1S,4S)-5-甲基-2,5-二氮-双环[2.2.1]庚-2-基)-3-氧基-丙烯基]-苯基}-N-羟基-丙烯酰胺、(E)-N-羟基 -3-{4-[(E)-3-氧基-3-(4-苯基-哌嗪-1-基)-丙烯基]-苯基}-丙烯酰胺、 (E)-3-[4-((E)-3-[1,4’]二哌啶基-1’-基-3-氧基-丙烯基)-苯基]-N-羟基-丙烯酰胺、(E)-N-羟基-3-{4-[(E)-3-氧基-3-(顺-3,4,5-三甲基-哌嗪-1-基)-丙烯基]-苯基}-丙烯酰胺、(E)-N-羟基-3-{4-[(E)-3-氧基-3-((1S,4S)-5-甲基-2,5-二氮-双环 [2.2.1]庚-2-基)-丙烯基]-苯基}-丙烯酰胺、(E)-N-羟基-3-{5-[(E)-3-氧基-3-(4-苯基-哌嗪-1-基)-丙烯基]-吡啶-2-基}-丙烯酰胺、(E)-N-羟基-3-{5-[(E)-3-(4-甲基-哌嗪-1-基)-3-氧基-丙烯基]-吡啶-2-基}-丙烯酰胺、(E)-N-羟基-3-{6-[(E)-3- 氧基-3-(4-苯基-哌嗪-1-基)-丙烯基]-吡啶-2-基}-丙烯酰胺、(E)-N-羟基 -3-{6-[(E)-3-(4-甲基-哌嗪-1-基)-3-氧基-丙烯基]-吡啶-2-基}-丙烯酰胺、 (E)-3-(6-{(E)-3-[4-(3-氯基-苯基)-哌嗪-1-基]-3-氧基-丙烯基}-吡啶-2-基)-N-羟基-丙烯酰胺、(E)-3-{6-[(E)-3-(4-苄酰基-哌嗪-1-基)-3-氧基-丙烯基]-吡啶-2- 基}-N-羟基-丙烯酰胺氢氯化物、(E)-3-(6-{(E)-3-[4-(2-氯基-苯基)-哌嗪-1- 基]-3-氧基-丙烯基}-吡啶-2-基)-N-羟基-丙烯酰胺氢氯化物、(E)-N-羟基 -3-{6-[(E)-3-氧基-3-(4-苯基-哌啶-1-基)-丙烯基]-吡啶-2-基}-丙烯酰胺氢氯化物、(E)-N-羟基-3-{6-[(E)-3-氧基-3-(4-嘧啶-2-基-哌嗪-1-基)-丙烯基]-吡啶-2- 基}-丙烯酰胺氢氯化物、(E)-3-(6-{(E)-3-[4-(4-氯基-苯基)-哌嗪-1-基]-3-氧基- 丙烯基}-吡啶-2-基)-N-羟基-丙烯酰胺氢氯化物,以及(E)-3-{6-[(E)-3-(4-苄基- 哌嗪-1-基)-3-氧基-丙烯基]-吡啶-2-基}-N-羟基-丙烯酰胺氢氯化物。另外的组蛋白去乙酰酶抑制剂,包含螺环衍生物,其透过Varasi等人被描述于美国专利申请公开第2011/039840号,并通过引用将其包括在内。组蛋白去乙酰酶抑制剂的前驱物透过Miller等人被描述于美国专利第8,227,636号,并通过引用将其包括在内。组蛋白去乙酰酶抑制剂透过Kozikowski等人被描述于美国专利第8,222,451号,并通过引用将其包括在内。组蛋白去乙酰酶抑制剂,包含双取代的苯胺化合物,其也透过Heidebrecht等人被描述于美国专利第 8,119,685号,并通过引用将其包括在内。组蛋白去乙酰酶抑制剂,包含芳基稠合的螺环化合物,其也透过Hamblett等人被描述于美国专利第8,119,852 号,并通过引用将其包括在内。
大麻素的使用透过Velasco Diez等人被公开于美国专利申请公开第 2011/0086113号,并通过引用将其包括在内。合适的大麻素包含但不限于四氢大麻酚及大麻二酚。
升糖素类似胜肽(GLP-1)受体促效剂的使用透过Karasik等人被描述于美国专利申请公开第2011/0046071号,并通过引用将其包括在内。一适当的 GLP-1受体促效剂是艾塞那肽(exendin-4)。
抗凋亡蛋白Bcl-2或Bcl-xL的抑制剂的使用透过Martin等人被描述于美国专利申请公开第2011/0021440号,并通过引用将其包括在内。
Stat3途径抑制剂的使用透过Li等人被描述于美国专利申请公开第 2010/0310503号,并通过引用将其包括在内。这些Stat3途径抑制剂包含但不限于2-(1-羟乙基)-萘并[2,3-b]呋喃-4,9-二酮、2-乙酰基-7-氯基-萘并[2,3-b] 呋喃-4,9-二酮、2-乙酰基-7-氟代-萘并[2,3-b]呋喃-4,9-二酮、2-乙酰值萘并 [2,3-b]呋喃-4,9-二酮,以及2-乙基-萘并[2,3-b]呋喃-4,9-二酮。
polo样激酶(Plk1)的抑制剂的使用透过Stengel等人被描述于美国专利申请公开第2010/0278833号,并通过引用将其包括在内。这些抑制剂包含但不限于噻吩-咪唑并吡啶,所述噻吩-咪唑并吡啶包含5-(6-氯基-1H-咪唑并[4,5-c] 吡啶-1-基)-3-{[2-(三氟代甲基)苄基]氧基}噻吩-2-羧酰胺、5-(1H-咪唑并[4,5-c] 吡啶-1-基)-3-{[2-(三氟代甲基)苄基]氧基}噻吩-2-羧酰胺、5-(3H-咪唑并[4,5-c] 吡啶-3-基)-3-{[2-(三氟代甲基)苄基]氧基}噻吩-2-羧酰胺、1-(5-胺甲酰基 -4-{[2-(三氟代甲基)苄基]氧基}-2-噻吩基)-N-(2-甲氧基乙基)-1H-咪唑并[4,5-c]吡啶-6-羧酰胺、1-(5-胺甲酰基-4-{[2-(三氟代甲基)苄基]氧基}-2-噻吩基)-N-(2-吗啉-4-基乙基)-1H-咪唑并[4,5-c]吡啶-6-羧酰胺、5-{6-[二乙胺基)甲基]-1H-咪唑并[4,5-c]吡啶-1-基}-3-{[2-(三氟代甲基)苄基]氧基}噻吩-2-羧酰胺、5-{6-[(环丙基胺基)甲基]-1H-咪唑并[4,5-c]吡啶-1-基}-3-{[2-(三氟代甲基) 苄基]氧基}噻吩-2-羧酰胺、5-{6-[(4-甲基哌嗪-1-基)甲基]-1H-咪唑并[4,5-c]吡啶-1-基}-3-{[2-(三氟代甲基)苄基]氧基}噻吩-2-羧酰胺,以及5-[6-(羟甲基)-1H-咪唑并[4,5-c]吡啶-1-基]-3-{[2-(三氟代甲基)苄基]氧基}噻吩-2-羧酰胺,但本发明并不局限于此。
GBPAR1活化剂的使用透过Arista等人被描述于美国专利申请公开第 2010/0261758号,通过引用将其包括。这些GBPAR1活化剂包含但不限于杂环酰胺。这些化合物包含但不限于N-(3,5-二氯苯基)-3-甲基-N-萘-2-基甲基- 异烟碱酰胺、(3,5-二氯苯基)-N-(2-甲氧基苄基)-3-甲基-异烟碱酰胺、3-甲基-N- 苯基-N-吡啶-3-基甲基-异烟碱酰胺、N-萘-2-基甲基-1-氧基-N-苯基-异烟碱酰胺、N-(3,5-二氯苯基)-3-甲基-N-(2-三氟代甲氧基苄基)-异烟碱酰胺、4-甲基- 恶唑-5-羧酸芐基-苯基胺、N-苄基-N-苯基异烟碱酰胺、N-苄基-N-p-甲苯基异烟碱酰胺、N-苄基-2-氟代-N-苯基异烟碱酰胺、N-苄基-3,5-二氯-N-苯基-异烟碱酰胺、N-苄基-2-氯基-N-苯基-异烟碱酰胺、N-苄基-2-氯基-6-甲基-N-苯基- 异烟碱酰胺、N-苄基-3-甲基-N-苯基-异烟碱酰胺、N-苄基-3-氯基-N-苯基-异烟碱酰胺、N-苄基-2,5-二氯-N-苯基-异烟碱酰胺、N-苄基-2-甲基-N-苯基-异烟碱酰胺、N-苄基-2-氰基-N-苯基-异烟碱酰胺、N-苄基-N-苯乙基-异烟碱酰胺、N-苄基-N-(2-氟代甲氧基-苯基)-异烟碱酰胺,以及N-苄基-N-(4-氯苯基)- 异烟碱酰胺。另外的GBPAR1活化剂透过Arista被描述于美国专利申请公开第2010/0048579号,并通过引用将其包括在内,包含哒嗪、吡啶及吡喃衍生物。
丝氨酸-息宁胺酸蛋白激酶及聚ADP核糖聚合酶(PARP)活性的调节剂的使用透过Chua等人被描述于美国专利申请公开第2009/0105233号,且透过 Drygin等人被描述于美国专利申请公开第2010/0173013号,这两者皆通过引用将其包括在内。所述丝氨酸-息宁胺酸蛋白激酶可以是但不限于CK2、 CK2α2、Pim-1、CDK1/细胞周期素B、c-RAF、Mer、MELK、DYRK2、Flt3、 Flt3(D835Y)、Flt4、HIPK3、HIPK2及ZIPK。
紫杉烷的使用透过Singh等人被描述于美国专利申请公开第 2010/0166872号,并通过引用将其包括在内。所述紫杉烷可以是但不限于紫杉醇或欧洲紫杉醇(docitaxel)。
二氢叶酸还原酶的抑制剂的使用透过Gant等人被描述于美国专利申请公开第2010/0150896号,并通过引用将其包括在内。这些二氢叶酸还原酶的抑制剂包含但不限于二胺基喹唑啉类。
芳香酶的抑制剂的使用透过Gant等人被描述于美国专利申请公开第 2010/0111901号,并通过引用将其包括在内。这些芳香酶的抑制剂包含但不限于三唑类。
苯并咪唑基抗癌剂的使用透过Goh等人被描述于美国专利申请公开第 2010/0098691号,并通过引用将其包括在内。所述苯并咪唑基抗癌剂可以是但不限于(E)-3-[1-(3-二甲胺基-2,2-二甲基-丙基)-2-异丙基-1H-苯并咪唑-5- 基]-N-羟基-丙烯酰胺、(E)-3-[2-丁基-1-(3-二甲胺基-2,2-二甲基-丙基)-1H-苯并咪唑-5-基]-N-羟基-丙烯酰胺、(E)-3-[1-(3-二甲胺基-2,2-二甲基-丙基)-2-(2- 甲基磺酰基-乙基)-1H-苯并咪唑-5-基]-N-羟基-丙烯酰胺、(E)-3-[1-(3-二甲胺基-2,2-二甲基-丙基)-2-乙氧基甲基-1H-苯并咪唑-5-基]-N-羟基-丙烯酰胺、 (E)-3-[1-(3-二甲胺基-2,2-二甲基-丙基)-2-异丁基-1H-苯并咪唑-5-基]-N-羟基- 丙烯酰胺、(E)-3-[1-(2-二乙胺基-乙基)-2-异丁基-1H-苯并咪唑-5-基]-N-羟基- 丙烯酰胺、(E)-3-[2-丁基-1-(2-二乙胺基-乙基)-1H-苯并咪唑-5-基]-N-羟基-丙烯酰胺、(E)-3-[2-丁-3-炔基-1-(3-二甲胺基-2,2-二甲基-丙基)-1H-苯并咪唑-5- 基]-N-羟基-丙烯酰胺、(E)-3-[2-丁-3-烯基-1-(3-二甲胺基-2,2-二甲基-丙基)-1H-苯并咪唑-5-基]-N-羟基-丙烯酰胺、(E)-3-[2-丁-3-烯基-1-(2-二乙胺基- 乙基)-1H-苯并咪唑-5-基]-N-羟基-丙烯酰胺、(E)-3-[2-丁-3-炔基-1-(2-二乙胺基-乙基)-1H-苯并咪唑-5-基]-N-羟基-丙烯酰胺、(E)-3-[1-(3-二甲胺基-2,2-二甲基-丙基)-2-(3,3,3-三氟代-丙基)-1H-苯并咪唑-5-基]-N-羟基-丙烯酰胺、 (E)-3-[1-(2-二乙胺基-乙基)-2-(3,3,3-三氟代-丙基)-1H-苯并咪唑-5-基]-N-羟基 -丙烯酰胺、(E)-3-[1-(2-二乙胺基-乙基)-2-乙氧基甲基-1H-苯并咪唑-5-基]-N- 羟基-丙烯酰胺、(E)-3-[1-(3-二甲胺基-2,2-二甲基-丙基)-2-甲基-1H-苯并咪唑 -5-基]-N-羟基-丙烯酰胺、(E)-3-[1-(2-二乙胺基-乙基)-2-(2,2-二甲基-丙基)-1H- 苯并咪唑-5-基]-N-羟基-丙烯酰胺、(E)-N-羟基-3-[1-(3-异丙基胺基-丙基)-2-(3,3,3-三氟代-丙基)-1-H-苯并咪唑-5-基]-丙烯酰胺、(E)-3-[2-(2,2-二甲基-丙基)-1-(2-异丙基胺基-乙基)-1H-苯并咪唑-5-基]-N-羟基-丙烯酰胺、 (E)-3-[1-(2-二异丙基胺基-乙基)-2-(2,2-二甲基-丙基)-1H-苯并咪唑-5-基]-N- 羟基-丙烯酰胺、(E)-3-[1-(2-二异丙基胺基-乙基)-2-异丁基-1H-苯并咪唑-5-基]-N-羟基-丙烯酰胺、(E)-3-[1-(3-二甲胺基-2,2-二甲基-丙基)-2-己-3-烯基 -1H-苯并咪唑-5-基]-N-羟基-丙烯酰胺、(E)-3-[1-(3-二甲胺基-2,2-二甲基-丙基)-2-(2,4,4-三甲基-戊基)-1H-苯并咪唑-5-基]-N-羟基-丙烯酰胺、(E)-3-[2-环己基-1-(3-二甲胺基-2,2-二甲基-丙基)-1H-苯并咪唑-5-基]-N-羟基-丙烯酰胺、 (E)-3-[2-双环[2.2.1]庚-5-烯-2-基-1-(3-二甲胺基-2,2-二甲基-丙基)-1H-苯并咪唑-5-基]-N-羟基-丙烯酰胺、(E)-3-[1-(2-二乙胺基-乙基)-2-己-3-烯基-1H-苯并咪唑-5-基]-N-羟基-丙烯酰胺、(E)-3-[1-(2-二异丙基胺基-乙基)-2-己-3-烯基 -1H-苯并咪唑-5-基]-N-羟基-丙烯酰胺、(E)-3-[2-己-3-烯基-1-(2-异丙基胺基- 乙基)-1H-苯并咪唑-5-基]-N-羟基-丙烯酰胺、(E)-3-[2-己-3-烯基-1-(3-异丙基胺基-丙基)-1H-苯并咪唑-5-基]-N-羟基-丙烯酰胺、(E)-3-[1-(2-乙胺基-乙基)-2-己-3-烯基-1H-苯并咪唑-5-基]-N-羟基-丙烯酰胺、(E)-3-[1-(2-二乙胺基- 乙基)-2-己基-1H-苯并咪唑-5-基]-N-羟基-丙烯酰胺、(E)-N-羟基-3-[1-(3-异丙基胺基-丙基)-2-(2,4,4-三甲基-戊基)-1H-苯并咪唑-5-基]-丙烯酰胺、 (E)-3-[2-(2,2-二甲基-丙基)-1-(3-异丙基胺基-丙基)-1H-苯并咪唑-5-基]-N-羟基-丙烯酰胺、(E)-3-[1-(2-二异丙基胺基-乙基)-2-(3,3,3-三氟代-丙基)-1H-苯并咪唑-5-基]-N-羟基-丙烯酰胺,以及(E)-N-羟基-3-[2-异丁基-1-(2-异丙基胺基- 乙基)-1H-苯并咪唑-5-基]-丙烯酰胺。
O6-甲基鸟嘌呤-DNA-甲基转移酶(MGMT)抑制剂的使用透过Liu等人被描述于美国专利申请第2010/0093647号,并通过引用将其包括在内。合适的 MGMT抑制剂包含但不限于O
CCR9抑制剂的使用透过Lehr等人被描述于美国专利申请公开第 2010/0075963号,并通过引用将其包括在内。这些CCR9抑制剂包含但不限于苄基磺酰基吲哚。
酸性鞘磷脂酶抑制剂的使用透过Baumann等人被描述于美国专利申请公开第2010/0022482号,并通过引用将其包括在内。通常,这些化合物为联苯衍生物。
拟胜肽大环类的使用透过Nash等人被描述于美国专利申请公开第 2009/0275519号,并通过引用将其包括在内。
胆烷酸胺化物的使用透过Schreiner等人被描述于美国专利申请公开第 2009/0258847号,并通过引用将其包括在内。这些胆烷酸胺化物包含但不限于取代的4-(3-羟基-10,13-羟甲基-十六氢-环戊(a)-菲-17-基)戊酸酰胺。
取代的氧氮磷环类的使用被描述于美国专利申请公开第2009/0202540 号,并通过引用将其包括在内。所述氧氮磷环类可以是但不限于异环磷酰胺及环磷酰胺。
抗TWEAK受体抗体的使用透过Culp被描述于美国专利申请公开第 2009/0074762号,并通过引用将其包括在内。所述TWEAK受体为所述肿瘤坏死因子受体超级家族的成员,且在许多实质肿瘤中,表现于癌细胞的表面上。
ErbB3结合蛋白的使用透过Zhang等人被描述于美国专利申请公开第 2008/0269133号,并通过引用将其包括在内。
一谷胱甘肽S-转移酶活化(GST-活化)抗肿瘤化合物的使用透过Brown等人被描述于美国专利申请公开第2008/0166428号,并通过引用将其包括在内。一优选的GST活化抗肿瘤化合物为坎佛司福酰胺(canfosfamide)。
取代的磷二酰胺化物的使用透过Ma等人被描述于美国专利申请公开第 2008/0125398号,并通过引用将其包括在内,其描述2-{[2-(取代胺基)乙基] 磺酰基}乙基N,N,N’,N’-四次(2-氯乙基)-磷二酰胺,且透过Lui等人被描述于美国专利申请公开第2008/0125397号,并通过引用将其包括在内,其描述 2-({2-氧基-2-[(吡啶-3-基甲基)胺基]乙基}磺酰基)乙基N,N,N’,N’-四次(2-氯乙基)磷二酰胺。取代的磷二酰胺化物的使用也透过Allen等人被描述于美国专利申请公开第2008/0039429号,并通过引用将其包括在内,其描述磺酰基乙基磷二酰胺及硫乙基磷二酰胺。
MEKK蛋白激酶的抑制剂的使用透过Sikorski等人被描述于美国专利申请公开第2006/0100226号,并通过引用将其包括在内。这些抑制剂包含但不限于2-硫代嘧啶酮,比如2-[3-(3,4-二氯-苄基胺基)-苄基磺酰基]-4-(3-甲氧基 -苯基)-6-氧基-1,6-二氢-嘧啶-5-羰腈、2-[3-(3,4-二氯-苄基胺基)-苄基磺酰基]-4-(3,4-二甲氧基-苯基)-6-氧基-1,6-二氢-嘧啶-5-羰腈,以及2-[3-(3,4-二氯- 苄基胺基)-苄基磺酰基-4-(4-甲氧基-3-噻吩-2-基-苯基)-6-氧基-1,6-二氢-嘧啶 -5-羰腈。
COX-2抑制剂的使用透过Masferrer等人被描述于美国专利申请公开第 2004/0072889号,并通过引用将其包括在内。合适的COX-2抑制剂包含但不限于塞来昔布、帕瑞考昔、德拉昔布、罗非考昔、依托考昔、伐地昔布及美洛西卡。
甲氰咪胍及N-乙酰半胱胺酸的使用透过Weidner被描述于美国专利申请公开第2003/0158118号,并通过引用将其包括在内。还可以使用甲氰咪胍或 N-乙酰半胱胺酸的衍生物。
