肽及其医学用途
文献发布时间:2023-06-19 11:32:36
技术领域
本发明涉及肽、包含所述肽的组合物及其作为血管生成(angiogenesis)和/或新血管生成(neoangiogenesis)的抑制剂的用途。此外,本发明涉及所述肽和所述组合物用于治疗与不正确的血管生成和/或新血管生成相关的疾病的用途。特别地,在本文中述及与VEGFR1相关的血管生成和/或新血管生成。
现有技术
鉴于抑制VEGFR-1的激活可以适用于相当严重和广泛范围的疾病,可以设想,将会强烈需要能够结合VEGFR-1并能够干扰配体VEGF-A、PlGF、VEGF-B和VEGF-A/PlGF异二聚体与VEGFR-1之间的相互作用的合成化合物。事实上,有利的是,合成化合物本质上不含生物来源的污染物,并且它们还可以以比重组来源的生物治疗剂低得多的成本生产。
为了中和配体,许多治疗方法使用单克隆抗体,因为它们是特征在于高特异性和亲和力的分子。然而,合成分子也有其优势,因为它们生产更容易且更便宜,更稳定,并且更容易递送。
在这方面,Ponticelli等人最近在2008年描述了一种选自肽文库的四聚体三肽,其中具有式(R-Glu)-(S-Cys(Bzl))-(S-Cha)的肽链被四聚化在三个赖氨酸的“核心”上(Tam,J.P.1988.Proc.Natl.Acad.Sci.USA 85:5409–5413)。
四聚体肽具有以下结构:
式(I)
Ponticelli等人报道的科学证据展示出上述四聚体肽在体外能够结合VEGFR1,并抑制PlGF、VEGF-A和VEGF-B的相互作用,IC
最后,该肽:
1)在体外显示出抗血管生成活性,干扰PlGF和VEGF-A的促血管生成活性;
2)在角膜中能够代替VEGF-A-sFlt1结合(在生理条件下不形成血管),从而使角膜无VEGF-A (VEGF-A free)并能够抑制新血管生成;
3)当腹膜内施用时,减少肿瘤生长、血管生成和动脉生成以及转移;和
4)当玻璃体内施用时,减少脉络膜新血管形成(Cicatiello等人2015)。
该肽的抗血管生成活性是由于抑制新血管的形成和抑制炎症细胞(优选地单核巨噬细胞)在新血管生成部位的募集的能力两者。
抗动脉生成活性是基于抑制平滑肌细胞在新血管生成部位募集的能力。
在本文中,本发明的作者惊讶地发现,通过在肽的C-末端插入化学基团,特别是特征在于侧链具有与硫醇或硫醚基团相当的空间位阻的氨基酸,人们显著提高了分子的活性。
事实上,上述修饰不损害与VEGFR1的选择性结合以及以剂量依赖性方式与VEGF-A和/或PlGF竞争结合VEGFR1的能力。相反,这些修饰能够在低于1000nM的浓度对PlGF或VEGF与VEGFR1之间的相互作用产生50%的抑制(IC
此外,本发明的作者惊讶地发现,当口服施用或通过灌胃施用时,在Ponticelli等人中描述的肽和本发明的肽都展示出显著的抑制脉络膜新血管形成的能力。因此,这些分子对于治疗(优选地通过口服施用)与血管生成(优选地VEGFR1依赖性血管生成)改变相关的疾病或在任何情况下由血管生成(优选地VEGFR1依赖性血管生成)改变引起的疾病是治疗有效的。
下面是本发明的详细描述,以及参考以下附图和定义的非限制性说明性实例。
-图1示出了iVR1和iVR1-Cys以及抗PlGF单克隆抗体对PlGF诱导的VEGFR-1磷酸化的抑制活性。通过蛋白印迹法对293-VEGFR-1细胞进行20ng/ml PlGF诱导的VEGFR-1磷酸化分析。iVR1-Cys和iVR1以5μM的浓度同时添加至PlGF。使用浓度为3.3nM的人类抗PlGF中和单克隆抗体作为抑制对照。PBS用作阴性对照。
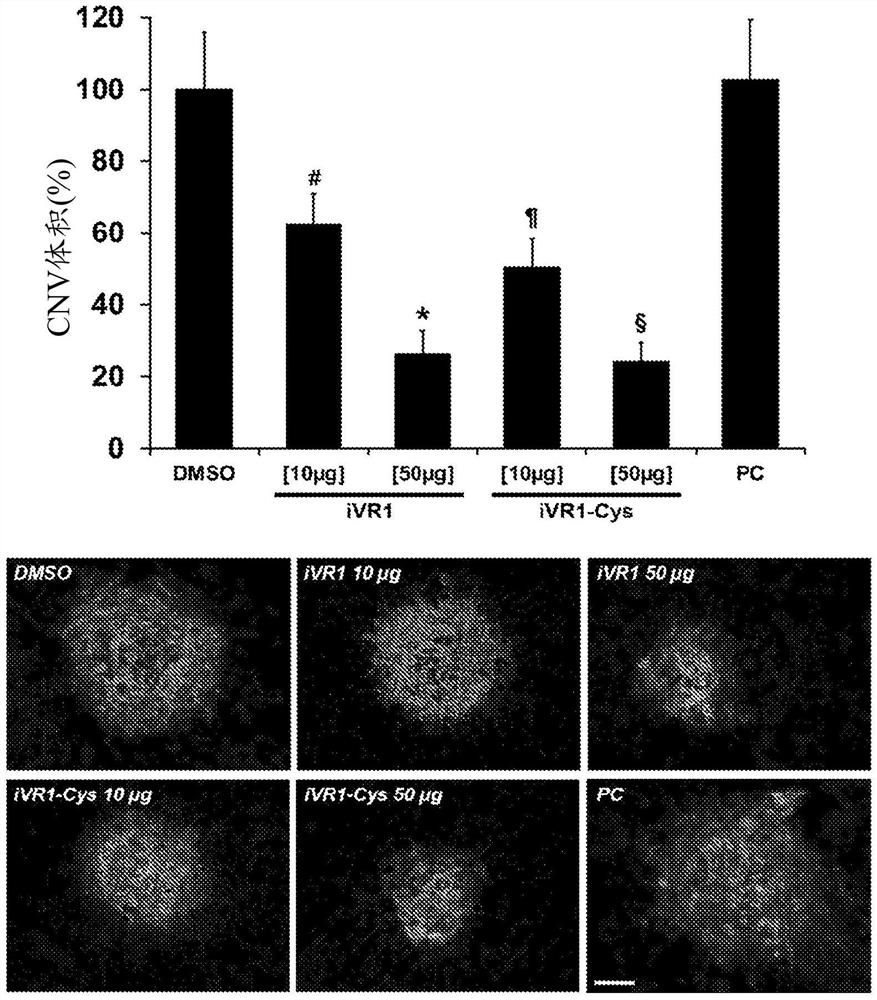
-图2示出,玻璃体内施用的iVR1-Cys以剂量依赖性方式抑制激光诱导的脉络膜新血管形成。与注射媒介物(DMSO)相比,单次玻璃体内注射10μg或50μg的iVR1-Cys引起的脉络膜新血管形成的剂量依赖性减少等于48.9%和75.9%。相同量的iVR1引起的CNV抑制等于37.8%和73.9%。对照肽(PC)未显示出抑制能力。对于iVR1 10μg和50μg在n=12个和15个斑点上进行新血管形成体积的量化,对于iVR1-Cys 10μg和50μg在n=10个和8个斑点上进行新血管形成体积的量化;对于PC在n=15个斑点上进行新血管形成体积的量化,并且对于DMSO在n=14个斑点上进行新血管形成体积的量化。数据表示为相对于对照的平均值±SEM。#p<0.05;*p>0.0002;
