成像装置
文献发布时间:2023-06-19 11:35:49
技术领域
本发明涉及一种成像装置,诸如激光打印机、复印机或传真机,其通过把利用电子照相方法或类似方法已经形成在图像承载构件上的调色剂图像转印到转印材料上来形成记录图像。
背景技术
通常,电子照相成像装置通过在用作图像承载构件的感光鼓的表面上形成显影剂图像(调色剂图像)并将显影剂图像转印到用作转印介质的转印材料上来形成图像。已经提出了各种显影剂补充系统。典型的示例是处理盒。根据一种处理盒系统,感光鼓和显影剂容器是一体的,并且当显影剂已经用完时,可以用新的处理盒更换处理盒。这是有利的,因为用户可容易地进行维护。
同时,当显影设备的调色剂用完时,向显影设备补充新的调色剂的调色剂补充系统也是已知的。例如,PTL 1描述了一种调色剂补充容器,其能够可拆卸地附接到成像装置上,并且当调色剂补充容器附接到成像装置上时,调色剂经由配备有输送螺杆的调色剂输送通道从调色剂补充容器输送到显影容器。
然而,已知的调色剂补充系统存在以下问题。也就是说,例如,必须提供配备有输送螺杆的调色剂输送通道,并且这已经导致装置在结构上更加复杂或者尺寸更大。
引用列表
专利文献
PTL 1:日本专利特开No.8-30084
发明内容
根据本发明的一个方面,提供了一种成像装置,包括:图像承载构件;显影剂承载构件;能够移动的搅拌构件;框架,所述框架支撑所述显影剂承载构件并构成显影剂容纳室,所述显影剂容纳室容纳待被供应到显影剂承载构件的显影剂;以及盖,所述盖可以在第一位置和第二位置之间移动。所述显影剂承载构件通过使用显影剂而使由曝光单元形成在图像承载构件上的静电潜像显影。所述显影剂容纳室具有附接端口,其中储存有显影剂的显影剂供应容器可拆卸地附接并定位到所述附接端口。所述第一位置是所述盖覆盖附接端口的位置,所述第二位置是使附接端口能从外部进入的位置。当盖处于所述第二位置并且显影剂供应容器附接到附接端口以允许显影剂供应容器的内部和显影剂容纳室彼此连通时,储存在显影剂供应容器中的显影剂由于显影剂的自重而移动到显影剂容纳室。在显影剂供应容器附接到附接端口的状态下,从重力的竖直方向看,显影剂供应容器的上部部分相对于成像装置中的所述第一位置位于向外的上侧;并且当从附接端口拆卸显影剂供应容器时,盖可以从所述第二位置移动到所述第一位置。
根据参考附图对示范性实施例的以下描述,本发明的其它特征将变得显而易见。
附图说明
图1A是示出根据一个实施例的成像装置和调色剂瓶的横截面结构的图。
图1B是示出根据该实施例的成像装置和调色剂瓶的横截面结构的另一张图。
图2A是示出根据一个实施例的从与显影辊的纵向方向正交的方向观察的显影设备和调色剂瓶的图。
图2B是示出用于检测容纳在显影剂容纳室中的剩余显影剂的量是否低于特定量的结构的图。
图3A是示出根据一个实施例的用于开口的帽的示例的图。
图3B是示出根据一个实施例的用于开口的帽的另一个示例的图。
图3C是示出根据一个实施例的用于开口的帽的另一个示例的图。
图4A是示出根据另一实施例的具有开口的显影设备的图。
图4B是示出根据另一实施例的具有开口的另一显影设备的图。
图5是示出根据一个实施例的显影电流检测系统的图。
图6是示出在一个实施例中使用的法拉第笼的结构的图。
图7是示出在一个实施例中如何确定调色剂摩擦起电降低的流程图。
图8是调色剂的示意图。
具体实施方式
现在将参考附图描述本发明的实施例。下面描述的实施例并不限制权利要求所阐述的本发明,并且并非实施例中描述的特征的所有组合对于本发明提供的解决方案都是必要的。
[第一实施例]
[成像装置的整体结构]
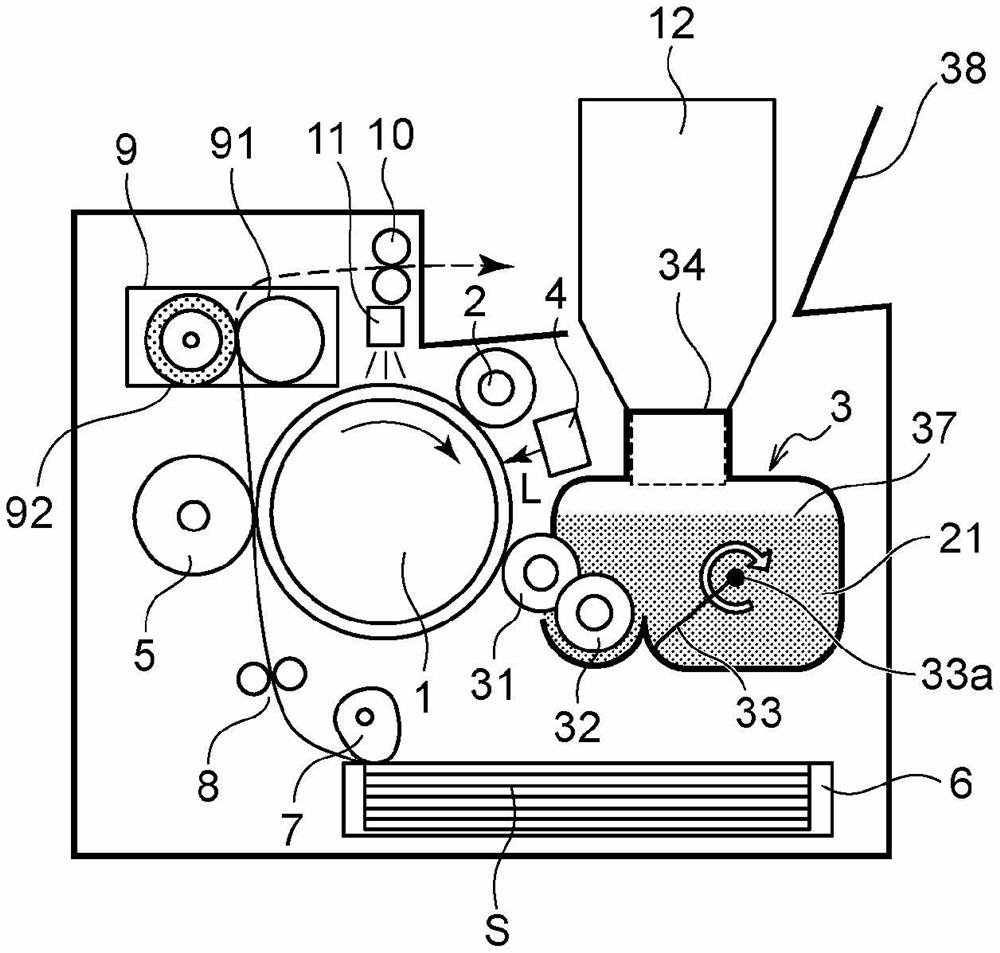
图1A示出了用作单色打印机的成像装置的示意性结构。
本实施例的成像装置包括感光鼓1,感光鼓是用作图像承载构件的圆筒形感光体。用作充电单元的充电辊2和用作显影单元的显影设备3围绕感光鼓1设置。从图的向下方向看,用作曝光单元的曝光设备4设置在充电辊2和显影设备3之间。转印辊5与感光鼓1压力接触。
调色剂在显影设备3的显影剂容纳室37中。本实施例的调色剂的粒径为6微米,并且其常规充电极性是负极性。
本实施例的感光鼓1是可带负电荷的有机感光体。感光鼓1包括鼓形铝基板和基板上的感光层,并且由驱动设备(未示出)驱动,从而以特定的处理速度沿图中箭头方向(顺时针)旋转。在本实施例中,处理速度等于感光鼓1的周向速度(表面移动速度)。