一抗IL-6受体抗体的使用透过Nakamura等人被描述于美国专利申请公开第2002/0131967号,并通过引用将其包括在内。所述抗体可以是一拟人化抗体。
一抗氧化剂的使用透过Chinery等人被描述于美国专利申请公开第 2001/0049349号,并通过引用将其包括在内。合适的抗氧化剂包含但不限于吡咯啶二硫代胺基甲酸、普布可(4,4’-(异丙基亚基二硫基)双(2,6-二-t-丁基苯酚)、维生素C、维生素E及6-羟基-2,5,7,8-四甲基口克唍-2-羧酸。
一微管蛋白聚合的异恶唑抑制剂的使用透过Sun等人被描述于美国专利第8,269,017号,并通过引用将其包括在内。合适的微管蛋白聚合的异恶唑抑制剂包含但不限于2-胺基-N-(2-甲氧基-5-[5-(3,4,5-三甲氧苯基)-异恶唑-4- 基)-苯基)乙酰胺盐酸盐、2-胺基-3-羟基-N-(2-甲氧基-5-[5-(3,4,5-三甲氧苯基) 异恶唑-4-基)-苯基)丙酰胺氢氯化物、2-胺基-N-(2-甲氧基-5-[5-(3,4,5-三甲氧苯基)异恶唑-4-基)-苯基)丙酰胺、2-胺基-N-(2-甲氧基-5-[5-(3,4,5-三甲氧苯基)-异恶唑-4-基)-苯基)-4-(甲硫基)丁酰胺氢氯化物、2-胺基-N-(2-甲氧基 -5-[5-(3,4,5-三甲氧苯基)-异恶唑-4-基)-苯基)丁酰胺、2-胺基-N-(2-甲氧基 -5-[5-(3,4,5-三甲氧苯基)-异恶唑-4-基)-苯基)-3-苯基丙酰胺氢氯化物、2-胺基 -N-(2-甲氧基-5-[5-(3,4,5-三甲氧苯基)-异恶唑-4-基)-苯基)-4-甲基戊酰胺氢氯化物、2-胺基-N-(2-甲氧基-5-[5-(3,4,5-三甲氧基-苯基)-异恶唑-4-基)-苯基)-3-(4-甲氧苯基)丙酰胺氢氯化物、1-{2-甲氧基-5-[5-(3,4,5-三甲氧基-苯基)- 异恶唑-4-基]-苯基胺甲酰基}-2-甲基-丙基-氯化铵、1-{2-甲氧基-5-[5-(3,4,5- 三甲氧苯基)-异恶唑-4-基]-苯基胺甲酰基}-2-甲基-丁基-氯化铵、2-羟基-1-{2- 甲氧基-5-[5-(3,4,5-三甲氧苯基)-异恶唑-4-基]-苯基胺甲酰基}-丙基-氯化铵、 2-(4-羟基-苯基)-1-{2-甲氧基-5-[5-(3,4,5-三甲氧苯基)-异恶唑-4-基]-苯基胺甲酰基}-乙基-氯化铵、C-{2-甲氧基-5-[5-(3,4,5-三甲氧苯基)-异恶唑-4-基]-苯基胺甲酰基}-C-苯基-甲基-氯化铵、2-(1H-吲哚-2-基)-1-{2-甲氧基-5-[5-(3,4,5- 三甲氧苯基)-异恶唑-4-基]-苯基胺甲酰基}-乙基-氯化铵、2-苯并呋喃-2-基 -1-{2-甲氧基-5-[5-(3,4,5-三甲氧苯基)-异恶唑-4-基]-苯基胺甲酰基}-乙基-氯化铵、2-羧基-1-{2-甲氧基-5-[5-(3,4,5-三甲氧苯基)-异恶唑-4-基]-苯基胺甲酰基}-乙基-氯化铵、3-羧基-1-{2-甲氧基-5-[5-(3,4,5-三甲氧苯基)-异恶唑-4-基]- 苯基胺甲酰基}-丙基-氯化铵、3-胺甲酰基-1-{2-甲氧基-5-[5-(3,4,5-三甲氧苯基)-异恶唑-4-基]-苯基胺甲酰基}-丙基-氯化铵、2-胺甲酰基-1-{2-甲氧基 -5-[5-(3,4,5-三甲氧苯基)-异恶唑-4-基]-苯基胺甲酰基}-乙基-氯化铵,以及 2-(3H-咪唑-4-基)-1-{2-甲氧基-5-[5-(3,4,5-三甲氧苯基)-异恶唑-4-基]-苯基胺甲酰基}-乙基-氯化铵。
哒嗪酮PARP抑制剂的使用透过Branca等人被描述于美国专利第8,268,827号,并通过引用将其包括在内。哒嗪酮PARP抑制剂包含但不限于 6-{4-氟代-3-[(3-氧基-4-苯基哌嗪-1-基)羰基]苄基}-4,5-二甲基-3-氧基-2,3-二氢哒嗪-1-正离子三氟代醋酸盐、6-{3-[(4-环己基-3-氧代哌嗪-1-基)羰基]-4-氟代苄基}-4,5-二甲基-3-氧基-2,3-二氢哒嗪-1-正离子三氟代醋酸盐、6-{3-[(4- 环戊基-3-氧代哌嗪-1-基)羰基]-4-氟代苄基}-4,5-二甲基哒嗪-3(2H)-酮、6-{4- 氟代-3-[(3-氧基-4-苯基哌嗪-1-基)羰基]苄基}-4,5-二甲基哒嗪-3(2H)-酮氢氯化物、4-乙基-6-{4-氟代-3-[(3-氧基-4-苯基哌嗪-1-基)羰基]苄基}哒嗪-3(2H)- 酮三氟代醋酸盐、6-{3-[(4-环己基-3-氧代哌嗪-1-基)羰基]-4-氟代苄基}-4-乙基哒嗪-3(2H)-酮三氟代醋酸盐、3-{4-氟代-3-[(4-甲基-3-氧代哌嗪-1-基)羰基] 苄基}-4,5-二甲基-6-氧基-1,6-二氢哒嗪-1-正离子三氟代醋酸盐、3-(4-氟代 -3-{[4-(4-氟代苄基)-3-氧代哌嗪-1-基]羰基}苄基)-4,5-二甲基-6-氧基-1,6-二氢哒嗪-1-正离子三氟代醋酸盐、6-(3-{[4-(2-氯苯基)-3-氧代哌嗪-1-基]羰基}-4- 氟代苄基)-4,5-二甲基-3-氧基-2,3-二氢哒嗪-1-正离子三氟代醋酸盐、 6-(3-{[4-(3-氯基-4-氟代苯基)-3-氧代哌嗪-1-基]羰基}-4-氟代苄基)-4,5-二甲基-3-氧基-2,3-二氢哒嗪-1-正离子三氟代醋酸盐,以及6-(3-{[4-(3,4-二氟代苯基)-3-氧代哌嗪-1-基]羰基}-4-氟代苄基)-4,5-二甲基-3-氧基-2,3-二氢哒嗪-1- 正离子三氟代醋酸盐。其他PARP抑制剂透过Moore等人被描述于美国专利第8,143,447号,并通过引用将其包括在内;这些化合物包含硝基苯甲酰胺衍生物。
极光蛋白激酶抑制剂的使用透过Mortimore等人被描述于美国专利第8,268,811号,并通过引用将其包括在内。所述极光蛋白激酶抑制剂包含但不限于噻唑类及吡唑类。极光蛋白激酶抑制剂的使用也透过Binch等人被描述于美国专利第8,129,399号,并通过引用将其包括在内;这些极光蛋白激酶抑制剂包含但不限于胺吡啶。
结合至摄护腺特殊性膜抗原的胜肽(PSMA)的使用透过Denmeade等人被描述于美国专利第8,258,256号,并通过引用将其包括在内。
CD19结合剂的使用透过McDonagh等人被描述于美国专利第8,242,252 号,并通过引用将其包括在内。这些CD19结合剂包含但不限于抗CD19抗体。
苯重氮基盐的使用透过Glick被描述于美国专利第8,242,109号,并通过引用将其包括在内。
类铎受体(TLR)促效剂的使用透过Howbert等人被描述于美国专利第 8,242,106号,并通过引用将其包括在内。合适的TLR促效剂包含但不限于(1E, 4E)-2-胺基-N,N-二丙基-8-(4-(吡咯啶-1-羰基)苯基)-3H-苯并[b]氮呯-4-羧酰胺。
架桥的双环磺酰胺的使用透过Lewis等人被描述于美国专利第8,242,103 号,并通过引用将其包括在内。
表皮生长因子受体(EGFR)激酶的抑制剂的使用透过Kuriyan等人被描述于美国专利第8,242,080号,并通过引用将其包括在内。通常,这些EGFR 激酶的抑制剂靶向非对称活化二聚体界面。
具有肌动蛋白结合活性的T2家族的核糖核酸的使用透过Roiz等人被描述于美国专利第8,236,543号,并通过引用将其包括在内。通常,在主动或被动核糖核酸形式中,所述核糖核酸结合肌动蛋白。
萜烯苯酸或其类似物的使用透过Lee等人被描述于美国专利第8,232,318 号,并通过引用将其包括在内。
一细胞周期素依赖性激酶的抑制剂的使用透过Shipps等人被描述于美国专利第8,227,605号;这些抑制剂包含但不限于2-胺基噻唑-4-羧酰胺。一细胞周期素依赖性激酶的抑制剂的使用也透过Mallams等人被描述于美国专利第7,700,773号,并通过引用将其包括在内;这些抑制剂包含但不限于吡唑并[1,5-a]吡啶、吡唑并[1,5-c]嘧啶及2H-吲唑化合物的4-氰基、4-胺基及4- 胺甲基衍生物,以及咪唑并[1,2-a]吡啶及咪唑并[1,5-a]吡嗪化合物的5-氰基、 5-胺基及5-胺甲基衍生物。
一p53及MDM2之间交互作用的抑制剂的使用透过Wang等人被描述于美国专利第8,222,288号,并通过引用将其包括在内。
所述受体酪氨酸激酶MET的抑制剂的使用透过Dinsmore等人被描述于美国专利第8,222,269号,并通过引用将其包括在内。这些所述受体酪氨酸激酶MET的抑制剂包含但不限于5H-苯并[4,5]环七[1,2-b]吡啶衍生物。所述受体酪氨酸激酶MET的抑制剂也透过Jewell等人被描述于美国专利第 8,207,186号,并通过引用将其包括在内。这些化合物包含但不限于苯并环七吡啶,包含5H-苯并[4,5]环七[1,2-b]吡啶衍生物。
拉戈唑或拉戈唑类似物的使用透过Williams等人被描述于美国专利第 8,217,076号,并通过引用将其包括在内。
所述蛋白激酶AKT的抑制剂的使用透过Furuyama等人被描述于美国专利第8,207,169号,并通过引用将其包括在内;这些抑制剂包含但不限于三唑吡啶并吡啶类,包含取代的[1,2,4]三唑[4’,3’:1,6]吡啶并[2,3-b]吡嗪。
2’-氟代-5-甲基-β-L-阿拉伯糖呋喃糖基尿苷或L-脱氧胸腺核苷的使用透过Cheng被描述于美国专利第8,207,143号,并通过引用将其包括在内。
调节HSP90活性的化合物的使用透过Ying等人被描述于美国专利第 8,188,075号,并通过引用将其包括在内。这些化合物包含但不限于取代的三唑类,包含3-(2-羟苯基)-4-(萘-1-基)-5-硫醇三唑、3-(2,4-二羟苯基)-4-[4-(2- 甲氧基乙氧基)-萘-1-基]-5-硫醇三唑、3-(2,4-二羟苯基)-4-(2-甲基-4-溴基苯基)-5-硫醇三唑、3-(3,4-二羟苯基)-4-(6-甲氧基-萘-1-基)-5-硫醇三唑、3-(3,4- 二羟苯基)-4-(6-乙氧基-萘-1-基)-5-硫醇三唑、3-(3,4-二羟苯基)-4-(6-丙氧基- 萘-1-基)-5-硫醇三唑、3-(2,4-二羟基-5-乙基-苯基)-4-(5-甲氧基-萘-1-基)-5-硫醇三唑、3-(3,4-二羟苯基)-4-(6-异丙氧基-萘-1-基)-5-硫醇三唑、3-(2,4-二羟苯基)-4-(2,6-二乙基苯基)-5-硫醇三唑、3-(2,4-二羟苯基)-4-(2-甲基-6-乙基苯基)-5-硫醇三唑、3-(2,4-二羟苯基)-4-(2,6-二异丙基苯基)-5-硫醇三唑、3-(2,4- 二羟苯基)-4-(1-乙基-吲哚-4-基)-5-硫醇三唑,以及3-(2,4-二羟苯基)-4-(2,3- 二氢-苯并[1,4]二恶英-5-基)-5-硫醇三唑。
JAK激酶或PDK激酶的抑制剂的使用透过Guerin等人被描述于美国专利第8,183,245号,并通过引用将其包括在内。所述JAK激酶包含JAK1、 JAK2、JAK3及TYK2。这些种类激酶的合适的抑制剂包含但不限于5-(1-甲基-1H-吡唑-4-基)-3-(6-哌嗪-1-基吡嗪-2-基)-1H-吡咯并[2,3-b]吡啶、5-(1-甲基 -1H-吡唑-4-基)-3-[6-(哌啶-4-基氧基)吡嗪-2-基]-1H-吡咯并[2,3-b]吡啶、 3-[6-(环己氧基)吡嗪-2-基]-5-(1-甲基-1H-吡唑-4-基)-1H-吡咯并[2,3-b]吡啶、 N-甲基-6-[5-(1-甲基-1H-吡唑-4-基)-1H-吡咯并[2,3-b]吡啶-3-基]-N-哌啶-4-基吡嗪-2-胺、3-[6-(哌啶-4-基氧基)吡嗪-2-基]-5-(1H-吡唑-4-基)-1H-吡咯并 [2,3-b]吡啶、3-{6-[(3R)-哌啶-3-基氧基]吡嗪-2-基}-5-(1H-吡唑-4-基)-1H-吡咯并[2,3-b]吡啶,以及3-{6-[(3S)-哌啶-3-基氧基]吡嗪-2-基}-5-(1H-吡唑-4- 基)-1H-吡咯并[2,3-b]吡啶。
第四型磷酸二酯酶(phosphodiesterase type IV,PDE4)的抑制剂的使用透过Muller等人被描述于美国专利第8,158,672号,并通过引用将其包括在内。所述PDE4的抑制剂包含氟代烷氧基取代的1,3-二氢异吲哚基化合物。
c-Met原致癌基因受体酪氨酸激酶的抑制剂的使用透过Zhuo等人被描述于美国专利第8,143,251号,通过引用将其包括。这些抑制剂包含但不限于三唑三嗪,包含[1,2,4]三唑[4,3-b][1,2,4]三嗪。C-Met原致癌基因受体酪氨酸激酶的抑制剂也透过Cui等人被描述于美国专利第8,106,197号,并通过引用将其包括在内;这些抑制剂包含胺基杂芳基化合物。
吲哚胺2,3-二氧合酶的抑制剂的使用透过Combs等人被描述于美国专利第8,088,803号,并通过引用将其包括在内;这些抑制剂包含但不限于1,2,5- 恶二唑衍生物。
抑制ATDC(TRIM29)表现的药剂的使用透过Simeone等人被描述于美国专利第8,088,749号,并通过引用将其包括在内。这些药剂包含经由RNA干扰的功能的寡核苷酸。
核受体和共活化胜肽的交互作用的拟蛋白抑制剂的使用透过Hamilton 等人被描述于美国专利第8,084,471号,并通过引用将其包括在内。这些抑制剂包含但不限于2,3’,3″–三取代的联三苯。
XIAP家族蛋白的拮抗物的使用透过Chen等人被描述于美国专利第 7,910,621号,并通过引用将其包括在内。这些拮抗物包含但不限于恩贝酸。
肿瘤靶向超抗原的使用透过Hedlund等人被描述于美国专利第7,763,253 号,并通过引用将其包括在内。
Pim激酶的抑制剂的使用透过Bearss等人被描述于美国专利第7,750,007 号,并通过引用将其包括在内。这些抑制剂包含但不限于咪唑并[1,2-b]哒嗪及吡唑并[1,5-a]嘧啶化合物。
CHK1或CHK2激酶的抑制剂的使用透过Tepe被描述于美国专利第 7,732,436号,并通过引用将其包括在内。这些抑制剂包含但不限于吲哚氮呯及其酸胺盐。
血管生成素蛋白4的抑制剂的使用透过Gerber等人被描述于美国专利第 7,740,846号,并通过引用将其包括在内。这些抑制剂包含但不限于抗体,包含单株抗体。
Smo的抑制剂的使用透过Balkovec等人被描述于美国专利第7,691,997 号,通过引用将其包括。Smo(Smoothened)是一通过刺猬蛋白信号传递的媒介物。适当的抑制剂包含但不限于5-(1,1-二氟代乙基)-3-(4-{4-甲基-5-[2-(三氟代甲基)苯基]-4H-1,2,4-三唑-3-基}双环[2.2.2]辛-1-基)-1,2,4-恶二唑、5-(3,3- 二氟代环丁基)-3-(4-{4-甲基-5-[2-(三氟代甲基)苯基]-4H-1,2,4-三唑-3-基}双环[2.2.2]辛-1-基)-1,2,4-恶二唑、5-(1-氟代-1-甲基乙基)-3-(4-{4-甲基-5-[2-(三氟代甲基)苯基]-4H-1,2,4-三唑-3-基}双环[2.2.2]辛-1-基)-1,2,4-恶二唑、2-(1,1- 二氟代乙基)-5-(4-{4-甲基-5-[2-(三氟代甲基)苯基]-4H-1,2,4-三唑-3-基}双环 [2.2.2]辛-1-基)-1,3,4-恶二唑、2-(3,3-二氟代环丁基)-5-(4-{4-甲基-5-[2-(三氟代甲基)苯基]-4H-1,2,4-三唑-3-基}双环[2.2.2]辛-1-基)-1,3,4-恶二唑,以及2-(1- 氟代-1-甲基乙基)-5-(4-{4-甲基-5-[2-(三氟代甲基)苯基]-4H-1,2,4-三唑-3-基} 双环[2.2.2]辛-1-基)-1,3,4-恶二唑。
烟碱型乙酰胆碱受体拮抗物的使用透过Cooke等人被公开于美国专利第 7,652,038号,并通过引用将其包括在内。烟碱型乙酰胆碱受体拮抗物包含但不限于梅坎米胺(mecamylamine)、六羟季铵(hexamethonium)、二氢-β-刺桐啶、 d-筒箭毒碱(d-tubocurarine)、潘必啶(pempidine)、氯化异桑大明 (chlorisondamine)、刺桐定碱(erysodine)、樟脑磺酸屈美沙芬(trimethaphan camsylate)、血安定(pentolinium)、雨伞节蛇毒(bungarotoxin)、琥珀胆碱、四乙基铵、咪噻吩(trimethaphan)、氯化异桑大明及曲美替定(trimethidinium)。
法呢基蛋白转移酶抑制剂的使用透过Zhu等人被描述于美国专利第 7,557,107号,并通过引用将其包括在内。这些法呢基蛋白转移酶抑制剂包含三环化合物。
腺苷A3受体拮抗物的使用透过Leung等人被描述于美国专利第 6,326,390号,并通过引用将其包括在内。这些腺苷A3受体拮抗物包含三环非黄嘌呤拮抗物及三唑并喹唑啉类。
Atadja等人所申请的美国专利申请公开第2010/0069458号,并通过引用将其包括在内,其公开下面附加治疗试剂的使用,其可以与一如上所述的烷基化己醣醇衍生物一起使用:
(1)ACE抑制剂包含但不限于贝那普利(benazepril)、enazepril、卡托普利(captopril)、依那普利(enalapril)、福辛普利(osinopril)、赖诺普利(lisinopril)、莫西普利(moexipril)、喹那普利(quinapril)、雷米普利(ramipril)、培哚普利 (perindopril)及群多普利(trandolapril);