-图3示出,口服施用的iVR1-Cys抑制激光诱导的脉络膜新血管形成。与媒介物相比,每天两次口服施用50mg/Kg的iVR1-Cys持续7天,引起脉络膜新血管形成减少45.9%。相同量的iVR1引起类似的CNV抑制(49.7%)。对于iVR1-Cys在n=18个斑点上进行新血管形成体积的量化,对于iVR1在n=20个斑点上进行新血管形成体积的量化,并且对于媒介物在n=10个斑点上进行新血管形成体积的量化。数据表示为相对于对照的平均值±SEM。*p=0.001和§p=0.007,对比DMSO。下方是代表CNV的图像。比例尺代表100μm。
在本文中,术语“VEGF”意指血管内皮生长因子。在人类中存在5种不同的血管内皮生长因子,VEGF-A、VEGF-B、VEGF-C、VEGF-D和PlGF,由5种不同的基因编码。都是糖基化的二聚体蛋白。
在本文中,术语“VEGF-A”意指血管内皮生长因子-A,以前也称为VPF(血管通透性因子)。它是VEGF家族中最强有力的因子,在生理性和病理性血管生成中起着决定性作用。通过选择性剪接获得的至少六种不同亚型已在人类中描述。它们都能够与两种受体相互作用,这两种受体被称为VEGFR-1和VEGFR-2。
在本文中,术语“PlGF”意指胎盘生长因子,其作用仅限于与病理状态相关的血管生成状况。在人类中已经描述了四种不同的亚型。都能够特异性地结合VEGFR-1。VEGF-A和PlGF在病理状况中以强的协同作用起作用,因为两者都与VEGFR-1相互作用,并且因为当两个相应的基因在同一细胞中表达时,它们能够产生能够与VEGFR-1相互作用或诱导VEGFR-1/VEGFR-2异二聚体化的VEGF-A/PlGF异二聚体。
在本文中,术语“VEGFR-1”意指VEGF受体1,也称为Flt-1。VEGFR-1具有细胞内酪氨酸-激酶结构域,而细胞外部分由七个IgG样结构域组成。VEGF-A、VEGF-B或PlGF导致受体二聚体化,随后通过酪氨酸-激酶结构域的自磷酸化激活。除了在内皮细胞中表达,VEGFR-1还在许多其他类型的细胞中表达,包括平滑肌细胞、单核巨噬细胞、成纤维细胞和内皮前体细胞。它在募集不同类型的有助于血管生成的细胞中起着重要作用。在本文中,术语“可溶性VEGFR-1”(sVEGFR-1)意指VEGF受体1的可溶性形式,也称为sFlt-1。它由VEGFR-1的前六个IgG样胞外结构域加上一个尾组成,并且由VEGFR-1基因通过选择性剪接产生。它通常由表达VEGFR-1全长形式的相同细胞表达,但角膜除外,在角膜中可溶性形式优先表达,这对于维持角膜处于无血管状态是决定性的。全长和可溶性人类VEGFR1的信使序列优选地分别为SEQ ID NO:1和SEQ ID NO:2,而全长人类VEGFR1的蛋白序列分别是SEQ ID NO:3和SEQ IDNO:4。特征在于与本文描述的序列具有80%-99.9%同一性的序列必须被认为是本说明书的一部分。
在本文中,术语“VEGFR-2”意指VEGF受体2,在人类中也称为KDR,并且在小鼠中也称为Flk-1。VEGFR-2被VEGF-A特异性地结合,并且具有类似于对VEGFR-1描述的结构域和激活机制。与受体1不同,它主要在内皮细胞中表达。它在刺激内皮细胞的增殖、迁移和分化中具有重要作用。
在本文中,术语“血管生成”意指从现有血管形成新血管的过程;在本文中,血管生成优选地被称为与各种类型的病理状况相关的新血管形成过程,所述病理状况优选地选自:
-新生血管性眼病,优选地选自:黄斑水肿、湿性型年龄相关性黄斑变性、糖尿病性视网膜病变、早产儿视网膜病变、视网膜中央静脉阻塞视网膜病变、玻璃体出血和视网膜脱离及其组合;和/或
-实体肿瘤和/或肿瘤转移,所述肿瘤优选地选自:白血病和淋巴瘤,优选地急性淋巴细胞性白血病、急性非淋巴细胞性白血病、慢性淋巴细胞性白血病、多发性骨髓瘤、霍奇金淋巴瘤、霍奇金病、婴儿或成人实体肿瘤、脑肿瘤、成神经细胞瘤、成视网膜细胞瘤、肾母细胞瘤、骨肉瘤和软骨肉瘤、肺肿瘤、结肠直肠癌、乳腺癌、前列腺癌、子宫癌、卵巢癌、泌尿系统癌症、膀胱癌、口腔肿瘤、胰腺肿瘤、黑素瘤和皮肤肿瘤、胃肿瘤、脑肿瘤、甲状腺肿瘤、喉肿瘤、肝肿瘤、睾丸肿瘤;和/或
-骨或关节疾病,优选地选自:类风湿性关节炎、滑膜炎、软骨和/或骨破坏、骨髓炎、滑膜组织肥大和/或增生、骨赘形成、赘生物和/或转移及其组合;和/或
-血管疾病,优选地选自:动脉粥样硬化、血管瘤、血管内皮瘤及其组合;和/或
-皮肤病,优选地选自:银屑病、疣、化脓性肉芽肿、毛发生长、卡波西肉瘤、创伤瘢痕瘤、变应性水肿、赘生物及其组合;和/或