充电辊2以特定的压力接触力接触感光鼓1,并形成充电部分。用作充电电压供应单元的充电高压电源(未示出)向感光鼓1施加特定的充电电压,并且由此将感光鼓1的表面均匀地充电到特定电势。在本实施例中,感光鼓1由充电辊2充负电。
本实施例的曝光设备4是激光扫描仪设备。曝光设备4输出对应于从外部设备(诸如主机)输入的图像信息的激光束,并扫描和曝光感光鼓1的表面。由于这种曝光,在感光鼓1的表面上形成对应于图像信息的静电潜像(静电图像)。曝光设备4不限于激光扫描仪设备,并且例如,可以替代地采用LED阵列,其包括沿感光鼓1的纵向方向排列的LED。
在本实施例中,将接触显影方法用作显影设备3的显影方法。在显影设备3中,由框架(壳体)支撑用作显影剂承载构件的显影辊31,该框架构成容纳调色剂的显影剂容纳室37。用作显影剂供应单元的调色剂供应辊32也由框架支撑。通过在显影辊31和感光鼓1之间的相对部分(显影夹持部)处使用由显影辊31传送的调色剂将形成在感光鼓1上的静电潜像显影成调色剂图像。在该过程中,显影电压从用作显影电压施加单元的显影高压电源(在图5中由附图标记103表示)施加到显影辊31。在本实施例中,通过反向显影过程使静电潜像显影。也就是说,使得具有与感光鼓1的充电极性相同的极性的调色剂粘附到充电的感光鼓1的由于曝光而使电荷已衰减的部分,并且静电潜像由此显影成调色剂图像。
此外,供应调色剂的调色剂供应辊32可旋转地抵靠显影辊31。在本实施例中,采用单组分非磁性接触显影方法。或者,可以采用双组分非磁性接触/非接触显影方法。此外,可以采用单组分磁性接触/非接触显影方法或双组分磁性接触/非接触显影方法作为显影方法。在本实施例中,将通过聚合方法形成的聚合调色剂用作显影剂的一个示例。
用作搅拌构件的搅拌叶片33安装在显影剂容纳室37内。在横截面中观察,当搅拌叶片33通过从驱动设备(未示出)提供的驱动力绕旋转轴33a旋转时,搅拌叶片33将调色剂送到显影辊31和调色剂供应辊32上。搅拌叶片33(搅拌构件)可以直接从驱动设备(未示出)接收驱动力。此外,搅拌叶片33(搅拌构件)可以接收来自驱动齿轮(未示出)的驱动力,该驱动齿轮传递来自驱动设备(未示出)的驱动力。如图所示,搅拌叶片33绕旋转轴33a顺时针旋转。更具体地,由搅拌叶片33推动和移动搅拌叶片33的旋转半径内的调色剂。将移动的调色剂的某些部分送到显影辊31和调色剂供应辊32上。例如由在显影辊31的纵向方向上延伸的板片状构件形成搅拌叶片33,并且在此种情况下,在横截面中看,该板片推动旋转半径内的调色剂,并且使得调色剂移动。然后,将移动的调色剂的某些部分送到显影辊31和调色剂供应辊32上。此外,如图所示,搅拌叶片33具有在与搅拌叶片33的旋转方向相交的方向上延伸的形状,并且朝向显影辊31和调色剂供应辊32进给调色剂。当调色剂瓶12通过重力相对于调色剂的移动方向附接到开口34(附接端口部分)时,搅拌叶片33仅仅是设置在感光鼓1上游和调色剂瓶12的供应端口下游的空间中的一个搅拌叶片。该空间中只有一个搅拌叶片33。在本实施例中,在该空间中仅设置一个搅拌叶片33意味着能够向显影辊31和调色剂供应辊32进给调色剂的搅拌叶片33基本上只有一个。
搅拌叶片33还具有循环在显影中未使用并从显影辊31剥离的调色剂的功能,使得显影剂容纳室37中的调色剂的退化程度均匀。搅拌叶片33还具有调平因其自身重量而从调色剂瓶12掉落并移动到显影剂容纳室37的调色剂的功能。通过用搅拌叶片33调平补充的调色剂可精确地检测剩余调色剂的量。
转印辊5可由弹性构件形成,诸如由聚氨酯橡胶、三元乙丙橡胶(EPDM)或丁腈橡胶(NBR)组成的海绵橡胶或类似物。
转印辊5压靠在感光鼓1上,以形成感光鼓1和转印辊5压力接触的转印部分。用作转印电压施加单元的转印高压电源(未示出)连接到转印辊5,并且在特定时间施加特定电压。
与形成在感光鼓1上的调色剂图像到达转印部分的时刻同步地,盒6中的转印材料S由纸张进给单元7进给,穿过阻力辊对8,并被传送到转印部分。感光鼓1上形成的调色剂图像由转印辊5转印到转印材料S上,其中通过转印高压电源向转印辊施加特定的转印电压。
在转印调色剂图像之后,将转印材料S传送到定影设备9。定影设备9是膜加热型定影设备,其配备有定影膜91和加压辊92,定影膜具有内置的定影加热器(未示出)和用于测量定影加热器的温度的内置热敏电阻(未示出),加压辊用于与定影膜91压力接触。通过加热和加压转印材料S来定影调色剂图像,并且转印材料S经由排出辊对10从装置排出。