(2)腺苷激酶抑制剂包含但不限于5-碘杀结核菌素(5-iodotubericidin);
(3)肾上腺皮质拮抗物包含但不限于米托坦(mitotane);
(4)AKT途径抑制剂(蛋白激酶B抑制剂)包含但不限于鱼藤素(deguelin)及 1,5-二氢-5-甲基-1-β-D-呋喃核糖苷-1,4,5,6,8-五氮杂苊-3-胺;
(5)血管生成抑制剂包含但不限于烟曲霉素fumagillin、紫草素Shikonin、曲尼司特Tranilast、乌索酸;苏拉明;沙利多迈(thalidomide)、来那度胺 lenalidomide;酞嗪类包含但不限于1-(4-氯苯胺基)-4-(4-吡啶甲基)酞嗪、1-(4- 甲基苯胺)-4-(4-吡啶甲基)酞嗪、1-(3-氯苯胺基)-4-(4-吡啶甲基)酞嗪、1-苯胺基-4-(4-吡啶甲基)酞嗪、1-苄基胺基-4-(4-吡啶甲基)酞嗪、1-(4-甲氧苯胺基)-4-(4-吡啶甲基)酞嗪、1-(3-苄氧基苯胺基)-4-(4-吡啶甲基)酞嗪、1-(3-甲氧苯胺基)-4-(4-吡啶甲基)酞嗪、1-(2-甲氧苯胺基}-4-(4-吡啶甲基)酞嗪、1-(4- 三氟甲基苯胺)-4-(4-吡啶甲基)酞嗪、1-(4-氟苯胺)-4-(4-吡啶甲基)酞嗪、1-(3- 羟基苯胺基)-4-(4-吡啶甲基)酞嗪、1-(4-羟基苯胺基)-4-(4-吡啶甲基)酞嗪、 1-(3-胺基苯胺基)-4-(4-吡啶甲基)酞嗪、1-(3,4-二氯苯胺基)-4-(4-吡啶甲基)酞嗪、1-(4-溴苯胺基)-4-(4-吡啶甲基)酞嗪、1-(3-氯基-4-甲氧苯胺基)-4-(4-吡啶甲基)酞嗪、1-(4-氰苯胺)-4-(4-吡啶甲基)酞嗪、1-(3-氯基-4-氟苯胺)-4-(4-吡啶甲基)酞嗪、1-(3-甲基苯胺)-4-(4-吡啶甲基)酞嗪,以及,其他的透过Bold等人公开于PCT专利申请公开第WO 98/035958号的酞嗪类,并通过引用将其整体包括在内,透过Altmann等人公开于PCT专利申请公开第WO 00/09495 号的异喹啉,并通过引用将其整体包括在内,包含1-(3,5-二甲基苯胺)-4-(吡啶-4-基甲基)-异喹啉;透过Bold等人公开于PCT专利申请公开第WO 00/59509号的酞嗪类,并通过引用将其整体包括在内,包含E-1-(3-甲基苯胺)-4-[(2-(吡啶-3-基)乙烯基]酞嗪、Z-1-(3-甲基苯胺)-4-[(2-(吡啶-3-基)乙烯基] 酞嗪、1-(3-甲基苯胺)-4-[(2-(吡啶-3-基)乙基]酞嗪、1-(3-甲基苯胺)-4-[{2-(吡啶-4-基)乙烯基]酞嗪、1-(4-氯基-3-三氟甲基苯胺)-4-[(2-(吡啶-3-基)乙基]酞嗪、1-(4-氯苯胺基)-4-[(2-(吡啶-3-基)乙基]酞嗪、1-(3-氯苄基胺基)-4-[(2-(吡啶-3-基)乙基]酞嗪、1-(4-氯基-3-三氟甲基苯胺)-4-[3-(吡啶-3-基)丙基]酞嗪、 1-(4-氯苯胺基)-4-[3-(吡啶-3-基)丙基]酞嗪、1-(3-氯基-5-三氟甲基苯胺)-4-[3-(吡啶-3-基)丙基]酞嗪,以及1-(4-叔-丁基苯胺)-4-[3-(吡啶-3-基)丙基] 酞嗪;以及单株抗体;
(6)血管生成抑制类固醇包含但不限于阿奈可他(anecortave)、特安皮质醇、氢化可体松、11α-表氢化可的松(11α-epihydrocotisol)、皮甾酮(cortexolone)、 17α-羟孕酮、皮质固酮(corticosterone)、脱氧皮质固酮(desoxycorticosterone)、睪固酮(testosterone)、雌固酮(estrone)及地塞米松;
(7)抗雄激素类包含但不限于尼鲁米特(nilutamide)及比卡鲁胺(bicalutamide);
(8)抗雌激素类包含但不限于托瑞米芬(toremifene)、来曲唑(letrozole)、睾内酯(testolactone)、阿那曲唑(anastrozole)、比卡鲁胺、氟他胺(flutamide)、依西美坦(exemestane)、他莫昔芬(tamoxifen)、氟维司群(fulvestrant)及雷洛昔芬(raloxifene);
(9)抗高血钙剂包含但不限于硝酸镓(III)水合物及裴米卓耐特二钠(pamidronate disodium);
(10)细胞凋亡诱导物包含但不限于2-[[3-(2,3-二氯苯氧基)丙基]胺基]-乙醇、藤黄酸(gambogic acid)、信筒子醌(embellin)及三氧化二砷(arsenic trioxide);
(11)ATI受体拮抗物包含但不限于缬沙坦(valsartan);
(12)极光激酶抑制剂包含但不限于binucleine 2;
(13)芳香酶抑制剂包含但不限于:(a)类固醇包含但不限于阿他美坦(atamestane)、依西美坦及福美斯坦(formestane);以及(b)非类固醇包含但不限于胺基苯乙呱啶酮、罗谷亚胺(roglethimide)、吡啶并苯乙呱啶酮、曲洛司坦(trilostane)、睾内酯、克康那唑、伏氯唑(vorozole)、法倔唑(fadrozole)、阿那曲唑及来曲唑;
(14)双膦酸盐包含但不限于羟乙磷酸(etidronic acid)、氯膦酸(clodronicacid)、替鲁膦酸(tiludronic acid)、阿仑棒酸(alendronic acid)、伊班膦酸(ibandronicacid)、利塞膦酸(risedronic acid)及唑来膦酸(zoledronic acid);
(15)布鲁顿(Bruton)酪氨酸激酶抑制剂包含但不限于土曲霉酸(terreic acid);
(16)磷酸酶抑制剂包含但不限于赛灭宁(cypermethrin)、第灭宁(deltamethrin)、氰戊菊酯(fenvalerate)及酪氨酸磷酸化抑制剂8(tyrphostin 8);
(17)CaM激酶II抑制剂包含但不限于5-异喹啉磺酸4-[(2S)-2-[(5-异喹啉基磺酰基)甲胺基]-3-氧基-3-(4-苯基-1-哌嗪基)丙基]苯基酯,以及N-[2-[[[3-(4-氯苯基)-2-丙烯基]甲基]胺基]甲基]苯基]-N-(2-羟乙基)-4-甲氧基-苯磺酰胺;
(18)CD45酪胺酸磷酸酶抑制剂包含但不限于[[2-(4-溴苯氧基)-5-硝基苯基]羟甲基]-膦酸;
(19)CDC25磷酸酶抑制剂包含但不限于2,3-双[(2-羟乙基)硫基]-1,4-萘醌;
(20)CHK激酶抑制剂包含但不限于脱溴hymenialdisine;
(21)靶向/递减一蛋白或脂肪激酶活性的化合物;或蛋白或脂肪磷酸酶活性;或进一步抗血管生成化合物包含但不限于蛋白酪氨酸激酶和/或丝氨酸和/或苏胺酸激酶抑制剂或脂肪激酶抑制剂,包含但不限于:
(a)化合物靶向,降低或抑制血管内皮生长因子受体(vascular endothelialgrowth factor receptor,VEGFR)或血管内皮生长因子(vascular endothelial growthfactor,VEGF)的活性,包含但不限于7H-吡咯并[2,3-d]嘧啶衍生物,包含:[6-[4-(4-乙基-哌嗪-1-基甲基)-苯基]-7H-吡咯并[2,3-d]嘧啶嘧啶-4-基]-(R)-1-苯基-乙基)-胺(称为AEE788)、BAY 43-9006,以及公开于 PCT专利申请公开第WO 00/09495号的异喹啉化合物,比如(4-叔-丁基- 苯基)-94-吡啶-4-基甲基-异喹啉-1-基)-胺;
(b)化合物靶向,降低或抑制所述血小板衍生生长因子受体 (platelet-derivedgrowth factor-receptor,PDGFR)的活性,包含但不限于: N-苯基-2-嘧啶-胺衍生物,比如,伊马替尼、SU101、SU6668及GFB-111;
(c)化合物靶向,降低或抑制所述纤维原细胞生长因子受体(fibroblast growthfactor-receptor,FGFR)的活性;
(e)化合物靶向,降低或抑制所述Trk受体酪氨酸激酶家族的活性;
(f)化合物靶向,降低或抑制所述Axl受体酪氨酸激酶家族的活性;
(g)化合物靶向,降低或抑制所述c-Met受体的活性;
(h)化合物靶向,降低或抑制所述Ret受体酪氨酸激酶的活性;
(i)化合物靶向,降低或抑制所述Kit/SCFR受体酪氨酸激酶的活性;
(j)化合物靶向,降低或抑制所述C-kit受体酪氨酸激酶的活性,包含但不限于伊马替尼;
(k)化合物靶向,降低或抑制c-Abl家族及其基因融合产物的成员的活性,比如BCR-Abl激酶,比如N-苯基-2-嘧啶-胺衍生物包含但不限于:伊马替尼、6-(2,6-二氯苯基)-2-[(4-氟代-3-甲苯基)胺基]-8-甲基-吡啶并 [2,3-d]嘧啶-7(8H)-酮(PD180970)、甲基-4-[N-(2′,5′-二羟基苄基)胺基] 苯甲酸盐(Tyrphostin AG957)、4-[[(2,5-二羟苯基)甲基]胺基]苯甲酸三环 [3.3.1.13,7]癸-1-基酯(阿德福辛(adaphostin)或NSC 680410)、6-(2,6-二氯苯基)-8-甲基-2-(3-甲基磺酰基苯胺基)吡啶并[2,3-d]嘧啶-7-酮(PD173955),以及达沙替尼(desatinib);
(l)化合物靶向,降低或抑制丝氨酸激酶/息宁胺酸激酶的蛋白激酶 (proteinkinase C,PKC)及Raf家族的成员、MEK、SRC、JAK、FAK、PDK 及Ras/MAPK家族成员或PI(3)激酶家族或PI(3)-激酶-相关激酶家族的成员,和/或所述细胞周期素依赖性激酶(cyclin-dependent kinase,CDK)家族的成员的活性,且特别是那些公开于美国专利第5,093,330号的星状孢菌素衍生物,比如但不限于米哚妥林;进一步化合物的例子比如包含 UCN-01、沙芬戈(safingol)、索拉非尼(sorafenib)、苔藓虫素1(Bryostatin 1)、呱立福辛(Perifosine)、伊莫福新(Ilmofosine)、3-[3-[2,5-二氢-4-(1-甲基-1H-吲哚-3-基)-2,5-二氧-1H-吡咯-3-基]-1H-吲哚-1-基]丙基硫代氨基亚胺酸酯(RO 318220)、3-[(8S)-8-[(二甲胺基)甲基]-6,7,8,9-四氢吡啶并 [1,2-a]吲哚-10-基]-4-(1-甲基-1H-吲哚-3-基)-1H-吡咯-2,5-二酮(RO 320432)、12-(2-氰基乙基)-6,7,12,13-四氢-13-甲基-5-氧基-5H-吲哚并 [2,3-a]吡咯并[3,4-c]咔唑(GO 6976)、Isis 3521、(S)-13-[(二甲胺基)甲基]-10,11,14,15-四氢-4,9:16,21-二次甲基-1H,13H-二苯并[e,k]吡咯并 [3,4-h][1,4,13]oxadiazacy clohexadecene-1,3(2H)-二酮(LY333531)、 LY379196、异喹啉化合物,如此的这些是公开于PCT专利申请公开第 WO 00/09495号;法呢基转移酶抑制剂包含但不限于替吡法尼(tipifarnib) 及洛那法尼(lonafarnib)、2-(2-氯基-4-碘基-苯胺基)-N-环丙基甲氧基-3,4- 二氟代-苯甲酰胺(PD184352),以及QAN697,一PI3K抑制剂;
(m)化合物靶向,降低或抑制蛋白酪氨酸激酶的活性,比如但不限于甲磺酸伊马替尼、酪氨酸磷酸化抑制剂(Tyrphostin)、嘧啶基胺基苯甲酰胺及其衍生物;酪氨酸磷酸化抑制剂优选为低分子量(M
(n)化合物靶向,降低或抑制受体酪氨酸激酶(EGFR、ErbB2、ErbB3、 ErbB4作为同质二聚体或异源二聚体)的表皮生长因子家族的活性,比如但不限于下述前案中一般地及特定地所公开那些化合物、蛋白或单株抗体:Traxler等人所申请的PCT专利申请公开第WO97/02266号(比如 (R)-6-(4-羟苯基)-4-[(1-苯基乙基)-胺基]-7H-吡咯并-[2,3-d]嘧啶)、 Zimmermann所申请的欧洲专利申请公开第EP 0564409号、Zimmermann 等人所申请的PCT专利申请公开第WO 99/03854号、Barker等人所申请的欧洲专利申请公开第EP 0520722号、Barker等人所申请的欧洲专利申请公开第EP 0566226号、Wissner等人所申请的欧洲专利申请公开第EP 0787722号、Arnold等人所申请的欧洲专利申请公开第EP 0837063号、Schnur等人所申请的美国专利第US5,747,498号、McMahon等人所申请的PCT专利申请公开第WO 98/10767号、Barker所申请的PCT专利申请公开第WO 97/30034号、Schnur所申请的PCT专利申请公开第WO 97/49688号、Bridges等人所申请的PCT专利申请公开第WO 97/38983 号、Schnur等人所申请的PCT专利申请公开第WO 96/30347号包含但不限于N-(3-乙炔基苯基)-6,7-双(2-甲氧基乙氧基)-4-喹唑啉胺(CP 358774或厄洛替尼)或Gibson等人所申请的PCT专利申请公开第WO 96/33980号包含但不限于N-(3-氯基-4-氟代-苯基)-7-甲氧基-6-(3-吗啉-4- 基丙氧基)喹唑啉-4-胺(吉非替尼);以及Barker等人所申请的PCT专利申请公开第WO 95/03283号包含但不限于化合物6-胺基-4-(3-甲苯基-胺基)-喹唑啉(ZM105180);单株抗体包含但不限于曲妥单抗(trastuzumab) 及西妥昔;以及其他小分子抑制剂包含但不限于:卡奈替尼(canertinib)、培利替尼(pelitinib)、拉帕替尼及7H-吡咯并-[2,3-d]嘧啶衍生物,其是透过Bold等人公开于PCT专利申请公开第WO 03/013541号;
(22)靶向、递减或抑制一蛋白或脂肪磷酸酶的活性的化合物包含但不限于磷酸酶1、磷酸酶2A、PTEN或CDC25的抑制剂,比如但不限于冈田井酸(okadaic acid)或其衍生物;
(23)诱发细胞分化过程的化合物包含但不限于视黄酸(retinoic acid)、α-生育酚(α-tocopherol)、γ-生育酚、δ-生育酚、α-三烯生育酚(α-tocotrienol)、γ-三烯生育酚及δ-三烯生育酚;
(24)cRAF激酶抑制剂包含但不限于3-(3,5-二溴-4-羟基亚苄基)-5-碘基-1,3-二氢吲哚-2-酮,以及3-(二甲胺基)-N-[3-[(4-羟基苯甲酰基)胺基]-4-甲苯基]- 苯甲酰胺;
(25)细胞周期素依赖性激酶抑制剂包含但不限于N9-异丙基-奥罗莫星;奥罗莫星(olomoucine);苯甲酸(purvalanol B)、roascovitine、肯泡隆(kenpaullone) 及1-丁醇(purvalanol A);
(26)半胱胺酸蛋白酶抑制剂包含但不限于N-[(1S)-3-氟代-2-氧基-1-(2-苯基]乙基)丙基]胺基]-2-氧基-1-(苯基甲基)乙基]-4-吗啉甲酰胺基;
(27)DNA崁入剂包含但不限于普卡霉素及放线菌素(dactinomycin);
(28)DNA股断裂剂包含但不限于博来霉素(bleomycin);
(29)E3连接酶抑制剂包含但不限于N-((3,3,3-三氟代-2-三氟代甲基)丙酰基)胺苯磺酰胺;
(30)EDG结合剂包含但不限于FTY720;
(31)内分泌激素包含但不限于亮丙瑞林(leuprolide)及醋酸美皆斯妥(megestrol acetate);
(32)法呢基转移酶抑制剂包含但不限于α-羟基法呢基膦酸、 2-[[(2S)-2-[[(2S,3S)-2-[[(2R)-2-胺基-3-硫醇丙基]胺基]-3-甲基戊基]氧基]-1-氧基-3-苯基丙基]胺基]-4-(甲磺酰基)-1-甲基乙基丁酸酯(2S),以及手霉素 A(manumycin A);
(33)Flk-1激酶抑制剂包含但不限于2-氰基-3-[4-羟基-3,5-双(1-甲基乙基)苯基]-N-(3-苯基丙基)-(2-E)-2-丙烯酰胺;
(34)Flt-3抑制剂包含但不限于N-苄酰基-星状孢菌素、米哚妥林,以及N-(2- 二乙胺基乙基)-5-[(Z)-(5-氟代-2-氧基-1H-吲哚-3-亚基)甲基]-2,4-二甲基-1H- 吡咯-3-羧酰胺(纾癌特(sunitinib));
(35)性腺释素促效剂包含但不限于阿巴瑞克(abarelix)、戈舍瑞林(goserelin)及醋酸戈舍瑞林(goserelin acetate);
(36)肝素酶抑制剂包含但不限于硫代磷酸甘露醇戊糖(PI-88);
(37)组蛋白去乙酰酶(histone deacetylase,HDAC)抑制剂包含但不限于透过Bair等人公开于PCT专利申请公开第WO 02/22577号的化合物,其包含但不限于N-羟基-3-[4-[[(2-羟乙基)[2-(1H-吲哚-3-基)乙基]-胺基]甲基]苯基]-2E-2- 丙烯酰胺、辛二酰苯胺异羟肟酸、4-(2-胺基-苯基胺甲酰基)-苄基]-胺甲酸吡啶-3-基甲基酯及其衍生物、丁酸、pyroxamide、曲古抑菌素A(trichostatin A)、 oxamflatin、组蛋白脱乙酰酶(apicidin)、缩酚酸胜肽、德普菌素(depudecin)、氯化筒箭毒碱(trapoxin)、HC毒素(HCtoxin),以及苯丁酸钠盐;