-在脂肪组织疾病,优选地肥胖症中观察到的血管生成;和/或
-糖尿病和/或其后果,优选地视网膜病变和/或糖尿病足;和/或
-造血系统疾病,优选地AIDS和/或卡波西肉瘤。
在本文中,术语“新血管生成”意指新的血管生成,优选地是指在先前没有新血管的组织中形成新血管和/或在已经血管化的组织中血管数量的增加;在本文中,新血管生成优选地依赖于VEGFR-1的活性。
在本文中,术语“血管形成(vascularization)”意指血管生成(angiogenesis),即它们被用作同义词。
在本文中,术语“新血管形成(neovascularization)”意指新血管生成(neoangiogenesis),优选地依赖于VEGFR-1的活性。
在本文中,术语“动脉生成”意指通过用平滑肌细胞覆盖血管来稳定新血管的过程。
在本文中,“抑制剂”意指能够通过结合受体本身和/或其可溶性配体来拮抗受体活性,从而阻止它们的相互作用的化学和/或生物实体。
在本文中,术语“有效剂量”意指在其中本发明中描述的活性物质的施用能够确定期望的生物效应的剂量区间。如本领域技术人员所熟知的,有效剂量可以根据以下因素而变化:健康状态、需要治疗的个体的身体状况、年龄、活性物质的配方、照顾患者的医生的评估、单个个体的系统有效地响应的能力、期望的响应程度、分类群(例如人类、非人类灵长类动物、灵长类动物等)以及其他相关因素。预计,本发明中描述的活性物质的有效剂量将落在一个足够宽的区间内,该区间可通过常规试验确定。一般来说,如Ragan-Shaw等人(FASEBJ.2008Mar;22(3):659-61)所报道的,并且还因此在本文中,当优选地全身施用,优选地通过肠途径全身施用,更优选地口服、舌下或直肠施用时,施用的有效剂量优选地在10mg/剂量和2000mg/剂量之间的范围内。可选地,当优选地玻璃体内施用时,施用的有效剂量在1mg/剂量和100mg/剂量之间的范围内。可选地,施用的有效剂量优选地在0.16mg/Kg体重至33.3mg/Kg体重之间的范围内。治疗方案提供单剂量或多剂量。
本发明的第一方面涉及肽,优选地多聚体肽,所述肽是分离的肽且特征在于以下通式(II):
{{{[Y1–Glu–Cys(Bzl)–Cha]2–Z1}i–Z2}j–Z3}z–Y2–Y3
(式II)
其中
-Y1是肽的氨基末端官能团(NH
在本文中,应该澄清的是,适用于定义本说明书的基团中存在的手性中心的绝对构型的D/L符号可以与按照文献中报道的规则的R/S符号互换,如本领域技术人员已知的。
表I
-Glu表示谷氨酸,优选地在氨基酸的Cα上呈绝对构型R(R-Glu)。
-Cys(Bzl)表示苄基半胱氨酸,优选地在含有苄基基团的氨基酸的Cα上呈绝对构型S(S-苄基-半胱氨酸/S-Cys(Bzl)),所述苄基基团与所述氨基酸侧链的硫连接。
-Cha表示环己基丙氨酸,优选地在氨基酸的Cα上呈绝对构型S(S-环己基丙氨酸/S-Cha)。
-Y2优选地选自:
1.三肽R-Glu–S-Cys(Bzl)–S-Cha,和
·α-氨基酸,优选地选自甘氨酸或特征在于至少一个硫醇或硫醚基团的α-氨基酸,所述特征在于至少一个硫醇或硫醚基团的α-氨基酸优选地选自表II中示出的那些及其组合。
该列表还应理解为包括这样的化学基团(优选地氨基酸),所述化学基团(优选地氨基酸)具有这样的空间位阻和/或化学性质(特别地在氨基酸的情况下的侧链),所述空间位阻和/或化学性质(特别地在氨基酸的情况下的侧链)模拟表II中列出的化学基团(优选地氨基酸)的空间位阻和/或化学性质(特别地在氨基酸的情况下的侧链)且其特征在于相似性,优选地至少70%的相似性,所述相似性用本领域技术人员已知的方法确定,例如,但不排他地,用Woong-Hee Shin等人,Molecules 2015,20,12841-12862中描述的方法。
表II
-Y3优选地选自:羧基基团、羧酰胺基团、N-甲基取代的羧酰胺或二取代的N,N-二甲基基团、羟基基团和氢。
-Z1、Z2和Z3优选地表示三官能团,优选地特征在于以下式(III):
其中k是整数,优选地包含在1和4之间,并且B优选地是氨基基团或羟基基团。所述三官能分子优选地呈R或S绝对构型。
优选地,Z1、Z2和Z3用于获得支链结构的目的。事实上,这种类型的结构通常用于按照为此目的的已知方法将肽多聚体化,例如当B是氨基基团时,可以使用Tam等人描述的方法(Tam J.P.,1988,PNAS,85,5409-5413)。