在感光鼓1的旋转方向上,曝光前设备11设置在转印部分的下游和带电部分的上游,曝光前设备用作擦除感光鼓1上的电荷的电荷擦除单元。曝光前设备11在感光鼓1进入带电部分之前擦除感光鼓1的表面电势,使得在带电部分稳定地进行放电。
通过以下过程去除没有转印到转印材料S上并残留在感光鼓1上的转印残留调色剂。转印残留调色剂是带正电荷的调色剂和带负电荷但电荷不足的调色剂的混合物。当转印后感光鼓1上的电荷被曝光前设备11擦除时,在充电期间发生均匀放电,因此转印残留调色剂再次带负电荷。随着感光鼓1旋转,在带电部分中再次带负电荷的残留的转印残留调色剂到达显影部分。现在,将通过区分位于感光鼓1的曝光部分的转印残留调色剂和位于感光鼓1的非曝光部分的转印残留调色剂来描述已经到达显影部分的转印残留调色剂的行为。在显影部分中,由于显影电压和感光鼓1的未曝光部分的电势之间的电势差,附着到感光鼓1的未曝光部分的转印残留调色剂迁移到显影辊31,并被回收到显影剂容纳室37中。回收到显影剂容纳室37的调色剂再次用于形成图像。相反,附着到感光鼓1的曝光部分的转印残留调色剂不会在显影部分中从感光鼓1迁移到显影辊31,而是与来自显影辊31的已显影的调色剂一起迁移到转印部分,转印到转印材料S上,并被从感光鼓1中移除。
在本实施例中,转印残留调色剂被回收到显影设备3中并被重复使用;可选地,可以通过使用抵靠感光鼓1的已知清洁刮刀来回收转印残留调色剂,使得不再使用转印残留调色剂。在任一情况下,本实施例的效果保持不受影响。然而,自然地,根据本实施例的结构,不需要临时存储回收的转印残留调色剂的回收容器,并且因此装置的尺寸可保持紧凑。此外,由于重复使用转印残留调色剂,所以可降低打印成本。
<通过利用显影剂的重量将显影剂从显影剂供应瓶补充到显影剂容纳室中>
安装在装置中的显影设备3具有开口34,该开口用作装配调色剂瓶(显影剂供应瓶)的端口。开口34相对于装置主体的外壳位于装置主体的内部,并且可通过开口34补充调色剂。调色剂瓶12的供给端口和开口34配置成使得调色剂瓶能够可拆卸地附接到开口34。用作最靠近开口34定位的移动构件的搅拌叶片33设置在构成显影剂容纳室37的框架内。结果,补充的调色剂在显影辊的纵向方向上的料位迅速变平,并且在补充调色剂之后可迅速开始打印操作。显影剂容纳室37的框架内的旋转和移动构件的其它示例是显影辊31和调色剂供应辊32。
在下面的描述中,使用了术语“调色剂瓶”;然而,调色剂瓶可附接到装置上并具有容纳要补充或供应到显影设备的显影剂(调色剂)的功能就足够了。因此,调色剂瓶可被称为“显影剂容器”或“显影剂供应容器”等。
这里,详细描述了在本实施例中使用的术语“附接”。如下面描述的图1A、图1B、图4A和图4B所示,“附接”是指其中相对于开口34在水平方向和竖直方向上设定调色剂瓶12的供给端口的位置的状态。在图1B中,将该位置设定为调色剂瓶12的末端装配到显影剂容纳室37中并抵靠显影剂容纳室。在图4B中,将该位置设定为调色剂瓶12的表面的一部分抵靠显影剂容纳室37。将调色剂瓶12附接到主体的机构不限于这种形式。可采用各种机构来设定调色剂瓶12的供应端口相对于开口34的位置。
此外,能够可拆卸地附接到成像装置的调色剂瓶的显影剂存储单元的结构通常由树脂形成;可选地,可以减小树脂的壁厚,并且可以使用柔性片材,该柔性片材容易因用户的抓握动作而变形。柔性片材的预期壁厚为例如约0.03至1毫米。通过减小片材的壁厚,可提供具有袋状显影剂容纳部分的显影剂补充容器。当纸张材料用于形成构成显影剂容纳部分的柔性片材时,可提供环境友好的显影剂补充容器。此外,调色剂瓶12的供应端口可以由树脂形成;或者,作为一种环保措施,可以使用高强度纸板来形成供应端口。
为了让用户将调色剂瓶12附接到成像装置,用户将盖38从第一位置移动到第二位置并打开盖38,使得用户可从外部进入用作附接端口的开口34。盖38配置成在第一位置和第二位置(也称为“打开位置”)之间移动。第一位置是在图像形成期间采取的位置,并且在第一位置处的盖38覆盖装置的内部(附接端口),如图1A所示。第二位置是使得用户能够从外部进入成像装置的内部的位置。当盖38处于第二位置时,用户可进入装置的内部(附接端口),并且可将调色剂瓶12附接到开口34或者从开口34拆下调色剂瓶12。当盖35附接到开口34时,用户从开口34移除盖35。图3A至图3C示出了用于开口34的帽35的一些示例。帽35可以具有任何形状或是任何类型的,只要该构件可密封开口34并将调色剂保持在显影剂容纳室37内。