(38)HSP90抑制剂包含但不限于:17-丙烯基胺;17-去甲氧基格尔德霉素 (17-demethoxygeldanamycin,17AAG);a格尔德霉素衍生物;其他的格尔德霉素相关化合物;根赤壳菌素(radicicol);以及5-(2,4-二羟基-5-异丙基-苯基)-4-(4-吗啉-4-基甲基-苯基)-恶唑-3-羧酸乙胺;
(39)IκBα抑制剂IKKs)包含但不限于3-[(4-甲苯基)磺酰基]-(2E)-2-丙烯腈;
(40)胰岛素受体酪氨酸激酶抑制剂包含但不限于羟基-2-萘基甲基膦酸;
(41)c-Jun N-端激酶抑制剂包含但不限于吡唑葱酮及表没食子儿茶素没食子酸酯;
(42)微管结合剂包含但不限于:硫酸长春花碱;硫酸氧化长春花碱(vincristinesulfate);长春地辛;长春瑞滨;多烯紫杉醇(docetaxel);紫杉醇;圆皮海绵内酯(discodermolides);秋水仙素;以及埃博霉素(epothilones)及其衍生物,比如埃博霉素B或其衍生物;
(43)丝裂原活化蛋白(mitogen-activated protein,MAP)激酶抑制剂包含但不限于N-[2-[[[3-(4-氯苯基)-2-丙烯基]甲基]胺基]甲基]苯基]-N-(2-羟乙基)-4-甲氧基-苯磺酰胺;
(44)MDM2抑制剂包含但不限于反-4-碘,4'-硼烷基-查酮;
(45)MEK抑制剂包含但不限于双[胺基[2-胺苯基)硫基]甲烯基]-丁二腈;
(46)甲硫胺酸胺基胜肽酶抑制剂包含但不限于苯甲酰胺(bengamide)及其衍生物;
(47)MMP抑制剂包含但不限于:放线酰胺素(Actinonin);表没食子儿茶素没食子酸酯;胶原拟胜肽及非拟胜肽抑制剂;四环素衍生物,比如异羟肟酸酯、巴马司他、马立马司他、普马司他(primomastat)、TAA211、N-羟基-2(R)-[[(4- 甲氧苯基)磺酰基](3-甲基吡啶)胺基]-3-甲基丁酰胺氢氯化物(MMI270B)及 AAJ996;
(48)NGFR酪氨酸激酶抑制剂包含但不限于Tyrphostin AG 879;
(49)p38 MAP激酶抑制剂包含但不限于3-(二甲胺基)-N-[3-[(4-羟基苯甲酰基)胺基]-4-甲苯基]-苯甲酰胺;
(50)p56酪氨酸激酶抑制剂包含但不限于9,10-二氢-3-羟基-1-甲氧基-9,10-二氧-2-蒽甲醛,以及Tyrphostin 46;
(51)PDGFR酪氨酸激酶抑制剂包含但不限于Tyrphostin AG 1296、Tyrphostin 9、2-胺基-4-(1H-吲哚-5-基)-1,3-丁二烯-1,1,3-三羰腈及伊马替尼;
(52)磷脂酰肌醇-3激酶抑制剂包含但不限于渥曼青霉素(wortmannin)及槲皮素二水合物;
(53)磷酸酶抑制剂包含但不限于斑蝥酸(cantharidic acid)、斑蝥素(cantharidin) 及(E)-N-[4-(2-羧基乙烯基)苄酰基]甘胺酰基-L-α-麸胺酰基-L-白氨酰胺;
(54)铂剂包含但不限于卡铂普来锭、顺铂、草酸铂、沙铂(satraplatin)及ZD0473;
(55)蛋白磷酸酶抑制剂包含但不限于:
(a)PP1抑制剂及PP2A抑制剂包含但不限于斑蝥酸及斑蝥素;
(b)酪胺酸磷酸酶抑制剂包含但不限于L-P-溴四咪唑草酸盐、苄基膦酸,以及(5R)-4-羟基-5-(羟甲基)-3-(1-氧基十六基)-2(5H)-呋喃酮;
(56)PKC抑制剂包含但不限于-[1-[3-(二甲胺基)丙基]-1H-吲哚-3-基]-4-(1H-吲哚-3-基)-1H-吡咯并-2,5-二酮、神经胺醇(sphingosine)、星状孢菌素、 Tyrphostin 51及金丝桃素(hypericin);
(57)PKC delta激酶抑制剂包含但不限于吕宋揪夹粉素(rottlerin);
(58)多胺合成抑制剂包含但不限于(RS)-2,5-二胺基-2-(二氟代甲基)戊酸(DMFO);
(59)蛋白酶体抑制剂包含但不限于阿克那霉素A(aclacinomycin A)、霉胶毒素(gliotoxin)及硼替佐米;
(60)PTP1B抑制剂包含但不限于(E)-N-[4-(2-羧基乙烯基)苄酰基]甘胺酰基 -L-α-麸胺酰基-L-白氨酰胺;
(61)蛋白酪氨酸激酶抑制剂包含但不限于:Tyrphostin AG 126、Tyrphostin AG1288、Tyrphostin AG 1295、格尔德霉素及染料木黄酮;
(62)SRC家族酪氨酸激酶抑制剂包含但不限于1-(1,1-二甲基乙基)-3-(1-萘基)-1H-吡唑并[3,4-d]嘧啶-4-胺,以及3-(4-氯苯基)-1-(1,1-二甲基乙基)-1H-吡唑并[3,4-d]嘧啶-4-胺;
(63)Syk酪氨酸激酶抑制剂包含但不限于白皮杉醇(piceatannol);
(64)Janus(JAK-2和/或JAK-3)酪氨酸激酶抑制剂包含但不限于Tyrphostin AG490,以及2-萘基乙烯基酮;
(65)Ras致癌基因异构体的抑制剂包含但不限于(2S)-2-[[(2S)-2-[(2S,3S)-2-[(2R)-2-胺基-3-硫醇丙基]胺基]-3-甲基戊基]氧基]-1-氧基-3-苯基丙基]胺基]-4-(甲磺酰基)-丁酸1-甲基乙基酯(L-744832)、 DK8G557,以及替吡法尼;
(66)类维生素A包含但不限于异视网酸(isotretinoin)及视网酸(tretinoin);
(67)核糖核苷酸还原酶抑制剂包含但不限于羟基尿素及2-羟基-1H-异吲哚 -1,3-二酮;
(68)RNA聚合酶II伸长抑制剂包含但不限于5,6-二氯-1-β-D-呋喃核糖苷苯并咪唑;
(69)S-腺核苷甲硫胺酸脱羧酶抑制剂包含但不限于5-脒基-1-四氢萘酮-2’-脒基腙,以及其他透过Stanek公开于美国专利第5,461,076号等人的化合物,并通过引用将其包括在内;
(70)丝胺酸/苏胺酸激酶激酶抑制剂包含但不限于索拉非尼及2-胺基嘌呤;
(71)靶向、递减或抑制丝氨酸/息宁胺酸mTOR激酶的活性或功能的化合物包含但不限于依维莫司(everolimus)、替西罗莫司(temsirolimus)、佐他莫司 (zotarolimus)、雷帕霉素、雷帕霉素的衍生物及类似物、雷帕霉素(deforolimus)、 AP23841、西罗莫司(sirolimus)及依维莫司;
(72)生长抑制素受体拮抗物包含但不限于奥曲肽及帕西瑞肽 (pasireotide)(SOM230);
(73)固醇生物合成抑制剂包含但不限于特比萘定(terbinadine);
(74)端粒酶抑制剂包含但不限于端粒酶素(telomestatin);以及
(75)位置异构酶抑制剂包含但不限于:
(a)位置异构酶I抑制剂包含但不限于拓扑替康、吉马替康(gimatecan)、伊立替康、喜树碱及其类似物、9-硝基喜树碱及大分子喜树碱共轭物 PNU-16614、大分子喜树碱共轭物(其是透过Angelucci等人描述于PCT 专利申请公开第WO 99/17804号)、10-羟基喜树碱醋酸盐、伊妥普赛埃达霉素氢氯化物、替尼泊苷、阿霉素、表阿霉素(epirubicin)氢氯化物、双羟蒽醌氢氯化物,以及道诺霉素氢氯化物;以及
(b)位置异构酶II抑制剂包含但不限于蒽环类,比如阿霉素,包含其脂质体制剂;道诺霉素,包含其脂质体制剂;表阿霉素;埃达霉素;奈莫柔比星(nemorubicin);双羟蒽醌;洛索蒽醌;伊妥普赛;以及去羟栀子甙(eniposide);
(76)VEGFR酪氨酸激酶抑制剂包含但不限于3-(4-二甲基胺基苄基茚基)-2- 吲哚酮;以及
(77)RANKL抑制剂包含但不限于狄诺塞麦(denosumab)。
当所述改良是通过化学敏化作用来进行的,所述化学敏化作用可以包含但不限于结合使用一作为化学致敏剂的烷基化己醣醇衍生物与一药剂,所述药剂是选自于由下列所组成的群组:
(a)位置异构酶抑制剂;
(b)伪核苷;
(c)伪核苷酸;
(d)胸苷合成酶抑制剂;
(e)信号传递抑制剂;
(f)顺铂或铂类似物;
(g)烷化剂;
(h)抗微管蛋白剂;
(i)抗代谢物;
(j)小蘗碱;
(k)芹菜素;
(l)秋水仙素或秋水仙素的类似物;
(m)染料木黄酮;
(n)伊妥普赛;
(o)阿拉伯糖基胞嘧啶;
(p)喜树碱;
(q)长春花生物碱类;
(r)5-氟代尿嘧啶;
(s)姜黄素;
(t)NF-κB抑制剂;
(u)迷迭香酸;以及
(v)丙脒腙。
当所述改良是通过化学增效作用来进行的,所述化学增效作用可以包含但不限于结合使用一作为化学增效剂的烷基化己醣醇衍生物与一药剂,所述药剂是选自于由下列所组成的群组:
(a)伪核苷;
(b)伪核苷酸;
(c)胸苷合成酶抑制剂;
(d)信号传递抑制剂;
(e)顺铂或铂类似物;
(f)烷化剂;
(g)抗微管蛋白剂;
(h)抗代谢物;
(i)小蘗碱;
(j)芹菜素;
(k)秋水仙素或秋水仙素的类似物;
(l)染料木黄酮;
(m)伊妥普赛;
(n)阿拉伯糖基胞嘧啶;
(o)喜树碱类;
(p)长春花生物碱类;
(q)位置异构酶抑制剂;
(r)5-氟代尿嘧啶;
(s)姜黄素;
(t)NF-κB抑制剂;
(u)迷迭香酸;
(v)丙脒腙;以及
(w)一生物治疗物。
在一选择方案中,当所述化学增效作用涉及一通过烷基化己醣醇衍生物的活性的烷化剂的化学增效作用,所述烷化剂可以是选自由BCNU、BCNU 片(格立得)、CCNU、苯达莫司汀(Treanda)、环己亚硝脲、ACNU及替莫唑胺 (Temodar)所组成的群组。
当受到化学增效作用的所述药剂是一生物治疗物,所述生物治疗物可以是但不限于一选自由癌思停、贺癌平、利妥昔及爱必妥所组成的群组的生物治疗物。
当所述改良是通过后治疗管理来进行的,所述后治疗管理可以是但不限于一选自于由下列所组成的群组的方法:
(a)一与疼痛管理相关的疗法;
(b)营养支持;
(c)给予一止吐剂;
(d)一抗呕吐疗法;
(e)给予一抗发炎剂;
(f)给予一解热剂;以及
(g)给予一免疫促进剂。
当所述改良是通过选择性医疗/后治疗支持来进行的,所述选择性医疗/ 后治疗支持可以是但不限于一选自于由下列所组成的群组的方法:
(a)催眠;
(b)针灸;
(c)身心疗法;
(d)一通过合成或提取产生的草本药物;以及
(e)应用人体运动学。
在一选择方案中,当所述方法是一通过合成或提取产生的草本药物,所述通过合成或提取产生的草本药物可以是选自于由下列所组成的群组:
(a)一NF-κB抑制剂;
(b)一天然的抗发炎剂;
(c)一免疫促进剂;
(d)一抗微生物药;以及
(e)一类黄酮、异黄酮或黄酮。
当所述通过合成或提取产生的草本药物是一NF-κB抑制剂,所述NF- κB抑制剂可以是选自于由小白菊内酯、姜黄素及迷迭香酸所组成的群组。当所述通过合成或提取产生的草本药物是一天然的抗发炎剂,所述天然的抗发炎剂可以是选自于由大黄酸及小白菊内酯所组成的群组。当所述通过合成或提取产生的草本药物是一免疫促进剂,所述免疫促进剂可以是一于紫锥花中发现或从紫锥花分离出来的产物。当所述通过合成或提取产生的草本药物是一抗微生物药,所述抗微生物药可以是小蘗碱。当所述通过合成或提取产生的草本药物是一类黄酮或黄酮,所述类黄酮、异黄酮或黄酮可以是选自于由芹菜素、染料木黄酮、芹菜素(apigenenin)、染料木黄酮、染料木苷、6″-O- 丙二酰基染料木苷、6″-O-乙酰基染料木苷、大豆黄酮、大豆苷、6″-O-丙二酰基大豆苷、6″-O-乙酰基染料木苷、黄豆黄素、黄豆黄苷、6″-O-丙二酰基黄豆黄苷及6-O-乙酰基黄豆黄苷所组成的群组。
当所述改良是通过一原料药产品改良来进行的,所述原料药产品改良可以是但不限于一选自于由下列所组成的群组的原料药产品改良:
(a)盐类形成;
(b)作为一均匀晶体结构的调剂;
(c)作为一纯异构物的调剂;
(d)提高纯度;
(e)具有低残留溶剂含量的调剂;以及
(f)具有低残留溶剂含量的调剂。
当所述改良是通过使用一稀释剂来进行的,所述稀释剂可以是但不限于一选自于由下列所组成的群组的稀释剂:
(a)一乳化液;
(b)二甲亚砜(DMSO);
(c)N-甲基甲酰胺(NMF);
(d)二甲基甲酰胺(DMF);
(e)二甲基乙酰胺(DMA);
(f)乙醇;
(g)苯甲醇;
(h)用于注射的含有葡萄糖的水;
(i)聚氧乙烯蓖麻油;
(j)环糊精;以及
(k)PEG。
当所述改良是通过使用一溶剂系统来进行的,所述溶剂系统可以是但不限于一选自于由下列所组成的群组的溶剂系统:
(a)一乳化液;
(b)DMSO;
(c)NMF;
(d)DMF;
(e)DMA;
(f)乙醇;
(g)苯甲醇;
(h)用于注射的含有葡萄糖的水;
(i)聚氧乙烯蓖麻油;
(j)PEG;以及
(k)盐系统。
当所述改良是通过使用一赋形剂来进行的,所述赋形剂可以是但不限于一选自于由下列所组成的群组的赋形剂:
(a)甘露醇;
(b)白蛋白;
(c)EDTA;
(d)亚硫酸氢钠;
(e)苯甲醇;
(f)碳酸盐缓冲液;
(g)磷酸盐缓冲液;
(h)PEG;
(i)维生素A;
(j)维生素D;
(k)维生素E;
(l)酯酵素抑制剂;
(m)细胞色素P450抑制剂;
(n)多重抗药性(MDR)抑制剂;
(o)有机树脂;
(p)洗涤剂;
(q)紫苏醇或其类似物;以及
(r)通道形成受体的活化剂。
合适的酯酵素抑制剂包含但不限于厄比内酯A(ebelactone A)及厄比内酯 B。
合适的细胞色素P450抑制剂包含但不限于1-胺基苯并三唑、N-羟基 -N’-(4-丁基-2-甲苯基)甲脒、酮康唑、甲氧沙林(methoxsalen)、甲吡酮 (metyrapone)、异烟棒曲霉素C(roquefortine C)、普罗地芬(proadifen)、2,3’,4,5’- 四甲基二苯乙烯,以及桃霉素(troleandomycin)。
合适的MDR抑制剂包含但不限于5’–甲氧基大风子素、INF 240、INF 271、INF 277、INF 392、INF 55、蛇根碱(reserpine)及GG918。MDR抑制剂被描述于M.Zloh&S.Gibbons,“MDR9抑制剂的分子相似性”,Int.J.Mol. Sci.5:37-47(2004),并通过引用将其包括在内。
合适的有机树脂包含但不限于一部分中和的聚丙烯酸,正如透过Rodgers 等人于美国专利第8,158,616号中所描述的,并通过引用将其包括在内。
合适的洗涤剂包含但不限于非离子型洗涤剂,比如一聚山梨醇酯或一泊洛沙姆,且透过
用以改善抗肿瘤剂的传输的紫苏醇或其类似物抗肿瘤剂的使用透过 Chen等人被描述于美国专利申请第2012/0219541号,并通过引用将其包括在内。
通道形成受体的活化剂的使用透过Bean等人被描述于美国专利申请公开第2010/0311678号,并通过引用将其包括在内。这样的通道形成受体的活化剂包含但不限于番椒晶素(capsaicin)、利多卡因(lidocaine)、丁香油 (eugenol)、arvanil(N-花生四烯酰香草胺)、大麻素(anandamide)、2-胺基乙氧基二苯基硼酸盐、树酯毒素(resiniferatoxin)、佛波醇12-苯乙酸盐13-乙酸酯 20-高香草酸(PPAHV)、奥伐尼(olvanil)、N-油酰基多巴胺、N-花生四烯酸多巴胺、6’-碘代仙人掌毒素(6’-iodoresiniferatoxin)(6’-IRTX)、C
当所述改良是通过使用一剂型来进行的,所述剂型可以是但不限于一选自于由下列所组成的群组的剂型:
(a)片剂;
(b)胶囊;
(c)外用凝胶;
(d)外用药膏;
(e)贴剂;
(f)栓塞剂;
(g)冻干剂量填充物;
(h)速释制剂;
(i)缓释制剂;
(j)控释制剂;以及
(k)液体胶囊。
于片剂、胶囊及外用凝胶、外用药膏或栓塞剂中药物组合物的制剂于本领域中是众所周知的,其比如透过Griffin等人描述于美国专利申请公开第 2004/0023290号,并通过引用将其包括在内。
作为贴剂(比如透皮贴剂)的药物组合物的制剂于本领域中是众所周知的,其比如透过Eros等人描述于美国专利第7,728,042号,并通过引用将其包括在内。
冻干剂量填充物于本领域中也是众所周知的。一通常的用于制备这样的冻干剂量填充物的方法,适用于二溴卫矛醇及其衍生物,其包含下面步骤:
(1)溶解所述药物至用于注射的水中,且预冷至10℃以下,稀释至具有用于注射的冷水的最终体积,以产出40mg/mL溶液;
(2)在无菌状态下将所述主体溶液过滤通过0.2-μm滤纸至一接收容器中,所述制剂及过滤应在1小时内完成;
(3)将标称1.0mL过滤后溶液填入于消毒的小玻璃瓶中;
(4)填充后,将所有小玻璃瓶放置于“冻干位置”中所插入的橡皮塞,且加载至所述预冷却冻干机中,设置冻干机的搁板温度于+5℃且维持1小时;然后搁板温度调整至-5℃且维持1小时,并且设置冷凝器至-60℃,并且开启;
(5)然后将所述小玻璃瓶冻结到-30℃或以下且维持不低于3小时,通常是4 小时;
(6)然后开启真空,搁板温度调整至-5℃,且初级干燥8小时;将所述搁板温度再次调整至-5℃且进行干燥至少5小时;