Z1、Z2和Z3可以以这样的方式组装,以获得具有多于一个基团Z1、Z2和Z3的式(II)结构,优选地包含1、3或7个三官能分子。
根据本发明的优选实施方案,Z1和/或Z2和/或Z3优选地通过酰胺键以形成支链结构的方式彼此连接。可选地,它们可以通过酯键彼此连接,例如当B优选地是羟基基团时。
-i优选地是4、2或1。
-j优选地是2、1或0。
-z优选地是1或0。
根据优选实施方案,当i=4时,j=2并且z=1。根据另一优选实施方案,当i=2时,j=1并且z=0。
根据另一优选实施方案,当i=1时,j=z=0。
如果j=0,则省略Z2基团,并且如果z=0,则省略Z3基团。
为了本发明的目的,特别优选的实施方案设想i等于2,j等于1并且Z2为0或省略(换句话说,Z3不出现,即Z3不存在)。
在本发明的特别优选的实施方案中,Z1、Z2和Z3是R-赖氨酸或S-赖氨酸(k=4),并且i优选地等于2。
本发明的多聚体肽的优选的式由下式(式IIa)表示:
(式IIa)
根据本发明的特别优选的实施方案,该肽是特征在于式(IIb)的四聚体肽:
(式IIb)
其中:
-Y1是氢原子;
-Y2是D-半胱氨酸;
-Y3是未被取代的伯酰胺基团;
-Z1、Z2和Z3如上定义;
-i等于2;
-j等于1;并且
-z等于零,即不存在。
为了方便起见,从此刻起,特征在于式IIb的肽的特别优选的实施方案将被称为iVR1-Cys。
上述肽显示出生物活性,优选地调节活性,更优选地抑制血管生成和/或新血管生成的活性,该活性与Ponticelli等人所述的肽的活性相比得到了改进,如以下实验结果中所报道和讨论的,在本文中,实验结果具有非限制性的说明目的。在本文中述及的血管生成和/或新生血管生成优选地是前面定义的VEGFR1依赖性的。
Ponticelli等人中描述的肽也是一种四聚体肽,其特征在于式(IIc):
(式IIc)
其中:
-Y1是氢原子;
-Y2是甘氨酸;
-Y3是未被取代的伯酰胺基团;
-Z1、Z2和Z3如上定义;
-i等于2;
-j等于1;并且
-z等于零。
为了方便起见,从此刻起,特征在于式IIc的肽的特别优选的实施方案将被称为iVR1。
本发明的作者惊讶地发现,通过修饰IVR1,特别是在羧基末端,优选地通过插入R-Glu–S-Cys(Bzl)–S-Cha基团或α-氨基酸进行修饰,人们获得特征在于改进的生物活性(优选地改进的调节能力,优选地通过抑制如以上定义的血管生成和/或新血管生成)的肽,所述α-氨基酸优选地选自特征在于至少一个硫醇或硫醚基团的α-氨基酸,所述特征在于至少一个硫醇或硫醚基团的α-氨基酸优选地选自表II中示出的那些及其组合。
事实上,如实施例中更详细显示和讨论的,iVR1-Cys展示出以剂量依赖性方式抑制PlGF和VEGF-A两者与VEGFR-1相互作用的能力,这种能力比iVR1有所改进。特别是,iVR1-Cys能够抑制PlGF与VEGFR-1相互作用的50%的浓度(IC
因此,iVR1-Cys显示的抑制能力是报道的iVR1的抑制能力的10倍大。
此外,本作者通过体内测定展示,对比媒介物和PC,iVR1引起脉络膜新血管形成的抑制为37.8%和39.3%(p<0.05),而对比媒介物和PC,iVR1-Cys引起的抑制为48.9%和51.0%(p<0.02)。因此,iVR1-Cys显示出比肽iVR1更大的抑制效力,因为它进一步减少了19.3%的新血管形成。
最后,当口服施用或通过灌胃施用时,通过实施例测试的两种肽与媒介物相比能够诱导对新血管形成的显著抑制。
后一个事实特别重要,因为尽管Ponticelli等人和Cicatiello等人2015已经展示了iVR1通过玻璃体内注射抑制脉络膜血管生成和新血管形成的能力,但绝对未预料通过不同途径(特别是通过灌胃)施用肽能够维持或甚至改进治疗效力,尤其是在高度复杂的器官如眼睛的情况下,以及由不受调控的(优选地增加的)血管生成/新血管生成引起的或在任何情况下与不受调控的(优选地增加的)血管生成/新血管生成相关的影响眼睛的疾病的情况下能够维持或甚至改进治疗效力。特别地,所述及的眼睛的新血管疾病优选地选自:黄斑水肿、湿性型年龄相关性黄斑变性、糖尿病性视网膜病变、早产儿视网膜病变、视网膜中央静脉阻塞、玻璃体出血和视网膜脱离及其组合。
根据这一证据,明确的是,通过口服途径或通过灌胃施用本发明的肽,对于治疗通常与血管生成/新血管生成相关的疾病,诸如例如癌症,也是治疗有效的。所述及的血管生成或新血管生成优选地是VEGFR1依赖性的。