用作可打开-关闭构件的盖38在图中的盖的左侧的铰接部上摆动,并覆盖或暴露装置的内部。然而,这种结构不是限制性的。例如,可以采用滑动门。可选地,可以采用具有两个门的双门,这两个门铰接到当盖打开时形成在成像装置主体中的开口的相应的相对侧。盖38可采用各种打开-关闭结构。
接下来,如图1B所示,在盖38处于打开位置(第二位置)时调色剂瓶12附接到开口34,调色剂通过其自重移动到显影剂容纳室37,并实现补充。更具体地,当调色剂瓶12附接到开口34时,显影剂容纳室37与调色剂瓶12的内部空间连通,并且存储在调色剂瓶12内部的调色剂通过其自重移动到显影剂容纳室37中。当在催促调色剂补充后将调色剂瓶12中的用于一次调色剂补充的预定量的调色剂(显影剂)或全部调色剂供应到显影剂容纳室37时,调色剂21的料位相对于重力方向高于旋转轴33a。换句话说,相对于重力方向,旋转轴33a在补充之后处于调色剂21的料位之下。这里,在补充调色剂之后,当沿着显影辊31的纵向方向观察时,显影剂容纳室37中的每个调色剂料位不完全相同。因此,在本实施例中,调色剂21的料位是指在补充调色剂之后显影剂容纳室37中调色剂21的料位的平均值。
如此,在本实施例中,由于重力导致调色剂从调色剂瓶12移动到显影剂容纳室37。另一种可想到的调色剂补充方法是这样的方法,该方法包括将调色剂补充瓶供应到调色剂补充通道,该调色剂补充通道不同于显影剂容纳室37并且配备有输送螺杆,并且通过使用输送螺杆经由调色剂补充通道将调色剂移动到显影剂容纳室37。然而,在此种情况下,由于调色剂补充通道,装置的尺寸增加。相反,根据本实施例的调色剂补充系统,可减小装置的尺寸。此外,为了向上述调色剂输送通道供应补充调色剂,需要时间来完成经由调色剂输送通道的调色剂输送,并且用户必须等待打印的重新开始。本实施例在这一点上也提供了改进。
如图1B所示,当调色剂瓶12附接到开口34时,调色剂瓶12在重力方向上的上部部分从装置主体的外壳朝向向外的上侧(重力方向上的上侧)突出。因此,调色剂瓶12不必完全容纳在成像装置中,并且可减小成像装置的尺寸。此外,由于调色剂瓶12在补充期间在重力的竖直方向上向外突出,所以当调色剂瓶12被从开口34拆卸并从装置移除时,盖38可以采取第一位置,所述第一位置是关闭位置。“关闭位置”是指在图像形成期间盖38所采取的位置,并且是指盖38覆盖开口34或成像装置内部的位置。
此外,如图1A所示,在搅拌叶片33处于倾斜状态时,操作可以停止,使得在调色剂补充期间,搅拌叶片33将补充的调色剂引导至显影辊31和调色剂供应辊32。如此,当搅拌叶片33用作调色剂引导构件时,调色剂可更快地补充到显影辊31。
调色剂瓶12的供应端口的形状和开口34的形状不限于图1A和图1B所示的形状,只要供应端口和开口34彼此能够可拆卸地附接即可。例如,在图4A中,开口34具有从显影剂容纳室37的表面突出的形状。突出部分的内壁延伸到显影剂容纳室37的内部。内壁向下引导调色剂瓶12的供应端口的表面(外周表面),并设定调色剂瓶12相对于显影剂容纳室37的位置。换句话说,由于调色剂瓶12的侧表面的部分与开口34的边缘抵靠,调色剂瓶12的向下运动受到限制。由图4A中的虚线表示延伸到内部的侧壁。
可选地,如图4B所示,调色剂瓶12可以具有与显影剂容纳室37的表面抵靠的调色剂瓶表面(部分),并且可以由这些表面之间的抵靠限制调色剂瓶12的向下运动。这些表面之间的抵靠也决定了调色剂瓶12在水平方向上的位置。
[装入调色剂瓶中的显影剂量]
现在将描述装入调色剂瓶12中的显影剂(调色剂)的量。可适当选择装入调色剂瓶12中的调色剂的量;然而,在本实施例中,装入调色剂瓶12中的调色剂的量可以是A[g]或更多且B[g]或更少。这里,A[g]是当显影设备3采取用于图像形成的姿态时,相对于包括显影辊31的最高位置(最高点)的水平面,包含在显影剂容纳室37的下侧上(下部部分中)的调色剂(显影剂)的量。换句话说,即使当在空的、调色剂耗尽的显影剂容纳室37上进行调色剂补充时,也可将调色剂补充到显影辊31由补充的调色剂覆盖的料位。当存储在图1A所示的调色剂瓶12内的调色剂21被供应到图1A所示的显影剂容纳室37时,调色剂瓶12内的所有调色剂21被供应到如图1B所示的显影剂容纳室37。
图2A示出了从与显影辊31的纵向方向正交的方向观察时显影设备3和调色剂瓶12之间的关系。显影剂容纳室37在纵向方向上延伸,并且具有足够的体积来容纳储存在调色剂瓶12内的所有调色剂21。