(7)开启冷凝器(设定于-60℃)及真空后,开始二次干燥,在二次干燥中,搁板温度控制在+5℃、1~3小时(通常为1.5小时),接续在25度下进行1~3小时(通常为1.5小时),最后在35-40度下进行至少5小时(通常为9小时),或直到所述产物被完全干燥;
(8)以过滤的惰性气体(比如氮气)来破除真空,在冻干机中用瓶塞塞住所述小玻璃瓶;
(9)从冻干机腔室取出所述小玻璃瓶,且以铝翻盖关闭密封,目视检查且以批准的卷标标记所有小玻璃瓶。
速释制剂透过van Dalen等人被描述于美国专利第8,148,393号,并通过引用将其包括在内。速释制剂比如可以包含传统的膜衣锭。
缓释制剂透过Wen等人被描述于美国专利第8,178,125号,并通过引用将其包括在内。缓释制剂比如可以包含微乳化液或液晶。
控释制剂透过Oshlack等人被描述于美国专利第8,231,898号,并通过引用将其包括在内。控释制剂比如可以包含一控释材料的基质。这样的控释材料可以包含亲水性和/或疏水性材料,比如树胶、纤维素醚、丙烯树脂、蛋白衍生的材料、蜡、虫胶及油类(比如氢化蓖麻油或氢化植物油)。然而,根据本发明,任何能够赋予芥基烷化剂的控释的药学上可接受疏水性或亲水性控释材料可以被使用。优选的控释聚合物包含烷基纤维素,比如乙基纤维素、丙烯酸及甲基丙烯酸聚合物及共聚合物,以及纤维素醚,特别是羟烷基纤维素(比如,羟丙甲基纤维素)及羧烷基纤维素。优选的丙烯酸及甲基丙烯酸聚合物及共聚合物包含甲基丙烯酸甲酯、甲基丙烯酸甲酯共聚物、乙氧基乙基甲基丙烯酸酯、氰基乙基丙烯酸甲酯、胺烷基丙烯酸甲酯共聚合物、聚丙烯酸、聚甲基丙烯酸、甲基丙烯酸烷基胺共聚合物、聚甲基丙烯酸甲酯、聚甲基丙烯酸酐、聚丙烯酸甲酯、聚丙烯酰胺、聚甲基丙烯酸酸酐,以及环氧丙基丙烯酸甲酯共聚合物。
当所述改良是通过使用剂量套组及包装来进行的,所述剂量套组及包装可以是但不限于剂量套组及包装,所述剂量套组及包装是选自于由下列所组成的群组:使用避免光照的棕色瓶,以及使用用以增加储存寿命稳定性的具有专用涂层的瓶塞。
当所述改良是通过使用一药物传递系统来进行的,所述药物传递系统可以是但不限于一选自于由下列所组成的群组的药物传递系统:
(a)口服剂型;
(b)纳米晶体;
(c)纳米颗粒;
(d)共溶剂;
(e)浆体;
(f)糖浆;
(g)生物溶蚀型聚合物;
(h)脂质体;
(i)缓释注射凝胶;
(j)微球;以及
(k)具有表皮生长因子受体结合胜肽的靶向组合物。
纳米晶体透过Hovey等人被描述于美国专利第7,101,576号,并通过引用将其包括在内。
用于药物传递的纳米颗粒透过Bosch等人被描述于美国专利第8,258,132 号,并通过引用将其包括在内。通常,这样的纳米颗粒具有一小于约1000 纳米(nm)的活性成分的平均粒径,更优选地,小于约400nm,且最优选地,小于约250nm。所述纳米颗粒可以以表面稳定剂来覆膜,比如,明胶、酪蛋白、卵磷脂(phosphatides)、葡聚糖、阿拉伯胶、胆固醇、西黄耆胶、硬脂酸、氯苄烷、硬脂酸钙、单硬脂酸甘油酯、鲸蜡硬脂醇、聚乙二醇乳化蜡、山梨醇酐酯、聚氧乙烯烷基醚(比如像是聚乙二醇1000的聚乙二醇醚)、聚氧乙烯蓖麻油衍生物、聚氧乙烯山梨醇脂肪酸酯(比如市售的
药学上可接受共溶剂透过Navratil等人被描述于美国专利第8,207,195 号,并通过引用将其包括在内,且包含但不限于水、甲醇、乙醇、1-丙醇、异丙醇、1-丁醇、异丁醇、t-丁醇、丙酮、甲基乙基酮、乙睛、乙酸乙酯、苯、甲苯、二甲苯(s)、乙二醇、二氯甲烷、1,2-二氯乙烷、N-甲基甲酰胺、N,N- 二甲基甲酰胺、N-甲基乙酰胺、吡啶、二氧杂环,以及二乙基醚。
使用于药物制剂的浆体透过Laxminarayan被描述于美国专利申请公开第2006/0229277号,并通过引用将其包括在内。
使用于药物制剂的糖浆透过Stoit等人被描述于美国专利第8,252,930号,并通过引用将其包括在内。如此的糖浆可以包含所述活性成分及一糖浆类型组成(比如糖或糖醇)以及一乙醇、水、甘油、丙二醇及聚乙二醇的混合物。如果需要,这样的液体制剂可以包含着色剂、调味剂、防腐剂、糖精及羧甲基纤维素或其他增稠剂。
生物溶蚀型聚合物透过Okumu等人被描述于美国专利第7,318,931号,并通过引用将其包括在内。一生物溶蚀型聚合物分解放在里面的一生物体,其是以聚合物的分子量随着时间下降来测量。聚合物分子量可以通过多种包含粒径排阻层析法(size exclusionchromatography,SEC)的方法来测定,且一般表示为重量平均分子量或数量平均分子量。如果,当磷酸盐缓冲液(phosphate buffered saline,PBS)的pH值为7.4及温度为37℃时,聚合物为生物溶蚀型,通过SEC测量的六个月期间内,它的重量平均分子量被降低至少25%。有用的生物溶蚀型聚合物包含聚酯,比如聚己内酯、聚乙醇酸、聚乳酸及聚羟丁酸;聚酸酐,比如聚己二酸酐及聚马来酸酐;聚二氧六环酮;多胺类;聚酰胺;聚氨酯;聚酯酰胺;聚原酸酯;聚缩醛;聚缩酮;聚碳酸酯;聚原酸碳酸酯;聚膦腈;聚苹果酸;聚胺基酸;聚乙烯吡咯啶;聚甲基乙烯基乙醚;聚草酸亚烷基酯;聚丁二酸烷基酯;多羟基纤维素;几丁质;几丁聚糖;以及共聚和物及其混和物。
脂质体作为药物传递媒介物是众所周知的。脂质体的制备透过Weng等人被描述于欧洲专利申请公开第EP 1332755号,并通过引用将其包括在内。脂质体可以包含能够靶向所述EGFR受体的短寡肽序列,正如透过Huang等人于美国专利申请公开第2012/0213844号中所描述的,并通过引用将其包括在内。或者,脂质体可以包含核定位信号/融合肽共轭物且形成标靶脂质体复合物,正如透过Boulikas于美国专利申请公开第2012/0183596号中所描述的,并通过引用将其包括在内。
持续释放注射凝胶于本领域中为已知的,且比如被描述于B.Jeong等人,“从PEG-PLGA-PEG三嵌段共聚合物的可生物降解注射温感性水胶的药物释放”,J.ControlledRelease 63:155-163(2000),并通过引用将其包括在内。
用于药物传递的微球的使用于本领域中是已知的,且比如被描述于H. Okada&H.Taguchi,“于药物传递中生物分解性微球”,Crit.Rev.Ther.Drug Carrier Sys.12:1-99(1995),并通过引用将其包括在内。
具有表皮生长因子受体结合胜肽的靶向组合物的使用透过Trikha等人被描述于美国专利申请公开第2010/0151003号,并通过引用将其包括在内。
当所述改良是通过使用一药物共轭形式来进行的,所述药物共轭形式可以是但不限于一选自于由下列所组成的群组的药物共轭形式:
(a)一聚合物系统;
(b)聚乳酸;
(c)聚甘醇酸;
(d)胺基酸;
(e)胜肽;
(f)多价连接符;
(g)免疫球蛋白;
(h)环糊精聚合物;
(i)修饰运铁蛋白;
(j)疏水性聚合物或疏水-亲水性聚合物;
(k)具有一磷甲酸钠偏酯的共轭物;
(l)具有加入一带电交联剂的细胞粘合剂的共轭物;以及
(m)具有通过一连接剂的β-葡萄醛酸的共轭物。
聚乳酸共轭物于本领域中是众所周知的,且比如被描述于R.Tong&C. Cheng,“喜树碱-聚乳酸共轭物及纳米共轭物的可控合成”,Bioconjugate Chem.21:111-121(2010),通过引用将其包括。
聚甘醇酸共轭物于本领域中也是众所周知的,且比如透过Elmaleh等人被描述于PCT专利申请公开第WO 2003/070823号,并通过引用将其包括在内。
多价连接符于本领域中为已知的,其比如透过Silva等人描述于美国专利申请公开第2007/0207952号,并通过引用将其包括在内。比如,多价连接符可以包含一用于与反应性半胱胺酸及多个亲核基团(比如NH或OH)或亲电基团(比如活化酯)反应的喜硫基团,其允许多个生物活性部分连接至所述连接基。
具有免疫球蛋白的共轭物透过Oguchi等人被公开于美国专利第 4,925,662号,并通过引用将其包括在内。通过使用交联剂制备所述共轭物,所述交联剂比如为碳二酰亚胺、戊二醛,或二乙基丙二酰胺。
环糊精聚合物、它的具有治疗活性剂的共轭物,以及它的与颗粒一起的投药透过Fetzer被描述于美国专利申请公开第2012/0213854号,并通过引用将其包括在内。
具有修饰运铁蛋白的共轭物透过Kamei等人被描述于美国专利申请公开第2011/0288023号,并通过引用将其包括在内。
具有疏水性聚合物或疏水-亲水性聚合物的共轭物透过Crawford等人被描述于美国专利申请公开第2011/0268658号,并通过引用将其包括在内。这些聚合物可以包含单胜肽、双胜肽或三胜肽。这些聚合物还可以包含聚乳酸 (polylactic acid,PLA)、聚乙醇酸(polyglycolic acid,PGA)、聚乳酸聚甘醇酸 (poly(lactic-co-glycolic)acid,PLGA)、聚己内酯(polycaprolactone,PCL)、聚二氧六环酮(polydioxanone,PDO)、聚酸酐、聚原酸酯或几丁聚糖。
具有一磷甲酸钠偏酯的共轭物透过Saha等人被描述于美国专利申请公开第2010/227831号,并通过引用将其包括在内。
具有加入一带电交联剂的细胞粘合剂的共轭物透过Chari等人被描述于美国专利第8,236,319号,并通过引用将其包括在内。
具有通过一连接剂的β-葡萄醛酸的共轭物透过Jeffrey被描述于美国专利第8,039,273号,并通过引用将其包括在内。
用于交联许多官能基的组合的合适的试剂于本领域中为已知的。比如,亲电基团可以与许多官能基反应,包含存在于蛋白或多胜肽中的那些。反应性胺基酸及亲电体的各种组合于本领域中为已知的,且可以被使用。比如, N-端半胱胺酸,包含硫醇基,可以与卤素或马来酰亚胺反应。这些被描述于 G.T.Hermanson,“生物嫁接技术”(Academic Press,San Diego,1996),pp. 146-150,并通过引用将其包括在内。这些被描述于G.T.Hermanson,“生物嫁接技术”(Academic Press,San Diego,1996),pp.146-150,并通过引用将其包括在内。半胱胺酸残留物的反应活性可以通过适当选择邻近的胺基酸残基来优化。比如,邻近于半胱胺酸残留物的组胺酸残基将增加所述半胱胺酸残留物的反应活性。其他反应性胺基酸及亲电试剂(electrophilic reagents)的组合于本领域中为已知的。比如,马来酰亚胺可以与胺基反应,比如赖氨酸的侧链的ε-胺基,特别是在较高的pH值范围。芳基卤化物也可以与这样的胺基起反应。卤乙酰基衍生物可以与组胺酸的咪唑基侧链氮、甲硫胺酸的侧链的硫醚基团、及赖氨酸的侧链的.ε(epsilon).-胺基起反应。许多其他亲电试剂已知会与赖氨酸的侧链的ε-胺基起反应,包含但不限于异硫氰酸盐、异氰酸酯、酰基迭氮化物、羟基琥珀酰亚胺酯类、氯化磺酰、环氧化合物、环氧乙烷、碳酸盐、亚氨酸酯、碳二酰亚胺及酸酐。这些被描述于G.T.Hermanson,“生物嫁接技术”(Academic Press,San Diego,1996),pp.137-146,并通过引用将其包括在内。此外,亲电试剂已知会与比如为天冬胺酸盐及麸胺酸的那些的羧酸酯侧链起反应,比如重氮烷类及重氮乙酰基化合物、羰基二咪唑以及碳二酰亚胺。这些被描述于G.T.Hermanson,"生物嫁接技术"(Academic Press, San Diego,1996),pp.152-154,并通过引用将其包括在内。此外,亲电试剂已知会与比如为在丝氨酸及息宁胺酸的侧链中的那些的羟基团起反应,包含反应性卤代烷衍生物。这些被描述于G.T.Hermanson,“生物嫁接技术” (Academic Press,San Diego,1996),pp.154-158,并通过引用将其包括在内。在另一选择性实施例中,亲电体及亲核基(亦即一具有亲电体的分子活性)的相对位置被逆转,以致所述蛋白具有一具有亲电基团的胺基酸残基,所述亲电基团是与亲核基起反应,且所述靶向分子包含其中一亲核基团。这包含具有羟胺(亲核基)的醛类(亲电体)的反应,如上所述,但比其反应更普遍;其他基团可以被作为亲电体及亲核基。合适的基团在有机化学中是众所周知的而不需进一步的详细说明。
额外的用于交联的反应性基团的组合于本领域中为已知的。比如,胺基可以与异硫氰酸盐、异氰酸酯、酰基迭氮化物、N-羟基琥珀酰亚胺 (N-hydroxysuccinimide,NHS)酯类、氯化磺酰、醛类、乙二醛类、环氧化合物、环氧乙烷、碳酸盐、烷化剂、亚氨酸酯、碳二酰亚胺及酸酐起反应。硫醇基可以与卤乙酰基或烷基卤衍生物、马来酰亚胺、氮丙啶、丙烯酰基衍生物、酰化剂或其他的通过氧化及形成混合的二硫化物的硫醇基起反应。羧基团可以与重氮烷类、重氮乙酰基化合物、羰基二咪唑、碳二酰亚胺起反应。羟基团可以与环氧化合物、环氧乙烷、羰基二咪唑、N,N’-二琥珀酰亚胺基碳酸盐、N-羟基琥珀酰亚胺基氯甲酸盐、过碘酸盐(用于氧化)、烷基卤素或异氰酸酯起反应。醛基团及酮基团可以与起反应肼类、形成希夫碱的试剂及其他的在还原性胺化反应或曼尼希(Mannich)缩合反应中的基团。尽管如此,其他的适用于交联反应的反应于本领域中为已知的。这样的交联试剂及反应被描述于G.T.Hermanson,“生物嫁接技术”(Academic Press,San Diego,1996),并通过引用将其包括在内。
当所述改良是通过使用一化合物类似物来进行的,所述化合物类似物可以是但不限于一选自于由下列所组成的群组的化合物类似物:
(a)侧链的变动以增加或减少亲脂性;
(b)另一化学官能性的增加以改变一选自于由反应活性、电子亲和力及结合能力所组成的群组的性质;以及
(c)盐类型的变动。
当所述改良是通过使用一前驱物系统来进行的,所述前驱物系统可以是但不限于一选自于由下列所组成的群组的前驱物系统:
(a)使用酵素敏感酯类;
(b)使用二聚体;
(c)使用希夫碱;
(d)使用吡哆醛复合物;
(e)使用咖啡因复合物;
(f)使用一氧化氮释放前驱物;
(g)使用具有纤维原细胞激活蛋白α-可分裂寡肽的前驱物;
(h)使用与一乙酰化剂或一胺甲酰化剂反应的产物的前驱物;
(i)使用己酸盐共轭物的前驱物;
(j)使用聚合物-剂的共轭物的前驱物;以及
(k)使用受到氧化还原活化的前驱物。
如本文所使用的术语“前驱物”指的是化合物,所述化合物于体内转形而生产一公开的化合物或一药学上可接受形式的化合物。在一些实施例中,一前驱物是一可在生理条件下或通过对如本文所述的生物活性化合物进行溶剂分解所转换的化合物。因此,所述术语“前驱物”指的是一药学上可接受的生物活性化合物的前导物。当投药至一目标对象时,一前驱物可能是无活性的,但其接续会在体内转换成活性化合物,比如通过水解(比如在血液或组织中水解)。在某些情况下,一前驱物在一从所述前驱物衍生的原始化合物之上具有改善的物理和/或传递性质。在一哺乳生物中,所述前驱物经常提供溶解度、组织兼容性或缓效释放的优点(H.Bundgard,Design of Prodrugs (Elsevier,Amsterdam,1988),pp.7-9,21-24),并通过引用将其包括在内。前驱物的讨论被提供于T.Higuchi等人,“作为新颖传递系统的前药”,ACS Symposium Series,Vol.14且被提供于E.B.Roche,ed.,Bioreversible Carriers in Drug Design(American Pharmaceutical Association&Pergamon Press, 1987),这两者皆通过引用将其包括在内。一前驱物的典型优势可以包含但不限于它的物理性质,比如用于肠胃外投药的水溶解度增加(在相较于原始化合物的生理pH上)、增加消化道的吸收,或用于长期储存的药物稳定性的增加。
当给予所述前驱物至一目标对象时,所述术语“前驱物”还意指包含任何体内释放活性化合物的共价键结合载体。如本文所述的治疗活性化合物的前驱物可以通过修改存在于治疗活性化合物中的一个或多个官能基(这样的一裂解所述修改的方式中,无论是在常规操作中或在体内,以产生原始治疗活性化合物)来制备。前驱物包含化合物,其中羟基团、胺基团或硫醇基团共价键结至任何基团,当活性化合物的前驱物给予一目标对象时,所述基团分别切割(cleave)成游离羟基团、游离胺基团或游离硫醇基团。前驱物的例子包含但不限于一具有可供反应的胺官能基的治疗活性剂的乙醇或乙酰胺的甲酸盐或苯甲酸盐衍生物、甲酰胺或苯甲酰胺衍生物等等。
比如,如果治疗活性剂或治疗活性剂的药学上可接受形式包含一羧酸官能基,一前驱物可以包含一通过以一基团取代羧酸基团的氢原子所形成的酯类,所述基团比如为C
同样地,如果公开的化合物或所述化合物的药学上可接受形式包含一乙醇官能基,一前驱物可以通过乙醇基团的氢原子的取代而形成,所述乙醇基团具有一基团,其比如为(C
如果一已公开的化合物或一药学上可接受形式的化合物包含一胺官能基,在具有一比如为R-羰基、RO-羰基、NRR′-羰基的基团的胺基中,一前驱物可以通过氢原子的取代而形成,其中R及R′各自地为(C
前驱物系统的使用被描述于T.