根据本发明的一种实施方案,肽可以被修饰以促进或改进递送,所述修饰优选地通过PEG化,或使用容器/穿梭体(shuttle)/载体系统,优选地脂质体、胶束、胶囊、乳剂、基质、凝胶等进行。
本发明的另一方面涉及一种组合物,该组合物包含如详细描述的肽和至少一种另外的药学上接受的成分。
该组合物优选地包含至少一种特征在于式IIa的肽,更优选地特征在于式IIb的肽,即iVR1-Cys。
在本文中,药学上接受的成分意指选自以下的化合物:赋形剂、稀释剂、载体、辅助剂(adjuvant)、防腐剂、抗生素、抗炎剂、油、维生素、抗氧化剂、螯合剂、增溶剂、粘度剂、惰性气体、表面活性剂、乳化剂、缓冲物质、免疫抑制剂、抗肿瘤剂及其组合。
例如,根据一种实施方案,组合物包含本发明的肽与以下的组合:至少一种抗血管生成/抗新血管生成分子、中和PlGF作用的抗体、至少一种抗VEGFR-1、抗VEGFR-2、抗VEGFR-3抗体、至少一种抗VEGF-A、抗VEGF-B、抗VEGF-C、抗VEGF-D、抗VEGF-E抗体及其组合。
本发明的另一方面涉及如以上所描述的肽,优选地特征在于式IIa的肽,更优选地特征在于式IIb的肽,即iVR1-Cys,用作药物。
本发明的另一方面涉及如以上所描述的肽,优选地特征在于式IIa的肽,更优选地特征在于式IIb的肽,即iVR1-Cys,或如以上所描述的包含所述肽的组合物,用于治疗与不正确的血管生成/新血管生成相关或由不正确的血管生成/新血管生成引起的病理状况,即其中血管生成/新血管生成不受调控的疾病;血管生成/新血管生成优选地已经增加,并且因此需要被抑制。
除了可用于治疗所述疾病外,如以上所描述的肽,优选地特征在于式IIa的肽,更优选地特征在于式IIb的肽,即iVR1-Cys,或如以上所描述的包含所述肽的组合物也可用于所述疾病的后续的进一步替代治疗处理。
如先前已经描述的,如以先前定义的血管生成/新血管生成优选地依赖于VEGFR1或VEGFR1途径或由VEGFR1或VEGFR1途径诱导/调节。
所述疾病/状况优选地选自:
-新生血管性眼病,优选地选自:黄斑水肿、湿性型年龄相关性黄斑变性、糖尿病性视网膜病变、早产儿视网膜病变、视网膜中央静脉阻塞视网膜病变、玻璃体出血和视网膜脱离及其组合;和/或
-实体肿瘤或液体肿瘤和/或肿瘤转移,所述肿瘤优选地选自:白血病和淋巴瘤,优选地急性淋巴细胞性白血病、急性非淋巴细胞性白血病、慢性淋巴细胞性白血病、多发性骨髓瘤、霍奇金淋巴瘤、霍奇金病、婴儿或成人实体肿瘤、脑肿瘤、成神经细胞瘤、成视网膜细胞瘤、肾母细胞瘤、骨肉瘤和软骨肉瘤、肺肿瘤、结肠直肠癌、乳腺癌、前列腺癌、子宫癌、卵巢癌、泌尿系统癌症、膀胱癌、口腔肿瘤、胰腺肿瘤、黑素瘤和皮肤肿瘤、胃肿瘤、脑肿瘤、甲状腺肿瘤、喉肿瘤、肝肿瘤、睾丸肿瘤;和/或
-骨或关节疾病,优选地选自:类风湿性关节炎、滑膜炎、软骨和/或骨破坏、骨髓炎、滑膜组织肥大和/或增生、骨赘形成、赘生物和/或转移及其组合;和/或
-血管疾病,优选地选自:动脉粥样硬化、血管瘤、血管内皮瘤及其组合;和/或
-皮肤病,优选地选自:银屑病、疣、化脓性肉芽肿、毛发生长、卡波西肉瘤、创伤瘢痕瘤、变应性水肿、赘生物及其组合;和/或
-在脂肪组织疾病,优选地肥胖症中观察到的血管生成;和/或
-糖尿病和/或其后果,优选地视网膜病变和/或糖尿病足;和/或
-造血系统疾病,优选地AIDS和/或卡波西肉瘤。
为了上述医学目的,本发明的肽和组合物可以任选地与用于治疗上述疾病的已知药物组合,或者也可以在用于治疗上述疾病的已知药物之前或之后使用。
此外,本发明的肽或组合物可以与用于治疗上述疾病的外科手术、放射治疗或化学治疗类型的已知治疗相关联。
本发明的肽或如以上所描述的包含所述肽的组合物可以配制成通过任何途径施用。施用途径优选地选自:全身途径,优选地口服途径、灌胃、舌下或直肠途径,局部、皮下、肌内、静脉内、动脉内、腹膜内、皮内和表皮内途径。
本发明的肽或组合物可以配制成固体,例如丸剂、片剂、颗粒剂、可溶性颗粒剂、小丸(pellet)、珠、锭剂等。可选地,本发明的肽或组合物可以配制成液体溶液,例如通过注射、吸入或雾化施用,或配制成滴剂或喷雾剂。
本发明的肽或如以上所描述的包含所述肽的组合物可以作为推注(bolus)施用。
本发明的肽或如以上所描述的包含所述肽的组合物可以通过医疗装置施用,例如通过支架、泵或贴片施用。
施用优选地可以是连续的,通过受控释放或通过恒定释放,优选地使用眼部药物递送装置。