此外,B[g]表示可装入显影剂容纳室37中的最大调色剂量和在装置开始催促用户补充调色剂时容纳在显影剂容纳室37中的剩余调色剂的阈值量之间的差值。图2B示出了用于检测容纳在显影剂容纳室37中的剩余显影剂的量是否低于特定量的结构。该结构包括光接收单元22,其接收从发光单元23发射的光。当容纳在显影剂容纳室37中的调色剂的量足够大时,来自发光单元23的光由调色剂阻挡,并且光接收单元22不接收光。当剩余调色剂的量低于特定量(特定体积)时,光接收单元22接收来自发光单元23的光,并且控制器101检测到剩余调色剂的量低于特定量。控制器101识别来自光接收单元22的经由图中未示出的信号线输入的输出信号,从而检测和感测剩余调色剂的量低于特定量。当在显影剂容纳室37中的搅拌叶片33旋转的同时检测剩余调色剂的量时,光被搅拌叶片33搅拌的调色剂阻挡的时长根据剩余调色剂的量而改变。控制器101可以通过光被阻挡的时长或光被接收的时长的差异来估计剩余调色剂的量。
当控制器101检测到光接收单元22已经接收到光时,控制器101经由图中未示出的输出I/F向外部设备发送催促调色剂补充的输出。换句话说,控制器101用作输出设备,当控制器101检测到剩余调色剂的量减少到低于特定量的量时,该输出设备发送催促调色剂补充的输出。外部设备的示例包括显示设备、扬声器和数据发送器。输出I/F可以是有线的或无线的。
应该注意的是,与B[g]一样,A[g]可以是在空的显影剂容纳室37中覆盖显影辊31所需的调色剂的量和在开始催促调色剂补充时显影剂容纳室37中剩余调色剂的量之间的差值。替代性地,与A[g]一样,B[g]可以是能够装入空的显影剂容纳室37中的调色剂的最大量。
A[g]和B[g]的设定不同。
如上所述,即使当被催促补充调色剂的用户通过将容纳在储存的调色剂瓶12中的所有显影剂补充到显影剂容纳室37中来补充调色剂时,显影剂容纳室37中的显影剂的量也没有达到显影剂容纳室37中可容纳的显影剂的最大量。因此,当用户在补充调色剂之后从成像装置移除调色剂瓶12时,可避免调色剂溢出。此外,在补充调色剂之后从成像装置移除调色剂瓶之后,将如图3A、图3B或图3C所示的帽35装配到显影剂容纳室37的开口34中。由于假定调色剂瓶是空的,所以可简化帽35的结构。
[保持所述装置处于去激活状态]
成像装置配备有用于检测盖38打开的光学传感器或机械传感器(图中未示出)。当控制器101接收到指示盖打开的信号时,控制器101不允许成像操作。即使从外部输入打印命令,也不允许涉及处理单元(诸如感光鼓1)的驱动的成像。作为检测盖打开的替代,可以检测调色剂瓶12的附接状态。换句话说,当传感器(图中未示出)检测到调色剂瓶12附接到开口34时,控制器101不允许涉及处理单元(诸如感光鼓1)的驱动的成像。成像装置可以通过检测安装在装置主体中的机械传感器被调色剂瓶12压下来检测调色剂瓶12的附接状态。此外,当存储单元(至少包括存储元件和电接触部分)安装在调色剂瓶12中时,存储读取设备安装到装置主体。在此种情况下,成像装置可以例如确定是否可以在存储读取设备和调色剂瓶12的存储单元之间执行特定的通信,然后基于该结果确定调色剂瓶12是否被附接。
如上所述,根据本实施例,显影剂补充系统可配置为更简单的结构,该结构涉及通过使用调色剂自身的重量将调色剂从调色剂瓶12移动到显影剂容纳室37。此外,可实现更加用户友好的调色剂补充。例如,在补充调色剂之后可快速恢复成像,并且可减少停机时间。此外,例如,不需要复杂的调色剂输送通道等,因此可减小成像装置的尺寸和成本。此外,例如,由于通过将调色剂瓶12附接到位于成像装置内部的开口34来补充调色剂,可避免调色剂补充型成像装置可能出现的问题,诸如调色剂飞散。
[第二实施例]
本实施例的成像装置的结构与第一实施例中的相同,因此省略其详细描述。在本实施例中,描述了在补充调色剂期间出现的图像问题及其对策。
本实施例提供了一种调色剂补充系统,其用于抑制补充雾化的发生。首先,描述了被称为“补充雾化”的现象,这是由于在补充调色剂期间补充的新调色剂和残留在显影设备3中的旧调色剂之间的性质差异而引起的。
[补充雾化]
现在将描述其中引起补充雾化的机理。具有尚未磨损的表面层的新调色剂可容易地保留电荷,因此由每单位重量调色剂保留的电荷量很大(在下文中,该量被称为“调色剂摩擦起电”)。相反,已经在显影部分等中受到重复压力的调色剂具有磨损的表面层,并且外部添加剂(诸如二氧化硅)嵌入调色剂基质(调色剂颗粒)中或从调色剂基质分离,从而降低调色剂的可充电性。可充电性降低的调色剂保留较少的电荷,并且调色剂摩擦起电较小。