当所述改良是通过使用一多重药物系统来进行的,所述多重药物系统可以是但不限于一选自于由下列所组成的群组的多重药物系统:
(a)多重抗药性的抑制剂;
(b)特异的抗药性抑制剂;
(c)特异的选择性酵素的抑制剂;
(d)信号传递抑制剂;
(e)甲异靛;
(f)伊马替尼;
(g)羟基尿素;
(h)达沙替尼;
(i)卡匹他滨;
(j)尼罗替尼;
(k)修复抑制剂;以及
(l)具有非重迭副作用的位置异构酶抑制剂。
多重抗药性抑制剂透过Inomata等人被描述于美国专利第6,011,069号,并通过引用将其包括在内。
特异的抗药性抑制剂被描述于T.Hideshima等人,“在人类多发性骨髓瘤细胞中蛋白酶体抑制剂PS-341抑制生长、诱导细胞凋亡及克服抗药性”, Cancer Res.61:3071-3076(2001),并通过引用将其包括在内。
特定酵素的选择性抑制剂被描述于D.Leung等人,”于复杂的蛋白质体中发现有效力的及选择性可逆型酵素抑制剂”,Nature Biotechnol.21:687-691 (2003),并通过引用将其包括在内。
抑制作用被描述于N.M.Martin,“DNA抑制作用及癌症疗法”,J.Photochem.Photobiol.B 63:162-170(2001),并通过引用将其包括在内。
当所述改良是通过生物治疗加强作用来进行的,所述生物治疗加强作用可以通过结合使用敏化剂/增效剂与一治疗试剂或技术而完成的,所述治疗试剂或技术是选自于由下列所组成的群组:
(a)生物反应修饰物;
(b)细胞激素;
(c)治疗抗体;
(d)治疗抗体;
(e)反义治疗;
(f)基因治疗;
(g)核醣酶;以及
(h)RNA干扰。
生物反应修饰物被描述于T.E.G.K.Murthy等人,“生物反应修饰物”,Int.J.Pharmtech Res.2:2152-2160(2010),并通过引用将其包括在内。
反义治疗比如被描述于B.Weiss等人,“用于研究及调节生物过程的反义RNA基因疗法(Antisense RNA Gene Therapy)”,Cell.Mol.Life Sci.55: 334-358(1999),并通过引用将其包括在内。
核醣酶比如被描述于S.Pascolo,“基于RNA的治疗”in Drug Discovery Handbook(S.C.Gad,ed.,Wiley-Interscience,Hoboken,NJ,2005),ch.27,pp. 1273-1278,并通过引用将其包括在内。
RNA干扰比如被描述于S.Pascolo,“基于RNA的治疗”in Drug DiscoveryHandbook(S.C.Gad,ed.,Wiley-Interscience,Hoboken,NJ,2005), ch.27,pp.1278-1283,并通过引用将其包括在内。
当所述生物治疗加强作用是结合使用具有治疗抗体的敏化剂/增效剂,所述治疗抗体可以是但不限于一治疗抗体,所述治疗抗体选自于由贝伐单抗 (bevacizumab)(癌思停)、利妥昔单抗(rituximab)(利妥昔)、曲妥单抗(贺癌平) 及西妥昔(爱必妥)所组成的群组。
当所述改良是通过使用抗生物治疗调控来进行的,所述抗生物治疗调控可以是但不限于用来抵抗还耐一治疗试剂或技术的抗TKI肿瘤,所述治疗试剂或技术是选自于由下列所组成的群组:
(a)生物反应修饰物;
(b)细胞激素;
(c)治疗抗体;
(d)治疗抗体;
(e)反义治疗;
(f)基因治疗;
(g)核醣酶;以及
(h)RNA干扰。
在另一选择方案中,当所述改良是通过使用抗生物治疗调控来进行的,所述抗生物治疗调控可以是但不限于用来抵抗还耐一治疗试剂或技术的与 AHI1基因的突变或异常调控相关的恶性肿瘤,所述治疗试剂或技术是选自于由下列所组成的群组:
(a)生物反应修饰物;
(b)细胞激素;
(c)治疗抗体;
(d)治疗抗体;
(e)反义治疗;
(f)基因治疗;
(g)核醣酶;以及
(h)RNA干扰。
当所述抗生物治疗调控是用来抵抗耐治疗抗体的肿瘤,所述治疗抗体可以是但不限于一治疗抗体,所述治疗抗体是选自于由贝伐单抗(癌思停)、利妥昔单抗(利妥昔)、曲妥单抗(贺癌平)及西妥昔(爱必妥)所组成的群组。
当所述改良是通过增强放射线疗法来进行的,所述放射线疗法的增强可以是但不限于一选自于由下列所组成的群组的放射线疗法增强试剂或技术: (a)使用低氧敏化剂;
(b)使用辐射敏化剂/保护剂;
(c)使用光敏化剂;
(d)使用辐射修复抑制剂;
(e)使用硫醇消耗剂;
(f)使用血管标靶剂;
(g)使用DNA修复抑制剂;
(h)使用放射株;
(i)使用放射性核种;
(j)使用放射性标帜抗体;以及
(k)使用近接疗法。
一烷基化己醣醇衍生物可以与用于治疗一抗TKI肿瘤的辐射一起组合使用。
低氧敏化剂被描述于C.C.Ling等人,“在不同辐射剂量率上低氧敏化剂的影响”,Radiation Res.109:396-406(1987),并通过引用将其包括在内。辐射敏化剂被描述于T.S.Lawrence,“辐射敏化剂及标靶治疗”,Oncology 17 (Suppl.13)23-28(2003),并通过引用将其包括在内。辐射保护剂被描述于S.B. Vuyyuri等人,“作为一新颖的用于预防粘膜炎的口服辐射保护剂的D-甲硫胺酸的评价”,Clin.Cancer Res.14:2161-2170(2008),并通过引用将其包括在内。光敏化剂被描述于R.R.Allison及C.H.Sibata,“肿瘤光动力治疗光敏剂:一临床回顾”,Photodiagnosis Photodynamic Ther.7:61-75(2010),并通过引用将其包括在内。辐射修复抑制剂及DNA修复抑制剂被描述于M. Hingorani等人,“辐射诱发DNA损伤的修复提高从复制缺陷型腺病毒载体的表现的评价”,Cancer Res.68:9771-9778(2008),并通过引用将其包括在内。硫醇消耗剂被描述于K.D.Held等人,“通过硫醇消耗剂富马酸二甲酯的哺乳动物细胞的辐照后敏化作用”,Radiation Res.127:75-80(1991),并通过引用将其包括在内。血管标靶剂被描述于A.L.Seynhaeve等人,“在有助于更好的肿瘤反应的鼠类黑色素瘤中脂质体的肿瘤坏死因子α介导均匀分布”,Cancer Res.67:9455-9462(2007)。
当所述改良是通过一新颖作用机制而进行的,所述新颖作用机制可以是但不限于一具有标靶或机制的治疗交互作用的新颖作用机制,所述标靶或机制是选自于由下列所组成的群组:
(a)聚ADP核糖聚合酶的抑制剂;
(b)影响血管的药剂;
(c)促进血管扩张的药剂;
(d)致癌基因靶向药物;
(e)信号传递抑制剂;
(f)引起EGFR抑制作用的药剂;
(g)引起蛋白激酶C抑制的药剂;
(h)引起磷脂酶C递减调节的药剂;
(i)引起jun递减调节的药剂;
(j)调节组织蛋白基因的表现的药剂;
(k)调节VEGF的表现的药剂;
(l)调节鸟胺酸脱羧酶的表现的药剂;
(m)调节jun D的表现的药剂;
(n)调节v-jun的表现的药剂;
(o)调节GPCRs的表现的药剂;
(p)调节蛋白激酶A的表现的药剂;
(q)调节蛋白激酶(除了蛋白激酶A)的表现的药剂;
(r)调节端粒酶的表现的药剂;
(s)调节摄护腺特殊基因的表现的药剂;以及
(t)调节组蛋白去乙酰酶的表现的药剂。
聚ADP核糖聚合酶的抑制剂包含veliparib(ABT-888)、AGO14699、 iniparib(BSI-201)、卡铂普来锭、吉西他滨、INO-1001、MK4827、烟碱酰胺、奥拉帕尼(olaparib)、紫杉醇、替莫唑胺及拓扑替康,且被描述于E.A.Comen 及M.Robson,“作为用于乳癌的治疗策略的聚ADP核糖聚合酶的抑制剂”, Oncology 24:55-62(2010),并通过引用将其包括在内。促进血管扩张的药剂包含左西孟旦(Levosimendan),且描述于W.G.Toller等人,“左西孟旦,一种新的影响肌收缩力的及血管扩张剂”,Anesthesiology 104:556-569(2006),并通过引用将其包括在内。EGFR抑制作用被描述于G.Giaccone及J.A. Rodriguez,“EGFR抑制剂:我们从肺癌治疗中学到了什么”,Nat.Clin.Pract. Oncol.11:554-561(2005),并通过引用将其包括在内。蛋白激酶C抑制被描述于H.C.Swannie及S.B.Kaye,“蛋白激酶C抑制剂”,Curr.Oncol.Rep.4: 37-46(2002),并通过引用将其包括在内。磷脂酶C递减调节被描述于A.M. Martelli等人,“于弗里德(Friend)细胞的核中磷酸肌醇的信号传递:磷脂酶Cβ递减调节与细胞分化是有关的”,Cancer Res.54:2536-2540(1994),并通过引用将其包括在内。Jun的递减调节(特别是c-Jun)被描述于A.A.P.Zada等人,“在CD44聚合后的急性骨髓性白血病细胞中c-Jun表现及细胞周期调节分子的递减调节”,Oncogene 22:2296-2308(2003),并通过引用将其包括在内。作为用于治疗性干预的标靶的组蛋白基因的作用被描述于B.Calabretta等人,“在人类恶性肿瘤骨髓细胞中G1-特定基因的变异表现”,Proc.Natl.Acad. Sci.USA 83:1495-1498(1986),并通过引用将其包括在内。作为用于治疗性预防的标靶的VEGF的作用被描述于A.Zielke等人,“在试验性肿瘤中人类嗜铬性细胞瘤的VEGF媒介血管生成是有关于恶性肿瘤且通过抗VEGF抗体而抑制”,Surgery 132:1056-1063(2002),并通过引用将其包括在内。作为用于治疗性预防的标靶的鸟胺酸脱羧酶的作用被描述于J.A.Nilsson等人,“于 Myc诱导的淋巴癌中靶向鸟胺酸脱羧酶预防肿瘤形成”,Cancer Cell 7: 433-444(2005),并通过引用将其包括在内。作为用于治疗性预防的标靶的泛素C的作用被描述于C.Aghajanian等人,“在晚期实质肿瘤恶性肿瘤中新颖蛋白酶体抑制剂PS341的I期临床试验”,Clin.Cancer Res.8:2505-2511 (2002),并通过引用将其包括在内。作为用于治疗性预防的标靶的Jun D的作用被描述于M.M.Caffarel等人,“在人类乳癌细胞上9-四氢大麻酚的抗增生效应中所涉及到的JunD”,Oncogene 27:5033-5044(2008),并通过引用将其包括在内。作为用于治疗性预防的标靶的v-Jun的作用被描述于M.Gao等人,“v-Jun及c-Jun的差别及拮抗作用”,Cancer Res.56:4229-4235(1996),并通过引用将其包括在内。作为用于治疗性预防的标靶的蛋白激酶A的作用被描述于P.C.Gordge等人,“在恶性肿瘤与正常乳房组织相比中蛋白激酶A 及蛋白激酶C的上升”,Eur.J.Cancer12:2120-2126(1996),并通过引用将其包括在内。作为用于治疗性预防的标靶的端粒酶的作用被描述于E.K. Parkinson等人,“端粒酶作为用于癌症化学疗法的新颖及潜在选择性标靶”, Ann.Med.35:466-475(2003),并通过引用将其包括在内。作为用于治疗性预防的标靶的组蛋白去乙酰酶的作用被描述于A.Melnick及J.D.Licht,“在血液恶性肿瘤中作为治疗标靶的组蛋白去乙酰酶”,Curr.Opin.Hematol.9: 322-332(2002),并通过引用将其包括在内。
当所述改良是通过使用选择性靶细胞群疗法来进行的,所述选择性靶细胞群疗法的使用可以是但不限于一选自于由下列所组成的群组的使用:
(a)用来抵抗辐射敏感细胞;
(b)用来抵抗辐射抗性细胞;以及
(c)用来抵抗能量耗尽细胞。
所述改良也能通过使用一具有电离辐射的烷基化组合来进行。
当所述改良是通过使用一用以提高烷基化己醣醇衍生物的活性的药剂来进行的,且所述用以提高所述烷基化己醣醇衍生物的活性的药剂可以是但不限于一选自于由下列所组成的群组的药剂:
(a)烟碱酰胺;
(b)咖啡因;
(c)粉防己碱;以及
(d)小蘗碱。
如上文所述,鉴于这些发现,所述AHI1基因的突变、过度表现或失调与许多恶性肿瘤是相关的,包含慢性骨髓细胞性白血病,一烷基化己醣醇衍生物还可以与一调节所述AHI1基因或所述AHI1蛋白的表现或活性的药剂一起使用。在此选择方案中,所述方法进一步包含:给予一调节所述AHI1基因或所述AHI1蛋白的表现或活性的药剂的治疗有效量。
在一选择方案中,调节AHI1基因或AHI1蛋白的表现或活性的所述药剂是一调节AHI1基因的表现的药剂。在所述AHI1基因的转录的水平上或在所述AHI1基因的转译的水平上,可以进行AHI1的表现的调控。
在一些实施例中,抑制性核苷酸是用来调节AHI1表现。这些包含短干扰RNA(shortinterfering RNA,siRNA)、微RNA(microRNA,miRNA)及合成发夹型RNA(synthetic hairpinRNA,shRNA);反义核酸;或互补 DNA(complementary DNA,cDNA)。在一些优选的实施例中,使用一siRNA 靶向AHI1表现。具有功能的干扰及通过双股RNA(比如siRNA)的内源性基因的表现已被证明在各种生物体中。比如请见,A.Fire等人,“在扁形虫 (Caenorhabditiselegans)中的通过双股RNA的有效及特殊的基因干扰”, Nature 391:806-811(1998);J.R.Kennerdell及R.W.Carthew,“dsDNA媒介基因干扰的使用以证明卷曲(frizzled)及卷曲2在无翅(Wingless)途径方面行动”, Cell 95:1017-1026(1998);F.Wianni及M.Zernicka-Goetz,“在早期小鼠发展中的具有通过双股RNA的基因功能的特殊干扰”,Nat.Cell Biol.2:70-75 (2000)。siRNAs可以包含含有自身互补序列或双股序列的发夹循环。siRNAs 通常具有少于100个碱基对及可以比如是大约30bps或更短,且可以通过本领域已知的方法来制造,包含使用互补DNA股或合成途径。这样的双股RNA 可以通过从模板的两个方向所读取的单股RNA的体外转录及正义及反义 RNA股的体外退火来合成。在以反向重复序列分隔的相反方向中,双股RNA 靶向AHI1还可以从cDNA载体构筑质体中选殖AHI1基因来合成。接着细胞转染,所述RNA被转录及互补股再粘合。双股RNA靶向AHI1基因可以通过适当的构筑质体的转染而引入至细胞(比如一白血病细胞或其他的肿瘤细胞)中。通常,在转译的水平上,介导siRNA、miRNA或shRNA所介导的 RNA干扰;换句话说,这些干扰RNA分子防止相应的mRNA分子的转译,而导致它们的降解。RNA干扰还可以在转录的水平上操作,阻止相对于这些干扰RNA分子的基因组的区域的转录。这些干扰RNA分子的结构和功能于本领域中是众所周知的,且比如被描述于R.F.Gesteland等人,eds,“RNA 世界”(3
对于这些方法,选殖至载体中及转染方法于本领域中也是众所周知的,且比如被描述于J.Sambrook及D.R.Russell,“分子选殖:实验室手册”(3
除了双股RNAs以外,其他的核酸试剂标靶AHI1还可以被使用在本发明的方法中,比如反义核酸。反义核酸是互补于特定标靶mRNA分子的至少一部分的DNA或RNA分子。在所述细胞中,所述单股反义分子杂交至那 mRNA,而形成一双股分子。在此双股形式中,所述细胞没有转译一mRNA。因此,反义核酸干扰mRNA至蛋白中的转译,并且,因此,具有一转录至那mRNA中的基因的表现。反义方法于体外已被用来抑制许多基因的表现。比如请见,C.J.Marcus-Sekura,“使用反义寡脱氧核醣核苷酸至研究基因表现的技术”,Anal.Biochem.172:289-295(1988);J.E.Hambor等人,“在人类细胞毒性T-细胞表现株中的用于反义RNA媒介基因抑制的EB病毒(Epstein-Barr Virus)游离态复制子的使用”,Proc.Natl.Acad.Sci.U.S.A.85:4010-4014 (1988);H Arima等人,“在通过硫代磷酸盐反义寡核苷酸的鼠类类巨噬细胞中介白质(Interleukin)-10产物的具体抑制”,AntisenseNucl.Acid Drug Dev. 8:319-327(1998);以及W.-F.Hou等人,“在PC12细胞的增殖及分化上反义脱氧寡核苷酸定向至个体调钙素基因转录体的影响”,Antisense Nucl.Acid DrugDev.8:295-308(1998),以上全部皆通过引用将其包括在内。反义技术进一步被描述于C.Lichtenstein及W.Nellen,eds.,“反义技术:一实用方法” (IRL Press,Oxford,1997),并通过引用将其包括在内。
AHI1核苷酸序列,比如源自于RNA的这些,其于本领域中为已知的,且包含纪录于基因库中NM_001134830.1、NM_001134831.1、 NM_001134832.1及NM_017651.4的基因序列;这些序列代表转录变体。基于已知的序列,使用本领域众所周知的方法,抑制性核苷酸(比如siRNA、 miRNA或sRNA)靶向PAR1可以容易地被合成。本发明示例性的siRNAs可以有29bps、25bps、22bps、21bps、20bps、15bps、10bps、5bps或任何这些数字之间的碱基对的整数。用于设计最优选抑制性siRNAs的工具包含可以从DNAengine Inc.(Seattle,WA)及Ambion,Inc.(Austin,TX)。
作为另一选择方案,所述抑制所述AHI1基因或所述AHI1蛋白的表现或活性的药剂可以是一抑制AHI1蛋白的活性的药剂。比如,这样的一药剂可以是一结合所述AHI1蛋白或一或多个特异地结合至所述AHI1蛋白的受体的抗体。这样抗体的制备于本领域中是众所周知的,且不需要进一步描述。一般地,尽管IgG抗体通常是优选的,但本发明抗体可以是比如为IgG、IgA、 IgD、IgE、IgM或IgY的任何一类。抗体可以是任何哺乳动物或鸟类起源,包含人类、鼠类(小鼠或大鼠)、驴、绵羊、山羊、兔、骆驼、马或鸡。在相同的选择方案中,所述抗体可以是双特异性。所述抗体可以通过任何类型的分子至所述抗体的共价连接来修改。比如,但不以此方式限制,所述抗体衍生物包含已被修改的抗体,比如,通过糖基化、乙酰化、聚乙二醇化、磷酸化、酰胺化、透过已知的防护/阻隔基团、蛋白酶切割、连接至一细胞配体或其他蛋白或者其他本领域已知的修改方式的衍生化。单株抗体可以使用各种本领域已知的技术来制备,包含融合瘤、重组体及噬菌体展示技术或其组合的使用。比如,单株抗体可以使用包含本领域已知及教导的那些的融合瘤技术来生产,比如,于Harlow等人,“抗体:一实验室手册”,(Cold Spring Harbor Laboratory Press,2nd ed.1988);Hammerling等人,单株抗体及T细胞融合瘤 563-681(Elsevier,N.Y.,1981)或通过其他本领域已知的标准方法。在此使用的术语“单株抗体”并不局限于通过融合瘤技术所生产的抗体。所述术语“单株抗体”指的是一来自于单一表现株的抗体,所述表现株包含任何真核的、原核的或噬菌体表现株,而不是通过其所生产的方法。比如,合适的抗体可以通过噬菌体展示或其他技术来生产。此外,而不以此方式限制,人类抗体可通过各种技术来制造,包含使用抗体库的噬菌体展示法,所述抗体库来自于人类免疫球蛋白序列且通过使用基因转殖小鼠,所述基因转殖小鼠不能表现功能的内源性免疫球蛋白,但其可以表达人类免疫球蛋白基因复合物。比如,人类重链及轻链免疫球蛋白基因复合物可以被随机地引入或通过同源重组至小鼠胚胎干细胞中。所述抗体也可以通过多核苷酸的表现编码这些抗体来生产。此外,本发明的抗体可以被稠合至标记序列,比如一为方便纯化的胜肽标记;合适的标记是一六组胺酸标记。所述抗体也可以通过本领域已知的方法而共轭至一诊断或治疗试剂。制备这样的共轭物的技术于本领域中是众所周知的。
其他这些单株抗体以及嵌合抗体、拟人化抗体及单链抗体的制备方法,其于本领域中为已知的。一般地,优选是投药至人类患者、人类抗体、嵌合抗体或拟人化抗体。