特别优选通过口服途径或灌胃施用。事实上,如前所述,当通过灌胃施用时,本发明的肽,包括iVR1,显示出在抑制血管生成/新血管生成方面也是有效的。它们显示出对抑制眼睛中的血管生成/新血管生成也是有效的;换句话说,当本发明的肽,包括iVR1,通过灌胃施用时,令人惊讶地观察到眼睛中血管生成/新血管生成的抑制。所述及的血管生成/新血管生成优选地是VEGFR1依赖性的。
根据这一科学证据,本发明的另一方面涉及通过口服或灌胃施用的本发明的肽,优选地至少一种特征在于式IIa的肽,更优选地特征在于式IIb的肽,即iVR1-Cys,和/或特征在于式IIc的肽,即iVR1,或包含所述肽的组合物,用于治疗由不正确的(优选地增加的)血管生成/新血管生成(优选VEGFR1依赖性血管生成/新血管生成)引起的疾病或在任何情况下与不正确的(优选地增加的)血管生成/新血管生成(优选VEGFR1依赖性血管生成/新血管生成)相关的疾病。
所述疾病/状况优选地选自:
-新生血管性眼病,优选地选自:黄斑水肿、湿性型年龄相关性黄斑变性、糖尿病性视网膜病变、早产儿视网膜病变、视网膜中央静脉阻塞视网膜病变、玻璃体出血和视网膜脱离及其组合;和/或
-实体肿瘤和/或肿瘤转移,所述肿瘤优选地选自:白血病和淋巴瘤,优选地急性淋巴细胞性白血病、急性非淋巴细胞性白血病、慢性淋巴细胞性白血病、多发性骨髓瘤、霍奇金淋巴瘤、霍奇金病、婴儿或成人实体肿瘤、脑肿瘤、成神经细胞瘤、成视网膜细胞瘤、肾母细胞瘤、骨肉瘤和软骨肉瘤、肺肿瘤、结肠直肠癌、乳腺癌、前列腺癌、子宫癌、卵巢癌、泌尿系统癌症、膀胱癌、口腔肿瘤、胰腺肿瘤、黑素瘤和皮肤肿瘤、胃肿瘤、脑肿瘤、甲状腺肿瘤、喉肿瘤、肝肿瘤、睾丸肿瘤;和/或
-骨或关节疾病,优选地选自:类风湿性关节炎、滑膜炎、软骨和/或骨破坏、骨髓炎、滑膜组织肥大和/或增生、骨赘形成、赘生物和/或转移及其组合;和/或
-血管疾病,优选地选自:动脉粥样硬化、血管瘤、血管内皮瘤及其组合;和/或
-皮肤病,优选地选自:银屑病、疣、化脓性肉芽肿、毛发生长、卡波西肉瘤、创伤瘢痕瘤、变应性水肿、赘生物及其组合;和/或
-在脂肪组织疾病,优选地肥胖症中观察到的血管生成;和/或
-糖尿病和/或其后果,优选地视网膜病变和/或糖尿病足;和/或
-造血系统疾病,优选地AIDS和/或卡波西肉瘤。
将本发明的肽或组合物施用于任何需要它的动物,优选地需要抑制VEGFR-1依赖性新血管生成的动物。
所述动物优选地为哺乳动物,更优选地所述动物为人类。
施用的如以上所描述的肽或组合物的有效剂量优选地范围为:
-在10mg/剂量和2000mg/剂量之间,其中优选地全身施用,优选地通过全身性肠内途径,更优选地口服、舌下或直肠施用;或者
-在1mg/剂量和100mg/剂量之间,其中优选地玻璃体内施用。
可选地,施用的有效剂量优选地在0.16mg/Kg体重和33.3mg/Kg体重之间的范围内。
治疗方案优选地提供单剂量或多剂量。
本发明的序列根据国际标准WIPO ST.25注释,并且其描述用程序Patent-In 3.5生成。序列的描述在此随附。
在本发明中,表III中标识的序列和具有从80%至99.9%范围内的同一性的序列应被认为是所描述的。
表III
实施例
测试PlGF或VEGF-A与VEGFR-1受体结合的测定是基于ELISA方法[Ponticelli等人,JBC.2008Dec 5;283(49):34250-9],并使用从R&DSystems获得的试剂进行。
人类重组受体VEGFR-1,特别是由与人类IgG Fc结构域融合的受体的七个胞外结构域组成的形式(R&D Systems,目录号321-FL),以在PBS pH 7.5中的0.5μg/ml的浓度在96孔微孔板的孔中(100μl/孔)在室温(RT)粘附16小时。
在使用由含有3%BSA的PBS pH 7.5组成的缓冲溶液封闭孔中的非特异性结合位点后,向粘附有受体的孔中添加PBET(PBS pH 7.5、BSA 0.1%、EDTA 5mM、Tween 0.004%)中的人类来源的5ng/ml重组PlGF(R&D Systems,目录号264-PG)或5ng/ml重组VEGF-A (R&DSystems,目录号293-VE)。
与配体(即PlGF或VEGF-A)同时地,以包含在780nM和50000nM之间的浓度添加分级剂量的iVR1、iVR1-Cys或对照肽(PC-[(S-Ser)-(S-Ala)-(S-Cha)三肽,具有与iVR1肽的结构相同的四聚体结构])。结合反应在37℃进行1小时,然后在室温进行1小时。