此外,当新调色剂和旧调色剂混合时,新调色剂和旧调色剂之间的可充电性差异的影响是显著的。也就是说,当新调色剂接触旧调色剂时,新调色剂的摩擦起电变得比单独使用新调色剂时高,并且旧调色剂的摩擦起电变得比单独使用旧调色剂时低。结果,旧调色剂的摩擦起电变得过低,并且电场几乎不能将旧调色剂保持在显影辊31上,从而导致起雾。
如在第一实施例中所述,由于根据成像装置的结构,补充的新调色剂与显影设备3中的旧调色剂直接接触,因此需要仔细解决补充雾化的发生。
[本实施例的特征]
在本实施例中,为了防止补充雾化,重要的是在补充调色剂期间尽可能消除旧调色剂和新调色剂之间的摩擦起电的差异。换句话说,需要补充新的调色剂,而不会过度降低旧调色剂的调色剂摩擦起电。在本实施例中,间接检测调色剂的摩擦起电,并且在调色剂摩擦起电还未过低时催促补充新的调色剂,以便抑制补充雾化的发生。更具体地,在本实施例中,测量特定量的调色剂显影期间的电流值(显影电流)以测量调色剂的电荷量,并且基于该结果确定是否催促补充新的调色剂。
[显影电流]
施加到显影辊31的显影电压和感光鼓1的曝光部分的电势之间的电势差通常等于或小于放电阈值。因此,显影期间流动的电流显著受到电荷(调色剂)运动的影响。因此,可通过估计每单位时间用于显影的调色剂的重量来预测每单位重量的调色剂电荷量(调色剂摩擦起电)。每单位时间用于显影的调色剂的重量可由每单位面积显影辊表面上调色剂的重量(以下称为“M/S”)和每单位时间显影的调色剂的面积来确定。每单位时间显影的调色剂的面积由显影的调色剂的纵向长度(换句话说,感光鼓1中曝光区域的纵向长度)以及成像装置的处理速度决定。因此,通过执行检测显影电流的特殊操作,每单位时间显影的调色剂的面积可保持在恒定值。换句话说,在本实施例中使用的成像装置中,调色剂的M/S变化很小,并且M/S基本恒定。因此,在显影过程中流动的电流可被认为等于每特定重量的调色剂的电荷量。此外,控制器101可根据每特定重量的电荷量来计算每单位重量的电荷量。将曝光强度设定为成像装置可输出的最大曝光剂量。以这种方式,由于显影辊31上的所有调色剂都被显影,所以可精确地测量调色剂的摩擦电。应注意,当调色剂的M/S变化较大时,可以用本领域已知的附着调色剂量传感器来测量调色剂的M/S,并且可以根据测量结果来确定调色剂的电荷量。
[用于测量显影电流的方法]
图5示出了检测系统,该检测系统在从显影高压电源103向显影辊31施加高电压时检测显影辊31中流动的电流。参考附图,当显影电压(例如,-350伏)从显影高压电源103施加到显影辊31时,电流检测电路102检测在显影辊31、感光鼓1和大地中流动的电流。将指示由电流检测电路102检测的电流值的信号输入控制器101,并且控制器101估计流动电流的大致大小并检测电流。
执行显影电流测量的时刻可是任意的;然而,在本实施例中,在安装成像装置时执行测量,然后每100页打印(每特定数量的已经通过该装置的纸张片材)时执行测量。然而,时刻不限于此。例如,每当从断电状态或省电模式激活成像装置时,可以测量显影电流。
当开始测量显影电流时,控制器101首先开始驱动各个单元,诸如感光鼓1、显影辊31和充电辊2。然后,在特定时刻,在来自控制器101的命令下通过曝光设备4曝光感光鼓1的表面,用调色剂显影形成的静电潜像,并测量显影电流。在本实施例中,由曝光设备4对感光鼓1的纵向方向上的216毫米的范围进行一秒钟的曝光(这对应于感光鼓1的表面的副扫描方向上的长度),并且当形成的静电潜像已经到达显影夹持部时,控制器101基于输入信号测量一秒钟期间的平均电流值I。
[确定是否需要补充调色剂]
根据发明人进行的研究,新调色剂的摩擦起电为约-40[μC/g]。此外,当通过向调色剂摩擦起电绝对值小于-20[μC/g]的劣化调色剂中添加新调色剂来进行补充时,发生补充雾化。换句话说,当新调色剂的摩擦起电是旧调色剂的摩擦起电的大约两倍大时,发生补充雾化。
在本实施例中,使用图6的透视图中所示的法拉第笼13测量调色剂摩擦起电。内部(图中的右侧)被置于减压状态,以便抽吸显影辊上的调色剂,并且通过安装调色剂过滤器133捕获调色剂。法拉第笼13还包括抽吸部分131和支架132。使用捕获的调色剂的质量M和用库仑计直接测量的电荷Q来计算每单位质量的电荷量Q/M[μC/g]。在本实施例中,当确定调色剂摩擦带电已经接近新调色剂的调色剂摩擦带电的一半时,发出催促补充调色剂的消息。