在另一选择方案中,调节AHI1蛋白的活性的所述药剂是一特异地结合所述AHI1蛋白的受体的拮抗物,包含血清素受体2C,但本发明并不局限于此。
在又一选择方案中,调节AHI1蛋白的活性的所述药剂是一特异地结合 AHI1蛋白的受体的抗体,包含血清素受体2C,但本发明并不局限于此。
在又一选择方案中,调节AHI1蛋白的活性的所述药剂是一抑制具有于 AHI1基因启动子中的结合位置的转录因子的药剂,包含RFX1、POU2F1、 POU2F1a、Nkx2-5、Evi-1及RSRFC4,但本发明并不局限于此。
本发明的另一方面是提供一种用以使不理想给药疗法的功效增加和/或副作用减少的组合物,所述不理想给药疗法为使用一烷基化己醣醇衍生物用于治疗一抗TKI肿瘤或一与AHI1基因的异常调控或突变相关的恶性肿瘤,所述组合物包含一选自于由下列所组成的群组的选择方案:
(i)一修饰烷基化己醣醇衍生物或者一烷基化己醣醇衍生物或一修饰烷基化己醣醇衍生物的衍生物、类似物或前驱物的治疗有效量,其中相较于一未修饰烷基化己醣醇衍生物,所述修饰烷基化己醣醇衍生物或者所述修饰烷基化己醣醇衍生物的衍生物、类似物或前驱物具有对于一抗TKI肿瘤或与AHI1 基因的异常调控或突变相关的恶性肿瘤的治疗增加疗效或减少副作用;
(ii)一组合物包含:
(a)一烷基化己醣醇衍生物、一修饰烷基化己醣醇衍生物或者一烷基化己醣醇衍生物或一修饰烷基化己醣醇衍生物的衍生物、类似物或前驱物的治疗有效量;以及
(b)至少一附加治疗试剂、受到化学敏化作用的治疗试剂、受到化学增效作用的治疗试剂、稀释剂、赋形剂、溶剂系统或药物传递系统,其中相较于一未修饰烷基化己醣醇衍生物,所述组合物具有对于一抗TKI肿瘤或与AHI1基因的异常调控或突变相关的恶性肿瘤的治疗增加疗效或减少副作用;
(iii)并入于一剂型之中的一烷基化己醣醇衍生物、一修饰烷基化己醣醇衍生物或者一烷基化己醣醇衍生物或一修饰烷基化己醣醇衍生物的衍生物、类似物或前驱物的治疗有效量,其中相较于一未修饰烷基化己醣醇衍生物,并入于所述剂型之中的一烷基化己醣醇衍生物、一修饰烷基化己醣醇衍生物或者一烷基化己醣醇衍生物或一修饰烷基化己醣醇衍生物的衍生物、类似物或前驱物具有对于一抗TKI肿瘤或与AHI1基因的异常调控或突变相关的恶性肿瘤的治疗增加疗效或减少副作用;
(iv)并入于一剂量套组及包装之中的一烷基化己醣醇衍生物、一修饰烷基化己醣醇衍生物或者一烷基化己醣醇衍生物或一修饰烷基化己醣醇衍生物的衍生物、类似物或前驱物的治疗有效量,其中相较于一未修饰烷基化己醣醇衍生物,并入于所述剂量套组及包装之中的一烷基化己醣醇衍生物、一修饰烷基化己醣醇衍生物或者一烷基化己醣醇衍生物或一修饰烷基化己醣醇衍生物的衍生物、类似物或前驱物具有对于一抗TKI肿瘤或与AHI1基因的异常调控或突变相关的恶性肿瘤的治疗增加疗效或减少副作用;以及
(v)受到一原料药产品改良的一烷基化己醣醇衍生物、一修饰烷基化己醣醇衍生物或者一烷基化己醣醇衍生物或一修饰烷基化己醣醇衍生物的衍生物、类似物或前驱物的治疗有效量,其中相较于一未修饰烷基化己醣醇衍生物,受到所述原料药产品改良的所述烷基化己醣醇衍生物、一修饰烷基化己醣醇衍生物或者一烷基化己醣醇衍生物或一修饰烷基化己醣醇衍生物的衍生物、类似物或前驱物具有对于一抗TKI肿瘤或与AHI1基因的异常调控或突变相关的恶性肿瘤的治疗增加疗效或减少副作用。
如上所述,所述烷基化己醣醇衍生物可以是但不限于卫康醇、卫康醇的衍生物或类似物、二乙酰二脱水卫矛醇、二乙酰二脱水卫矛醇的衍生物或类似物、二溴卫矛醇,或二溴卫矛醇的衍生物或类似物。
在一选择方案中,所述组合物具有对于一抗TKI肿瘤的治疗增加疗效或减少副作用。
在一选择方案中,所述组合物具有对于与AHI1基因的异常调控或突变相关的恶性肿瘤的治疗增加疗效或减少副作用。
在一选择方案中,所述组合物包含一并用药,包含:
(i)一烷基化己醣醇衍生物、一修饰烷基化己醣醇衍生物或者一烷基化己醣醇衍生物或一修饰烷基化己醣醇衍生物的衍生物、类似物或前驱物;以及
(ii)一附加治疗试剂,其选自于由下列所组成的群组:
(a)位置异构酶抑制剂;
(b)伪核苷;
(c)伪核苷酸;
(d)胸苷合成酶抑制剂;
(e)信号传递抑制剂;
(f)顺铂或铂类似物;
(g)烷化剂;
(h)抗微管蛋白剂;
(i)抗代谢物;
(j)小蘗碱;
(k)芹菜素;
(l)氨萘非特;
(m)长春花生物碱类;
(n)5-氟代尿嘧啶;
(o)姜黄素;
(p)NF-κB抑制剂;
(q)迷迭香酸;
(r)丙脒腙;
(s)粉防己碱;
(t)一JAK2抑制剂;
(u)一STAT5抑制剂;以及
(v)一Src抑制剂。
在这些选择方案中,当所述附加治疗试剂是一烷化剂,所述烷化剂可以是但不限于一选自由BCNU、BCNU片、CCNU、苯达莫司汀(Treanda)及替莫唑胺(Temodar)所组成的群组的烷化剂。
在另一选择方案中,所述组合物包含一并用药,包含:
(i)一烷基化己醣醇衍生物、一修饰烷基化己醣醇衍生物或者一烷基化己醣醇衍生物或一修饰烷基化己醣醇衍生物的衍生物、类似物或前驱物;以及
(ii)一BH3类似物。
在另一选择方案中,所述组合物包含一并用药,包含:
(i)一烷基化己醣醇衍生物、一修饰烷基化己醣醇衍生物或者一烷基化己醣醇衍生物或一修饰烷基化己醣醇衍生物的衍生物、类似物或前驱物;以及
(ii)一调节AHI1基因的表现或调节AHI1蛋白的活性的药剂。
在又一选择方案中,所述组合物包含一并用药,包含:
(i)一烷基化己醣醇衍生物、一修饰烷基化己醣醇衍生物或者一烷基化己醣醇衍生物或一修饰烷基化己醣醇衍生物的衍生物、类似物或前驱物;
(ii)一调节AHI1基因的表现或AHI1蛋白的活性的药剂;以及
(iii)一BH3类似物。
在另一选择方案中,所述组合物包含:
(i)一烷基化己醣醇衍生物、一修饰烷基化己醣醇衍生物或者一烷基化己醣醇衍生物或一修饰烷基化己醣醇衍生物的衍生物、类似物或前驱物;以及
(ii)一受到化学敏化作用的治疗试剂,其选自于由下列所组成的群组:
(a)位置异构酶抑制剂;
(b)伪核苷;
(c)伪核苷酸;
(d)胸苷合成酶抑制剂;
(e)信号传递抑制剂;
(f)顺铂或铂类似物;
(g)烷化剂;
(h)抗微管蛋白剂;
(i)抗代谢物;
(j)小蘗碱;
(k)芹菜素;
(l)秋水仙素或秋水仙素的类似物;
(m)染料木黄酮;
(n)伊妥普赛;
(o)阿拉伯糖基胞嘧啶;
(p)喜树碱;
(q)长春花生物碱类;
(r)5-氟代尿嘧啶;
(s)姜黄素;
(t)NF-κB抑制剂;
(u)迷迭香酸;以及
(v)丙脒腙,其中所述烷基化己醣醇衍生物、一修饰烷基化己醣醇衍生物或者一烷基化己醣醇衍生物或一修饰烷基化己醣醇衍生物的衍生物、类似物或前驱物作为一化学致敏剂。
在又一选择方案中,所述组合物包含:
(i)一烷基化己醣醇衍生物、一修饰烷基化己醣醇衍生物或者一烷基化己醣醇衍生物或一修饰烷基化己醣醇衍生物的衍生物、类似物或前驱物;以及
(ii)一受到化学增效作用的治疗试剂,其选自于由下列所组成的群组:
(a)伪核苷;
(b)伪核苷酸;
(c)胸苷合成酶抑制剂;
(d)信号传递抑制剂;
(e)顺铂或铂类似物;
(f)烷化剂;
(g)抗微管蛋白剂;
(h)抗代谢物;
(i)小蘗碱;
(j)芹菜素;
(k)秋水仙素或秋水仙素的类似物;
(l)染料木黄酮;
(m)伊妥普赛;
(n)阿拉伯糖基胞嘧啶;
(o)喜树碱类;
(p)长春花生物碱类;
(q)位置异构酶抑制剂;
(r)5-氟代尿嘧啶;
(s)姜黄素;
(t)NF-κB抑制剂;
(u)迷迭香酸;
(v)丙脒腙;以及
(w)一生物治疗物。
其中所述烷基化己醣醇衍生物、一修饰烷基化己醣醇衍生物或者一烷基化己醣醇衍生物或一修饰烷基化己醣醇衍生物的衍生物、类似物或前驱物作为一化学增效剂。
在这些选择方案中,其中所述附加治疗试剂是一生物治疗物,所述生物治疗物可以是但不限于一选自由癌思停、贺癌平、利妥昔及爱必妥所组成的群组的生物治疗物。
在又一选择方案中,所述烷基化己醣醇衍生物、一修饰烷基化己醣醇衍生物或者一烷基化己醣醇衍生物或一修饰烷基化己醣醇衍生物的衍生物、类似物或前驱物受到一原料药产品改良,其中所述原料药产品改良是选自于由下列所组成的群组:
(a)盐类形成;
(b)作为一均匀晶体结构的调剂;
(c)作为一纯异构物的调剂;
(d)提高纯度;
(e)具有低残留溶剂含量的调剂;以及
(f)具有低残留溶剂含量的调剂。
在又一选择方案中,所述组合物包含:一烷基化己醣醇衍生物、一修饰烷基化己醣醇衍生物或者一烷基化己醣醇衍生物或一修饰烷基化己醣醇衍生物的衍生物、类似物或前驱物;以及一稀释剂,其中所述稀释剂是选自于由下列所组成的群组:
(a)一乳化液;
(b)二甲亚砜(DMSO);
(c)N-甲基甲酰胺(NMF);
(d)二甲基甲酰胺(DMF);
(e)二甲基乙酰胺(DMA);
(f)乙醇;
(g)苯甲醇;
(h)用于注射的含有葡萄糖的水;
(i)聚氧乙烯蓖麻油;
(j)环糊精;以及
(k)PEG。
在又一选择方案中,所述组合物包含:一烷基化己醣醇衍生物、一修饰烷基化己醣醇衍生物或者一烷基化己醣醇衍生物或一修饰烷基化己醣醇衍生物的衍生物、类似物或前驱物;以及一溶剂系统,其中所述溶剂系统是选自于由下列所组成的群组:
(a)一乳化液;
(b)DMSO;
(c)NMF;
(d)DMF;
(e)DMA;
(f)乙醇;
(g)苯甲醇;
(h)用于注射的含有葡萄糖的水;
(i)聚氧乙烯蓖麻油;
(j)PEG;以及
(k)盐系统。
在又一选择方案中,所述组合物包含:一烷基化己醣醇衍生物、一修饰烷基化己醣醇衍生物或者一烷基化己醣醇衍生物或一修饰烷基化己醣醇衍生物的衍生物、类似物或前驱物;以及一赋形剂,其中所述赋形剂是选自于由下列所组成的群组:
(a)甘露醇;
(b)白蛋白;
(c)EDTA;
(d)亚硫酸氢钠;
(e)苯甲醇;
(f)碳酸盐缓冲液;
(g)磷酸盐缓冲液;
(h)PEG;
(i)维生素A;
(j)维生素D;
(k)维生素E;
(l)酯酵素抑制剂;
(m)细胞色素P450抑制剂;
(n)多重抗药性(MDR)抑制剂;
(o)有机树脂;
(p)洗涤剂;
(q)紫苏醇或其类似物;以及
(r)通道形成受体的活化剂。
在又一选择方案中,所述烷基化己醣醇衍生物、修饰烷基化己醣醇衍生物或者一烷基化己醣醇衍生物或修饰烷基化己醣醇衍生物的衍生物、类似物或前驱物是被并入于一剂型之中,所述剂型是选自于由下列所组成的群组:
(a)片剂;
(b)胶囊;
(c)外用凝胶;
(d)外用药膏;
(e)贴剂;
(f)栓塞剂;
(g)冻干剂量填充物;
(h)速释制剂;
(i)缓释制剂;
(j)控释制剂;以及
(k)液体胶囊。
在又一选择方案中,所述烷基化己醣醇衍生物、修饰烷基化己醣醇衍生物或者一烷基化己醣醇衍生物或修饰烷基化己醣醇衍生物的衍生物、类似物或前驱物是被并入于一剂量套组及包装之中,所述剂量套组及包装是选自于由避免光照的棕色瓶及用以增加储存寿命稳定性的具有专用涂层的瓶塞所组成的群组。
在又一选择方案中,所述组合物包含:(i)一烷基化己醣醇衍生物、修饰烷基化己醣醇衍生物或者一烷基化己醣醇衍生物或修饰烷基化己醣醇衍生物的衍生物、类似物或前驱物;以及(ii)一药物传递系统,其中所述药物传递系统是选自于由下列所组成的群组:
(a)口服剂型;
(b)纳米晶体;
(c)纳米颗粒;
(d)共溶剂;
(e)浆体;
(f)糖浆;
(g)生物溶蚀型聚合物;
(h)脂质体;
(i)缓释注射凝胶;
(j)微球;以及
(k)具有表皮生长因子受体结合胜肽的靶向组合物。
在本发明一合成物的又一选择方案中,所述治疗试剂是一修饰烷基化己醣醇衍生物,且所述修饰是选自于由下列所组成的群组:
(a)侧链的变动以增加或减少亲脂性;
(b)另一化学官能性的增加以改变一选自于由反应活性、电子亲和力及结合能力所组成的群组的性质;以及
(c)盐类型的变动。
在本发明一合成物的又一选择方案中,所述治疗试剂是一烷基化己醣醇衍生物、修饰烷基化己醣醇衍生物或者一烷基化己醣醇衍生物或修饰烷基化己醣醇衍生物的衍生物或类似物,且所述治疗试剂存在于一药物共轭形式中的组合物中,其中所述药物共轭形式是一选自于由下列所组成的群组的药物共轭形式:
(a)一聚合物系统;
(b)聚乳酸;
(c)聚甘醇酸;
(d)胺基酸;
(e)胜肽;
(f)多价连接符;
(g)免疫球蛋白;
(h)环糊精聚合物;
(i)修饰运铁蛋白;
(j)疏水性聚合物或疏水-亲水性聚合物;
(k)具有一磷甲酸钠偏酯的共轭物;
(l)具有加入一带电交联剂的细胞粘合剂的共轭物;以及
(m)具有通过一连接剂的β-葡萄醛酸的共轭物。
在本发明一合成物的又一选择方案中,所述治疗试剂是一烷基化己醣醇衍生物、修饰烷基化己醣醇衍生物或者一烷基化己醣醇衍生物或修饰烷基化己醣醇衍生物的衍生物或类似物,且所述治疗试剂处于一前驱物系统的形式,其中所述前驱物系统是选自于由下列所组成的群组:
(a)酵素敏感酯类;
(b)二聚体;
(c)希夫碱;
(d)吡哆醛复合物;
(e)咖啡因复合物;
(f)一氧化氮释放前驱物;
(g)具有纤维原细胞激活蛋白α-可分裂寡肽的前驱物;
(h)与一酰化剂或一胺甲酰化剂反应的产物;
(i)己酸盐共轭物;
(j)聚合物-剂的共轭物;以及
(k)受到氧化还原活化的前驱物。
在本发明一合成物的又一选择方案中,所述治疗试剂是一烷基化己醣醇衍生物、修饰烷基化己醣醇衍生物或者一烷基化己醣醇衍生物或修饰烷基化己醣醇衍生物的衍生物、类似物或前驱物,且所述组合物进一步包含至少一附加治疗试剂以形成一多重药物系统,其中所述至少一附加治疗试剂是选自于由下列所组成的群组:
(a)一多重抗药性的抑制剂;
(b)一特异的抗药性抑制剂;
(c)一特异的选择性酵素的抑制剂;
(d)一信号传递抑制剂;
(e)一修复酵素的抑制剂;以及
(f)一具有非重迭副作用的位置异构酶抑制剂。
在本发明一合成物的又一选择方案中,所述组合物包含一治疗试剂及一如上述的BH3类似物,所述治疗试剂为一烷基化己醣醇衍生物、修饰烷基化己醣醇衍生物或者一烷基化己醣醇衍生物或修饰烷基化己醣醇衍生物的衍生物、类似物或前驱物。
当本发明药物组合物包含一前驱物,使用本领域已知的常规技术可以辨认一化合物的前驱物及活性代谢物。比如请见,Bertolini等人,J.Med.Chem., 40,2011-2016(1997);Shan等人,J.Pharm.Sci.,86(7),765-767;Bagshawe, Drug Dev.Res.,34,220-230(1995);Bodor,Advances in Drug Res.,13,224-331 (1984);Bundgaard,前驱物的设计(Elsevier Press 1985);Larsen,前驱物的设计及应用、药物设计及发展(Krogsgaard-Larsen等人,eds.,Harwood Academic Publishers,1991);Dear等人,J.Chromatogr.B,748,281-293(2000);Spraul 等人,J.Pharmaceutical&Biomedical Analysis,10,601-605(1992);以及Prox 等人,Xenobiol.,3,103-112(1992)。
当于本发明的药物化合物中的所述药物活性化合物具有一足够酸性官能基、一足够碱性官能基或一足够酸性官能基及一足够碱性官能基两者,这些基团(group)或基团(groups)可以相应地与许多无机或有机碱以及无机及有机酸的任一个起反应,以形成一药学上可接受盐类。示例性的药学上可接受盐类包含通过具有无机或有机酸或者无机碱的药物活性化合物的反应所制备的那些盐类,比如包含硫酸盐、焦硫酸盐、重硫酸盐、亚硫酸盐、重亚硫酸盐、磷酸盐类、单氢磷酸盐、二氢磷酸盐、偏磷酸盐、焦磷酸盐、氯化物、溴化物、碘化物、乙酸盐、丙酸盐、癸酸盐、辛酸盐、丙烯酸酯、甲酸盐类、异丁酸盐、己酸盐、庚酸盐、丙炔酸盐、草酸盐、丙二酸盐、琥珀酸盐、辛二酸盐、癸二酸盐、富马酸、马来酸盐、丁炔-1,4-二酸盐、己炔-1,6-二酸盐、苯甲酸盐类、氯苯甲酸盐类、甲基苯甲酸盐类、二硝基苯甲酸盐类、羟基苯甲酸盐类、甲氧基苯甲酸盐类、酞酸盐、磺酸盐、二甲苯磺酸盐、苯乙酸盐、苯丙酸盐、苯丁酸盐、柠檬酸盐、乳酸盐、β-羟丁酸盐、甘醇酸盐、酒石酸、甲烷-磺酸盐、丙烷磺酸盐、萘-1-磺酸盐、萘-2-磺酸盐及扁桃酸盐的盐类。如果所述药物活性化合物具有一个或多个碱性官能基,所述期望的药学上可接受盐类可以通过任何本领域可用的合适方法来制备,比如,具有无机酸的游离碱的处理,比如氢氯酸、氢溴酸、硫酸、硝酸、磷酸等等,或者具有有机酸的游离碱的处理,比如醋酸、马来酸、琥珀酸、扁桃酸、富马酸、丙二酸、丙酮酸、草酸、乙醇酸、水杨酸、吡喃糖苷酸(比如葡萄糖醛酸或半乳糖醛酸)、α-羟基酸(比如柠檬酸或酒石酸)、胺基酸(比如天冬胺酸或谷氨酸)、芳香酸(比如苯甲酸或肉桂酸)、或磺酸(比如对-甲苯磺酸或乙磺酸)等等。如果所述药物活性化合物具有一个或多个酸性官能基,所述期望的药学上可接受盐类可以通过任何本领域可用的合适方法来制备,比如具有无机或有机碱的游离酸的处理,比如一胺类(初级、二级或三级)、一碱性金属氢氧化物或碱土金属氢氧化物等等。合适盐类的说明性例子包含来自于胺基酸的有机盐类,比如甘氨酸及精氨酸、氨、初级、二级及三级胺,以及环状胺(比如哌啶、吗啉及哌嗪);以及无机盐类,其是来自于钠、钙、钾、镁、锰、铁、铜、锌、铝及锂。
在药剂为固体的情况下,本领域的技术人员可以理解,本发明的化合物及盐类可以存在于不同晶型或多型型式,所有的这些皆意指是在本发明及指定公式的范围之内。
包含于本发明药物组合物的单位剂量的特定药物活性剂的量,比如为如上所述的卫康醇或卫康醇的衍生物或类似物,将取决因素而有所不同,所述因素比如为特定化合物、疾病情况及其严重性、目标对象在需要治疗时的特性(比如重量),但仍然可以由本领域技术人员定期地测定。通常,这样的药物组合物包含一药物活性剂的治疗有效量及一惰性药学上可接受载体或稀释剂。通常,在适于投药的选择途径的单位剂型中,制备这些组合物,比如口服投药或肠胃外投药。在通过根据常规方法结合一作为一具有适当的药物载体或稀释剂的活性成分的这样一药物活性剂的治疗有效量所制备的传统剂型中,如上所述的药物活性剂可以被投药。这些工序可以涉及混合、粒化、压缩及溶解适合所需制剂的成分。采用的药物载体可以是固体或液体。示例性的固体载体是乳糖、蔗糖、滑石、明胶、琼脂、果胶、阿拉伯胶、硬脂酸镁、硬脂酸等等。示例性的液体载体为糖浆、花生油、橄榄油、水等等。同样地,所述载体或稀释剂可以包含时间延迟或本领域已知的缓释材料,比如单硬脂酸甘油酯或甘油二硬脂酸酯单独或与蜡一起、乙基纤维素、羟丙甲基纤维素、甲基丙烯酸甲酯等等。
可以使用各种药物形式。因此,如果一固体载体被使用,在粉末或小颗粒形式中或在片剂或锭剂的形式中,所述制剂可以被制成片剂、放置于一硬明胶胶囊中。固体载体的量可能会有所不同,但一般是从约25mg到约1g。如果一液体载体被使用,在安瓿瓶(ampoule)或小玻璃瓶或者非水性液体悬浮液中,所述制剂将为糖浆、乳化液、软明胶胶囊、无菌可注射溶液或悬浮液的形式。
为了获得稳定的水溶性剂量形式,如上所述的药物活性剂的药学上可接受盐类溶解于一有机或无机酸(比如0.3M的琥珀酸或柠檬酸的溶液)的水性溶液中。如果可溶性盐类型不是可用的,所述药剂可以被溶解于一合适的共溶剂或共溶剂的组合中。在浓度范围为总体积的0-60%中,合适的共溶剂的例子包含但不限于乙醇、丙二醇、聚乙二醇300、聚山梨醇酯80、甘油等等。在一示例性实施例中,一式(I)的化合物溶解于DMSO,并以水稀释。在一适当的水性媒介物(比如水或等渗压盐水或葡萄糖溶液)中,所述组合物也可以是一活性成分的盐类型溶液的形式。
这将可以理解的是,根据正在使用的特定复合物、制定特定组合物的配方、投药及特定位置、受体(host)及疾病的模式和/或正在治疗的症状,本发明组合物中使用的药剂的实际剂量会有所不同。于本发明药物组合物中活性成分的实际剂量水平能够被改变,以便于得到一活性成分的量,所述活性成分是有效地实现用于特定目标对象、组合物及投药的模式的期望治疗反应,对于目标对象没有毒性。选定的剂量水平取决于各种药物动力学因子,包含所述特定的治疗试剂的活性、所述投药的途径、投药的时间、正在使用的特定化合物的排出率、病情的严重性、影响所述目标对象的其他健康因素,以及所述目标对象的肝及肾功能的状态。其也取决于治疗的持续时间;其他药物、化合物和/或材料与使用的特定治疗试剂结合使用;以及正在治疗的目标对象的年龄、体重、病情、一般健康状况及以往病史;和类似的因素。最优选剂量的测定方法被描述于现有技术中,比如:雷明顿:药学的科学和实践,Mack Publishing Co.,20