在结合和/或竞争步骤结束时,以PBET中300ng/ml的浓度向孔中添加抗人类-PlGF生物素化多克隆抗体(R&D Systems,目录号BAF264)或抗人类-VEGF-A(R&D Systems,目录号BAF293)。在37℃孵育1小时,然后在室温孵育1小时后,向孔中添加HRP缀合的抗生物素蛋白-链霉抗生物素蛋白系统(Vectastain elite ABC试剂盒)和HRP底物(邻苯二胺-Sigma,目录号P1526)。通过确定490nM处的吸光度进行量化。
肽的任何抑制活性以剩余结合的%来表示,将四聚体肽存在的情况下获得的PlGF或VEGF-A与受体结合的数据与四聚体肽不存在时获得的数据进行比较。iVR1代表抑制PlGF/VEGFR-1或VEGF-A/VEGFR-1相互作用的阳性对照。
结果在表IV和表V中给出,并表明,
因此,iVR1-Cys具有的抑制能力为iVR1的抑制能力的约10倍高,并且因此预期iVR1-Cys在涉及血管生成/新血管生成抑制的相同体外和体内实验方案中可以以10倍低的剂量使用,以获得与用iVR1获得的效果相同的效果。
PC未提供抑制。
表IV–PlGF/VEGFR-1相互作用的剂量依赖性抑制
表V–VEGF-A/VEGFR-1相互作用的剂量依赖性抑制
具有式(II)但Y2不同于D-半胱氨酸的四聚体肽抑制VEGF-A/VEGFR-1结合的能力通过以上描述的结合测定来评估。肽的Y2和相应的抑制VEGF-A/VEGFR-1相互作用的IC
表VI–抑制VEGF-A/VEGFR-1相互作用的IC
进行了PlGF诱导的受体VEGFR-1磷酸化的测定,以评价肽iVR1-Cys的抑制能力并将其活性与iVR1的活性进行比较。
为了激活VEGFR-1,使用了过表达该受体的细胞系,称为293-VEGFR-1,其通过稳定转染HEK-293细胞获得(Errico,M.等人2004JBC,279:43929-43939)。
为此目的,培养293-VEGFR-1细胞,直到达到亚汇合,并且然后通过将细胞在无血清培养基中保持/孵育至少16小时,使其“饥饿”。
在饥饿步骤结束时,去除培养基,并用100μM Na
然后(1)在37℃在用于饥饿的培养基中用单独的20ng/ml PlGF刺激细胞10分钟,和(2)在5μM浓度的肽的存在下用PlGF刺激细胞。
使用浓度为3.3nM的抗人类PlGF中和单克隆抗体(Thrombogenics)作为抑制对照。PBS用作阴性对照。
在孵育结束时,用冷的100μM Na
1:500稀释的抗p-VEGFR-1抗体(R&D Systems,目录号AF4170)用于检测磷酸化的VEGFR-1,同时通过使用1:500稀释的抗VEGFR-1抗体(Sigma-Aldrich,目录号V4262)检测受体的非磷酸化形式来进行归一化。
如图1中示出的,肽iVR1-Cys以结合测定(参见实施例1)中确定的其IC
激光诱导脉络膜新血管形成的实验模型需要对布鲁赫膜(Bruch’smembrane)产生损伤,布鲁赫膜的损伤使脉络膜与视网膜色素上皮(RPE)分开。这种损伤是由激光诱导的烧灼引起的,烧灼导致布鲁赫膜穿孔,从而激活脉络膜视网膜血管化,即从脉络膜开始侵入上覆的视网膜组织的新血管的生长。这种小鼠模型总括了渗出型人类年龄相关性黄斑变性(AMD)的主要特征,并且事实上通常用作AMD的临床前模型。它使得能够评估感兴趣的分子的抗血管生成活性。
为了能够使小鼠的眼底可视化并利用激光诱导损伤,按照下述实验程序,使用了Micron IV集成系统。
首先,通过施加0.5%托吡卡胺滴眼液诱导动物瞳孔扩张。然后通过腹膜内注射氯胺酮和赛拉嗪溶液(分别为80mg/Kg和10mg/Kg)对动物进行麻醉。镇静后,将动物放置在台(stand)上,并将2.5%羟丙基甲基纤维素水溶液施加在两只眼睛上。它具有防止角膜脱水和通过将Micron IV的照相机镜头与溶液接触来改进眼底的可视化的双重功能(类似于在具有浸没物镜的显微术中使用的程序)。
为了用激光诱导损伤,首先将激光定向器(laser pointer)激活和聚焦,以便使用RPE层作为参考来施加激光束。施加激光束的区域必须远离视网膜的主要血管,以防止可能的出血。通过施加激光束后立即形成的泡证实了在布鲁赫膜层的灼烧效率。激光束的施加条件是功率为200mW,持续100msec。
根据文献中呈现的数据,Lambert等人(Nature Protocols,2013,8:2197)在文章中做了很好的总结,已知该实验模型中的最大新血管形成是在损伤后7天获得的。
使用C57Bl6/J小鼠,每组n=5只。在用激光诱导损伤的程序结束时,立即进行玻璃体内注射,并使用带有32g针头的Hamilton注射器施用在1μL DMSO中的10μg和50μg的iVR1-Cys或iVR1和50μg的PC。作为对照,注射了DMSO本身。