在下面的描述中,通过使用图7中的流程图来描述成像装置的操作。
[步骤1]
当安装成像装置时(当显影剂是新的时),在控制器101的命令下,曝光设备4在由充电辊2充电的感光鼓1的表面上形成静电潜像。曝光1秒的情况下,静电潜像在纵向方向上的尺寸为216毫米(对应于副扫描方向上的长度)。
[步骤2]
当安装成像装置时(当显影剂是新的时),控制器101在先前的静电潜像通过显影夹持部的一秒钟内检测来自电流检测电路102的信号,测量显影电流,并获得当使用新的调色剂时流过的显影电流I
[步骤3至5]
接下来,当控制器101确定已经打印了100页时,控制器101再次测量显影电流。在步骤4中,执行与步骤1中相同的过程,并且在步骤5中,控制器101获得显影电流I
[步骤6]
接下来,控制器101通过基于检测结果计算I
当I
因为调色剂的摩擦起电足够高,所以不发送调色剂补充通知,并且过程返回到步骤3。I
当Ii/I
由于调色剂的摩擦起电接近新调色剂的摩擦起电的一半,所以过程进行到步骤7,并且发出催促补充调色剂的通知。I
[例外过程]
当光学传感器(未示出)在打印100张片材之前检测到调色剂耗尽时,不测量显影电流,过程进行到步骤7,并且发出补充调色剂的通知。
如上所述,根据本实施例,由于可以通过测量显影电流来检测调色剂摩擦起电,所以可在旧调色剂的调色剂摩擦带电过度降低之前补充新调色剂,从而可避免补充雾化的发生。
[第三实施例]
本实施例的成像装置的结构与第一实施例中的相同,因此省略其详细描述。如在第二实施例中所述,已经在显影部分等中受到重复压力的调色剂表现出劣化的可充电性。由此种劣化的调色剂(具有退化的电荷特性的调色剂)产生的图像力较小,并且显影辊31难以承载此种劣化的调色剂。即使劣化的调色剂保留在显影辊31上,劣化的调色剂也不容易静电移动到感光鼓1上,并且具有高调色剂摩擦起电的新调色剂优先用于显影并移动到感光鼓1上。结果,具有劣化的可充电性的劣化调色剂积聚。此外,难以通过静电力来控制具有劣化的可充电性的调色剂,从而容易导致背景起雾,也就是说,其中调色剂在感光鼓1的表面上的背景部分(暗电势部分)中显影的现象。尽管构成雾化的劣化调色剂被排出到外部,但是大部分劣化调色剂积聚,并且随着调色剂补充的重复,积聚的劣化调色剂的量增加。应该避免此种情况。
在本实施例中,描述了这样的显影剂,其抑制了第二实施例中描述的补充雾化的发生并且可减少具有劣化的可充电性的所积聚的劣化调色剂的量的增加。通过将下述显影剂应用于图1A和图1B所示的调色剂补充系统,可实现具有更少图像缺陷的优良的调色剂补充系统。
[改进调色剂的描述]
在本实施例中,使用改进的调色剂来形成显影剂,该显影剂可抑制由打印引起的调色剂摩擦起电的变化,从而防止具有劣化的充电特性的劣化调色剂的积聚,并防止在补充调色剂期间发生补充雾化。更具体地,将包括调色剂颗粒的调色剂用作调色剂,该调色剂颗粒含有粘合剂树脂和着色剂。这种调色剂在最大载荷为2.0×10
将在最大载荷为2.0×10
在最大载荷为2.0×10
将硬度调节到上述特定硬度范围内的方法之一是涉及用具有合适硬度的材料(诸如无机材料)形成调色剂表面层,然后控制表面层的化学结构或宏观结构以使得表面层具有合适的硬度的方法。
能够表现出上述特定硬度的材料的具体示例是有机硅聚合物。可通过直接键合到有机硅聚合物中的硅原子上的碳原子的数量、碳链的长度等来调节有机硅聚合物的硬度。调色剂颗粒可以具有这样的表面层,所述表面层包含有机硅聚合物,并且直接键合到有机硅聚合物中的硅原子上的碳原子的平均数量可以是每个硅原子一个或更多并且三个或更少,因为可容易地将硬度调节到上述特定硬度。直接键合到有机硅聚合物中的硅原子上的碳原子的数量优选为每个硅原子一个或更多且两个或更少,以及更优选为每个硅原子一个。
通过调节化学结构来调节马氏硬度的方法的示例包括调节表面层材料的交联度和聚合度。通过调节宏观结构来调节马氏硬度的方法的示例包括调节表面层上的不规则部的形状和调节连接在突起之间的网络结构。当在表面层中使用有机硅聚合物时,可以通过在有机硅聚合物的预处理过程中调节pH、浓度、温度、时间等来进行这种调节。此外,可通过调节由有机硅聚合物形成的表面层附着在调色剂的核心颗粒上的时刻、形式、浓度、反应温度等来进行调节。