于病患中的典型的每日剂量、每日给予一次或两次,比如,可以每天给予3000mg两次(总剂量为6000mg)。在一实施例中,所述剂量是介于约1000 至约3000mg之间。在另一实施例中,所述剂量是介于约1500至约2800mg 之间。在其他实施例中,所述剂量是介于约2000至约3000mg之间。
在所述目标对象中的血浆浓度可以是介于约100μM至约1000μM之间。在一些实施例中,所述血浆浓度可以介于约200μM至约800μM之间。在其他实施例中,所述浓度为约300μM至约600μM之间。在其它实施例中,所述血浆浓度可以介于约400μM至约800μM之间。前驱物的投药通常剂量为体重水平,其是化学相当于完全活化型的重量水平。
本发明组合物可以使用用于制备药物组合物的通常已知的技术来制造,比如,通过如混合、溶解、粒化、糖衣锭形成、悬浮、乳化、封装、捕获或冻干的传统技术。在使用一或多个生理学上可接受载体的传统方式中,药物组合物可以被配制,其可以选自于赋形剂以及有利于处理活性化合物到可用于药学上的制剂中的助剂。
适当的制剂是取决于所选择的投药的途径。对于注射,本发明药剂可以被配制于水性溶液中,优选是在生理兼容缓冲液中,比如Hanks的溶液、 Ringer的溶液或生理盐缓冲液。对于粘膜投药,适于渗透屏障的渗透剂被使用于所述制剂中。这样的渗透剂于本领域中通常是已知的。
对于口服投药,所述化合物可以通过结合活性化合物与本领域已知的药学上可接受载体而容易地配制。这种载体能够使得本发明化合物被配制成片剂、丸剂、糖衣锭、胶囊、液体、凝胶、糖浆、浆体、溶液、悬浮液等等,而用于治疗病患的口服给药。在具有活性成分(药剂)的掺合物中,用于口服使用的药物制剂可以使用一固体赋形剂来得到,如果需要获得片剂或糖衣锭核,随意地研磨所得到的混合物,且于添加合适的助剂后加工颗粒的混合物。合适的赋形剂包含:比如为糖类的填充物,包含乳糖、蔗糖、甘露醇或山梨醇;以及纤维素制品,比如玉米淀粉、小麦淀粉、大米淀粉、马铃薯淀粉、明胶、树胶、甲基纤维素、羟丙基甲基纤维素、羧甲基纤维素钠或聚乙烯吡咯啶(PVP)。如果需要,可以添加崩散剂,比如交联的聚乙烯吡咯烷酮、琼脂或海藻酸或其盐类(比如海藻酸钠)。
糖衣锭核拥有合适涂层。为了这个目的,可以使用浓缩的糖溶液,其可以随意地包含阿拉伯胶、聚乙烯吡咯烷酮、羧乙烯聚合物(Carbopol)凝胶、聚乙二醇和/或二氧化钛、漆溶液及合适的有机溶剂或溶剂混和物。染料或颜料可以被添加于所述片剂或糖衣锭涂层,而用于识别或表示不同组合的治疗活性剂。
可口服用的药物制剂包括明胶制成的压入式(push-fit)胶囊,以及明胶制成的软、密封胶囊和塑化剂(比如甘油或山梨醇)。在具有填充物(比如乳糖)、结合剂(比如淀粉)和/或润滑剂(比如滑石或硬脂酸镁),以及任选的稳定剂的掺合物中,所述压入式胶囊可以包含所述活性成分。在软胶囊中,在合适的液体中,可以溶解或悬浮所述治疗活性剂,所述液体比如为脂肪油、液态石蜡或液态聚乙二醇。此外,可以添加稳定剂。所有用于口服投药的制剂应该在适于这样的投药的剂量中。对于口腔投药,所述组合物可采取的形式为以常规方式所配制的片剂或锭剂。
用于肠胃外投药的药物制剂可以包含水性溶液或悬浮液。合适的亲脂性溶剂或媒介物包含如芝麻油或合成脂肪酸酯的脂肪油,比如油酸乙酯或甘油三酯。水性注射悬浮液可以包含增加所述悬浮液的粘度的物质,比如羧甲基纤维素钠、山梨醇或葡聚糖。随意地,所述悬浮液可以包含合适的增加所述组合物的溶解度或分散性的稳定剂或调节剂以允许制备高度浓缩的溶液,或者可以包含悬浮或分散药剂。用于口服使用的药物制剂可以通过结合药物活性剂与固体赋形剂来得到,如果需要获得片剂或糖衣锭核,随意地研磨所得到的混合物,且于添加合适的助剂后加工颗粒的混合物。合适的赋形剂特别是比如为糖类的填充物,包含乳糖、蔗糖、甘露醇或山梨醇;纤维素制品,比如玉米淀粉、小麦淀粉、大米淀粉、马铃薯淀粉、明胶、紫云英树胶、甲基纤维素、羟丙基甲基纤维素、羧甲基纤维素钠和/或聚乙烯吡咯啶(PVP)。如果需要,可以添加崩散调节剂,比如交联聚乙烯吡咯烷酮、琼脂或海藻酸或其盐类(比如海藻酸钠)。
其他的比如为稳定剂的成分,比如,比如为柠檬酸钠、维生素C棕榈酸酯(ascorbylpalmitate)、没食子酸丙酯、还原药剂、维生素C、维生素E、亚硫酸氢钠、丁基羟基甲苯、BHA、乙酰半胱胺酸、单硫甘油、苯基-α-萘胺或卵磷脂的抗氧化剂可以被使用。并且,比如为EDTA的螯合剂可以被使用。传统在此领域的药物组合物及制剂的其他成分,比如于片剂或丸剂中的润滑剂、着色剂或者调味剂可以被使用。并且,传统的药物赋形剂或载体可以被使用。所述药物赋形剂可以包含但不必限于碳酸钙、磷酸钙、各种醣类或类型的淀粉、纤维素衍生物、明胶、植物油、聚乙二醇及生理兼容溶剂。其他药物赋形剂于本领域中是众所周知的。示例性的药学上可接受载体包含但不限于任何溶剂和/或所有溶剂,包含水性及非水性溶剂、分散介质、涂层、抗菌剂和/或抗真菌剂,和/或等渗压和/或吸收延迟药剂等等。这样的介质和/ 或用于药学上活性物质的药剂的使用于本领域中是众所周知的。除非任何传统的介质、载体或药剂与活性成分或成分是不相容的,其在本发明组合物中的使用是被预期的。附属活性成分也可以成为所述组合物的一部分,尤其是如上所述。制剂应该满足如FDA生物制剂标准局或其他调节组织调节药物所要求的无菌、发热性、一般安全性及纯度标准。
对于鼻内或通过吸入的投药,在从加压容器或喷雾器与使用合适的推进剂而以气溶胶喷雾的形式呈现中,方便地输送本发明使用的所述化合物,比如,二氯二氟代甲烷、三氯氟代甲烷、二氯四氟代乙烷、二氧化碳或其他合适的气体。在一加压气溶胶的情况下,所述剂量单位可以通过提供一阀门来释放一计量的量来确定。用于吸入或吹入等等的明胶的胶囊及药筒可以被配制含有一化合物及合适的粉末基质(比如乳糖或淀粉)的粉末混合物。
所述化合物可配制成通过注射的肠胃外投药,比如,通过单次注射或持续性注入。用于注射的制剂可以存在于单位剂型中,比如,在安瓿瓶或在多剂量容器,且具有一添加的防腐剂。在油性或水性的媒介物中所述组合物可以采取如悬浮液、溶液或乳化液这样的形式,且可以含有比如为悬浮、稳定和/或分散药剂的配制药剂。
在水溶性形式中,用于肠胃外投药的药物制剂包含活性化合物的水性溶液。此外,治疗活性剂的悬浮液可以被制备成适当的油性注射悬浮液。合适的亲脂性溶剂或媒介物包含如芝麻油或合成脂肪酸酯的脂肪油,比如油酸乙酯或甘油三酯,或者脂质体。水性注射悬浮液可以包含增加所述悬浮液粘度的物质,比如羧甲基纤维素钠、山梨醇或葡聚糖。任意地,所述悬浮液可以包含合适的稳定剂或药剂,其为增加所述化合物的溶解度以允许制备高度浓缩的溶液。
或者,所述活性成分可以是以一合适的媒介物构成的粉末形式,比如,使用前的无菌无热原水。所述化合物还可以被配制于直肠的组合物(比如栓塞剂或保留灌肠)中,比如,包含比如为可可脂或其他甘油酯的传统的栓剂基质。
除了如上所述的制剂以外,所述化合物也可以被配制成长效制剂。这样的长效制剂可以通过植入(比如,皮下地或肌肉地)或通过肌肉注射来投药。因此比如,所述化合物可以与合适的聚合性或疏水性材料(比如在可接收油中作为一乳化液)或离子交换树脂形成新配方,或者形成难溶性衍生物,比如,形成难溶性盐类。
一用于疏水性化合物的示例性药物载体是一共溶剂系统,其包含苯甲醇、非极性表面活化剂、水溶性有机聚合物及一水相。所述共溶剂系统可以是一 VPD共溶剂系统。VPD是一由3%w/v苯甲醇、8%w/v非极性表面活化剂聚山梨醇酯80及65%w/v聚乙二醇300组成的溶液(以无水乙醇定量体积)。所述VPD共溶剂系统(VPD:5W)包含VPD及5%葡萄糖水溶液,两者的稀释比例为1:1。此共溶剂系统充分溶解疏水性化合物,且本身于全身性给药之上产生低毒性。当然,没有破坏其溶解度及毒性性质的情况下,共溶剂系统的比例可以变化很大。此外,共溶剂成分的性质可以被改变:比如,可以使用其他低毒性非极性表面活化剂来取代聚山梨醇酯80;聚乙二醇的分率大小可以被改变;其他生物兼容性聚合物可以取代聚乙二醇,比如聚乙烯吡咯烷酮;以及其他的糖类或多糖类可以被葡萄糖所取代。
或者,可以使用其他用于疏水性药物化合物的传递系统。脂质体及乳化液是用于疏水性药物的传递媒介物或载体的已知例子。虽然通常损失更大的毒性,但某些有机溶剂(比如二甲亚砜)也可以被使用。此外,所述化合物可以使用一持续释放系统来输送,比如含有所述治疗试剂的固体疏水性聚合物的半透性矩阵。各种持续释放材料已经被建立,且是本领域技术人员已知的。持续释放胶囊可以依据其学性质释放所述化合物几个星期至超过100天。根据治疗试剂的化学性质及生物稳定性,可以使用蛋白稳定化的附加策略。
所述药物组合物还可以包含合适的固体相或凝胶相载体或赋形剂。这样的载体或赋形剂的例子包含碳酸钙、磷酸钙、糖类、淀粉、纤维素衍生物、明胶及如聚乙二醇的聚合物。
可以通过各种本领域已知的方法给予一药物组合物。投药的途径和/或模式依据期望结果而不同。根据所述投药的途径,所述药物活性剂可以被一材料覆盖,以从酸和可使药剂失去活性的化合物的作用来保护所述靶向组合物或其他治疗试剂。可以使用传统的药物实践来提供合适的用于此药物组合物的投药的制剂或组合物至目标对象。可以使用任何适当的投药途径,比如为静脉注射、肠胃外投药、腹膜内投药、经皮投药、皮下注射、肌肉注射、尿道内投药或口服投药,但本发明并不局限于此。根据恶性肿瘤或其他疾病、疾患或者症状被治疗的严重性,以及其他条件影响目标对象的治疗,所述药物组合物的全身性传递(systemic delivery)或局部性传递(localized delivery)可以被使用在治疗的过程中。如上所述的药物组合物可以与旨在治疗特定疾病或症状的附加治疗试剂一起投药,其可以是相同的疾病或症状(所述药物组合物旨在治疗可能是相关的疾病或症状,或甚至可能是无关的疾病或症状)。
本发明药物组合物可以根据本领域中众所周知及经常进行的方法来制备。比如请见,雷明顿:药学的科学和实践,Mack Publishing Co.,20
本发明药物组合物在很多情况上通常是给予目标对象。单剂量之间的时间间隔可以是每周、每月或每年。时间间隔也可以是治疗反应或其他本领域众所周知的参数所指示的不规则。或者,所述药物组合物可以为一持续释放制剂,在这种情况下,低频率的投药是必需的。在药物组合物内包含的药物活性剂的议题中,剂量及频率根据半衰期而不同。投药的剂量及频率可以根据治疗是否为预防性的或治疗性的而不同。在预防性应用中,在长时间内相对不频繁的时间间隔中,给予相对较低的剂量。一些目标对象可以持续接受于其有生之年的治疗。在治疗应用中,在治疗应用中,于相对较短时间间隔上相对高的剂量有时是必须的,直到疾病的恶化被减少或终止,且优选是直到所述目标对象显示部分或完全改善疾病症状。此后,所述目标对象可以被给予一预防性制度。
正如上面所述,为了本申请的目的,治疗被监视是通过观察一或多个关于治疗疾病、疾患或症状的改善症状,或是通过观察一或多个关于治疗疾病、疾患或症状的改善临床指标。
持续释放制剂或控释制剂于本领域中是众所周知的。比如,所述持续释放或控释制剂可以是(1)一口服基质持续释放或控释制剂、(2)一口服多层持续释放或控释锭剂调配物、(3)一口服多颗粒持续释放或控释制剂、(4)一口服渗透持续释放或控释制剂、(5)一口服咀嚼持续释放或控释制剂或(6)一皮肤的持续释放或控释贴剂调配物。
药物控制释放的药物动力学原则比如被描述于B.M.Silber等人,“药物控制释放的药物动力学/药效学基础”in Controlled Drug Delivery: Fundamentals andApplications(J.R.Robinson&V.H.L.Lee,eds,2d ed.,Marcel Dekker,New York,1987),ch.5,pp.213-251,并通过引用将其包括在内。
本领域中的普通技术人员可以通过改质如上所述的制剂而容易地制备包含本发明药物活性剂的用于控释或持续释放的制剂,比如根据公开于V.H.K. Li等人的原则,“于持续释放及控释系统的设计上药物性质的影响及药物投药的途径”in Controlled DrugDelivery:Fundamentals and Applications(J.R. Robinson&V.H.L.Lee,eds,2d ed.,Marcel Dekker,New York,1987),ch.1,pp. 3-94,并通过引用将其包括在内。这个制备过程通常会将所述药物活性剂的物化特性考虑在内,比如水溶解度、分配系数、分子大小、稳定性以及非特异结合至蛋白及其他生物大分子。这个制备过程也将生物因子考虑在内,比如用于药物活性剂的吸收、分布、代谢、作用期间、副作用的可能存在及安全边际。因此,在本技术领域的普通技术人员可以修改所述制剂成为一具有以上特定应用所描述的理想特性的制剂。
Nardella所申请的美国专利第6,573,292号、Nardella所申请的美国专利第6,921,722号、Chao等人所申请的美国专利第7,314,886号及Chao等人所申请的美国专利第7,446,122号,其公开于治疗许多疾病及症状(包含癌症) 中各种药物活性剂及药物组合物的使用方法,以及测定这样的药物活性剂及药物组合物的治疗有效性的方法,以上全部皆通过引用将其包括在内。
通常,卫康醇的治疗有效量大约为40mg/m
通常,通过一选自于由静脉及口服所组成的群组的途径给予所述卫康醇。优选地,所述卫康醇为静脉注射。二乙酰二脱水卫矛醇或二溴卫矛醇可以使用相似的途径。
所述方法可以进一步包含:给予一电离辐射的治疗有效量的步骤。
本发明是通过下面实施例来说明的。此实施例仅用于说明本发明的目的,而非意在限制本发明。
例子
使用卫康醇以抑制肿瘤细胞的生长
材料与方法:
细胞株及培养条件:所有细胞置入于具有10%FBS(胎牛血清,fetal bovineserum;Invitrogen/Gibco)的DMEM(Dulbecco’s Modified Eagle’s medium;Invitrogen/Gibco)中且放置于37℃、5%CO
药物:100mM的储备溶液使用前被保持在-20℃。卫康醇(DAG;在图中,具有DAG的结果显示为“VAL”)是通过德玛药物公司而提供的。100mM的储备溶液被制备是通过溶解所述冻干粉末至所述注射液小瓶中的无菌磷酸盐缓冲液(phosphate buffered saline,PBS)中,并且使用前被保持在20℃。
生长试验:每一细胞株(3000cells/well)被接种于96孔盘(BD Falcon)内的 100μL培养基中,并隔夜培养。然后,于新鲜培养基中以DAG(浓度为0.1-100 μM)处理细胞72小时。在具有核染料Hoechst 33342(1μg/mL)(Sigma-Aldrich) 的2%多聚甲醛(Sigma-Aldrich)中固定所述细胞。以PBS温和洗涤后,所述细胞被保持在新鲜的PBS中,且在高内涵筛选(high content screening,HCS) (ThermoFisher Scientific)分析之前,所述盘被保持于4℃、黑暗中。每孔20 个视野数(view fields)进行扫描及分析。生长抑制计算的是一不具有溶剂及药物的控制的百分比;单以溶剂处理的样品作为一参照。每次处理三重复,且所述实验被重复一次。
结果
图1显示于所述细胞株MDA-MB-231上卫康醇(浓度0.1~100μM)的作用。此为一雌性素非依赖性人类乳癌细胞株(T.Hiraga等人,“在骨转移中 MDA-MB-231人类乳癌细胞中双膦酸盐伊班膦酸盐促进细胞凋亡”,Cancer Res.61:4418-4424(2001),并通过引用将其包括在内)。
图2显示于所述细胞株HCC1143上卫康醇(浓度0.1~100μM)的作用。 HCC1143是一种三阴性乳癌细胞株,其对PARP或TKIs是不敏感的,且是众所周知的积极性癌症转移细胞(D.Tryfonopoulos等人,“Src:一用于治疗三阴性乳癌的潜在标靶”,Ann.Oncol.22:2234-2240(2011),并通过引用将其包括在内)。
图3显示于所述细胞株K562上卫康醇(浓度0.1~100μM)的作用。K562 是一骨髓性白血病细胞株(C.B.Lozzio及B.B.Lozzio,“具有阳性费城染色体的人类慢性骨髓性白血病细胞株”,Blood 45:321-224(1975),并通过引用将其包括在内);原生型K562本身缺乏BIM缺失多型性,且对于TKIs维持灵敏度(K.P.Ng等人,“在癌症中一通常的BIM缺失多型性调和先天抗药性且对酪氨酸激酶抑制剂反应较差”,Nat.Med.18:521-528(2012),并通过引用将其包括在内)。
图4显示于所述细胞株K562-AHI-1上卫康醇(浓度0.1~100μM)的作用。所述细胞株K562-AHI-1带有一突变,其中的AHI1的表现是失调的。所述 AHI-1突变赋予TKI伊马替尼抗性至K562细胞。
结果
针对所有细胞株的测试,卫康醇表现出一高度的细胞毒性。尤其是,在 K562-AHI-1内AHI1中,对于K562而言,通过突变的引入没有明显地减弱卫康醇的细胞毒杀作用。此外,在三阴性乳癌细胞株HCC1143上,卫康醇表现出一高度的细胞毒性,所述三阴性乳癌细胞株HCC1143是对PARP或TKIs 不敏感的,且是众所周知的积极性癌症转移细胞。
本发明的优点
本发明提供多个用于治疗TKI抗性恶性肿瘤的有效方法及组合物,特别在具有一生殖细胞多型性的目标对象上,所述生殖细胞多型性于BIM基因中引起了一选择性剪接,所述BIM基因引起BIM的选择性剪接异构体的生长,所述BIM是缺乏涉及细胞凋亡的促进期的重要BH3区域。本发明还提供多个用于治疗与在AHI1基因中具有异常调控或突变的细胞相关的恶性肿瘤的有效方法及组合物。这些方法及组合物可以治疗这样的恶性肿瘤而不依靠胸腺核苷激酶的抑制作用。其耐受性良好且不会造成重大的副作用。
在具有阻断酪氨酸激酶抑制剂的有效性的生殖细胞多型性的目标对象中,或具有与在AHI1基因中具有异常调控或突变的细胞相关的恶性肿瘤(特别是高度增生疾病)的目标对象中,对于用于治疗许多疾病及症状的药物的制备,本发明的组合物及方法具有产业利用性,且作为药物组合物具有产业利用性。
本发明的方法权利要求提供具体的方法步骤,其超出一般自然法则的应用,且除了权利要求中所叙述或暗示的自然法则的具体应用之外,要求那些实践所述方法的步骤采用不同于在本领域中现有公知的那些步骤,从而约束权利要求到其中所叙述的具体应用。在某些情况下,这些权利要求被导向于使用现有药物的新途径。
本发明在此举例式的叙述,可在缺少任何本文中非特定公开的元素,限制下合适地实行。因此,举例来说,所述术语「包含」、「包括」、「含有」等,应被广义地与不带限制地解读。另外,本文中使用的术语及表达方式,是用于描述而非限制,此术语及表达方式的使用目的不在于排除其他未来显示及描述的相等物质,或其他相关部分,应认知在此发明所请求的范围中,不同的改变是有可能的。因此,应了解虽然本发明已在最优选实施例及选择性特征公开,本发明在此公开的改变及变异,可借助在此领域具通常知识者,且此改变及变异考虑本发明在此所公开的范围。本发明已在此作广义地及一般性的解释。每个落入属名公开范围的窄义的物种及亚属族群,也形成发明的一部分。这些包括每个发明所描述的属名,所述属名以但书或否定限制排除任何属名中的内容,不论所排除的内容是否特定存在于本文中。
另外,发明中的特色及方向,以马库西形式(Markush group)的术语叙述,在此领域受教育者,会判断本发明也以马库西形式中任何单独成员或子群体成员的术语来叙述。也应了解以上叙述的目的在于举例而非限制。回顾以上叙述时,许多实施例对在此领域具通常知识者来说是显而易见的。因此本发明的范围不应该参考以上叙述决定,而是参考附加的权利请求,以及此权利请求所给予同等物质的全部范围而决定。所有文章及参考文献的公开,包含专利出版物,其是通过引用将其包括在内。
- 使用卫康醇、二乙酰二脱水卫矛醇、二溴卫矛醇或类似物或其衍生物治疗具有基因多型性或AHI1失调或突变患者的抗酪氨酸激酶抑制剂的恶性肿瘤的方法
- 使用卫康醇或其衍生物治疗抗酪氨酸激酶抑制剂的恶性肿瘤的方法