七天之后,处死动物,并且摘取眼睛,并以4%多聚甲醛固定。随后,在立体显微镜下去除由角膜、虹膜和晶体组成的眼睛前段。剩下的部分,定义为“眼杯”或后段,由巩膜、脉络膜、RPE和视网膜组成,在0.7%FITC-单叶加纳籽(Griffonia simplicifolia)同工凝集素B4(Vector Laboratories,Burlingame,CA)的存在下孵育16小时。经过一系列洗涤后,去除视网膜,并在RPE/脉络膜上进行四次切割,使得RPE/脉络膜能够封固在载玻片上,以便在荧光显微镜下观察。对脉络膜新血管形成的体积进行量化。为了评估每个斑点(spot)的体积,在RPE细胞的水平从上表面到最深的焦平面采集了一系列图像(Z轴叠加(Z-Stacks),约20-25个图像),每个图像具有1μm的厚度。荧光的体积通过ImageJ程序(NIH,Bethesda,MD)测量,取每个单个平面的荧光面积之和。
对于iVR1 10μg和50μg在n=12个和15个斑点上进行CNV的量化,对于iVR1-Cys 10μg和50μg在n=10个和8个斑点上进行CNV的量化;对于PC在n=15个斑点上进行CNV的量化,并且对于DMSO在n=14个斑点上进行CNV的量化。图2中给出的结果表明,这两种肽都能够引起对新血管形成的剂量依赖性抑制。在较高剂量(50μg),获得了强有力的、显著的且相当的新血管形成抑制能力:对比媒介物和PC,iVR1-Cys-75.9%和-74.6%(p>0.002);对比媒介物和PC,iVR1-73.9%和-76.5%(p>0.0002)。
在10μg的剂量,对比媒介物和PC,iVR1引起新血管形成的抑制为37.8%和39.3%(p<0.05),而对比媒介物和PC,iVR1-Cys引起新血管形成的抑制为48.9%和51.0%(p<0.02)。因此,在低浓度,肽iVR1-Cys展示出比肽iVR1更大的抑制效力,因为它进一步减少了19.3%的新血管形成。因此,在所使用的较高剂量,肽的抑制能力可能达到了最大阈值。
对于口服施用(灌胃)的实验,按照前述实验程序,在C57Bl6/J小鼠中诱导脉络膜新血管形成,每组n=5只动物。在诱导损伤后,动物从麻醉恢复后,立即开始施用肽iVR1和iVR1-Cys以及媒介物,在实验方案规定的7天内每天两次。基于先前腹膜内施用肽iVR1获得的数据(Cicatiello等人2015,Oncotarget,6,10563–10576),肽以50mg/Kg的剂量施用。
为了能够进行口服施用,将肽溶解在DMSO中,并且然后与Nutilis食品增稠剂混合,从而得到由9份Nutilis和1份DMSO组成的最终混合物。
这些物质的制备浓度使得每个单次施用可以使用200μl DMSO混合物中的9:1Nutilis/物质,使用合适的针头直接施用到动物的胃中用于灌胃,针头的开口为20号。在对照组中,施用200μl 9:1的Nutilis/DMSO混合物。
在实验结束时,处死动物,取出眼睛并解剖以分离RPE-脉络膜,并通过免疫荧光分析确定CNV的体积,如以下所描述的。
对于iVR1-Cys在n=18个斑点上进行CNV的量化,对于iVR1在n=20个斑点上进行CNV的量化,并且对于媒介物在n=10个斑点上进行CNV的量化。
结果在图3中给出,并展示出,
肽iVR1-Cys在50mM磷酸盐缓冲溶液pH 7.3中的10%血清(胎牛血清,FCS)中在168h时的稳定性如Ponticelli等人描述的确定,依赖于其中描述的基于RP-HPLC色谱术的方法[Ponticelli等人,J Biol Chem.2008Dec 5;283(49):34250-9]。
参考曲线通过将化合物以0.1μmol/L和1000μmol/L之间的递增浓度溶解在DMSO中以获得完全溶解来构建。然后,以10μmol/L的初始浓度与10%FCS接触的分子的残留浓度通过以下来确定:在时间t=0时抽取3个等分试样,然后在最初的12小时内每小时抽取3个等分试样,并且然后在24h、72h、120h和168h抽取3个等分试样。将等分试样用0.1M乙酸以1∶1稀释,以分离任何结合到白蛋白的肽,离心以去除任何沉淀的物质,并在Ponticelli等人报道的条件下通过RP-HPLC进行分析。在等分试样中检测到的残留肽的量表示为相对于初始量的百分比,绘制为随时间变化的表。结果在表VII中示出为三次测定的平均值±标准差(SD)。
表VII
- 肽及其医学用途
- 一种海参肽特殊医学用途配方食品及其制备方法