在本实施例中可以采用以下方法。首先,制备调色剂的核心颗粒,并将其分散在水性介质中以获得核心颗粒分散液。核心颗粒包含粘合剂树脂和着色剂。可以在这样的浓度下进行分散,所述浓度使得核心颗粒的固体含量相对于核心颗粒分散液的总量按质量计为10%或更高且40%或更低。核心颗粒分散液的温度可以调节至35℃或更高。核心颗粒分散液的pH可以调节至其中抑制有机硅化合物缩合的pH。其中抑制有机硅聚合物的缩合的pH根据材料而变化,并且优选将pH调节至其中缩合被抑制最多的pH±0.5以内。同时,可以预先水解有机硅化合物。例如,在有机硅化合物的预处理中,有机硅化合物在单独的容器中水解。当有机硅化合物的量为100质量份时,用于水解的进料浓度优选为40质量份或更多且500质量份或更少的水,并且更优选为100质量份或更多且400质量份或更少的水,诸如离子交换水或反渗透(RO)水,其中已从该水中去除离子化合物。示例性的水解条件是2至7的pH、15℃至80℃的温度和30至600分钟的时间。
将所获得的水解液和核心颗粒分散液混合,并将pH调节至适于缩合的值(优选为6至12或1至3,以及更优选为8至12)。结果,在缩合有机硅化合物的同时,可在调色剂的核心颗粒表面上形成表面层。可以在35℃或更高温度下进行60分钟或更长时间的表面层的缩合和附着。可通过在将pH调节到适合缩合的值之前调节将温度保持在35℃或更高的时间来调节表面的宏观结构;然而,为了容易地获得特定的马氏硬度,保持时间优选为3分钟或更长且120分钟或更短。
通过上述方法,可减少反应残留物,并且可在表面层上形成不规则部(凹凸/凹和凸)。此外,由于可在突起之间形成网络结构,因此获得具有上述特定马氏硬度的调色剂变得容易。图8示出了调色剂46的示例。表面层46b覆盖调色剂核心颗粒46a。表面层46b具有凹凸形状。
当表面层包含有机硅聚合物时,有机硅聚合物的附着率优选为90%或更高且100%或更低。该比率更优选为95%或更高。当附着率在此范围内时,通过耐久性和使用导致的马氏硬度的变化很小,并且可保留电荷。用于测量有机硅聚合物附着率的方法如下所述。
[表面层]
当调色剂颗粒具有表面层时,表面层是覆盖调色剂核心颗粒并存在于调色剂颗粒的最外表面上的层。含有有机硅聚合物的表面层明显比常规调色剂颗粒硬。因此,从定影性的观点来看,未设置有表面层的部分可以形成在调色剂颗粒表面的一些部分中。
然而,含有机硅聚合物的表面层的厚度为2.5nm或更小的分割轴的数量的比率(在下文中,该比率也可以被称为表面层具有2.5nm或更小厚度的部分的比率)优选为20.0%或更小。这种条件近似于这样的结构,在所述结构中调色剂颗粒的至少80.0%的表面由包含有机硅聚合物并且厚度为2.5nm或更大的表面层形成。换句话说,当满足该条件时,包含有机硅聚合物的表面层充分覆盖核心表面。该比率更优选为10.0%或更低。可通过使用透射电子显微镜(TEM)的横截面观察来测量该比率,并且在下面给出测量的细节。
[含有机硅聚合物的表面层]
当调色剂颗粒具有包含有机硅聚合物的表面层时,可以包括由式(1)表示的亚结构。
R-SiO
在式(1)中,R代表具有1至6个碳原子的烃基。
在具有由式(1)表示的结构的有机硅聚合物中,硅原子的四个键中的一个键键合到R上,并且其余的三个键键合到O原子上。每个O原子的两个键都键合到Si上,换句话说,形成了硅氧烷键(Si-O-Si)。当从有机硅聚合物的角度考虑Si原子和O原子时,三个O原子被提供给两个Si原子,因此其表示为-SiO
在通过调色剂颗粒的四氢呋喃(THF)不溶物部分的
如上所述,亚结构-SiO
此外,由式(1)表示的亚结构获得的耐久性和式(1)中R的疏水性和可充电性抑制了容易渗出的低分子量(Mw为1000或更低)树脂、低玻璃转变温度(40℃或更低)树脂以及在某些情况下相对于表面层存在于内侧的脱模剂的渗出。
可通过用于形成有机硅聚合物的有机硅化合物的类型和量、反应温度、反应时间、反应溶剂以及为形成有机硅聚合物而进行的水解的pH、加成聚合和缩合聚合来控制由式(1)表示的亚结构的峰面积比。
在由式(1)表示的亚结构中,R可以是具有1至6个碳原子的烃基。以这种方式,容易稳定电荷量。特别地,苯基或具有1至5个碳原子的脂族烃基因其优异的环境稳定性而更为优选。
在本实施例中,为了进一步提高可充电性并抑制雾化,R更优选为具有1至3个碳原子的烃基。当可充电性优异时,转印性优异,并且转印残留调色剂的量较少;因此,减少了鼓、充电构件和转印构件的污染。
具有1至3个碳原子的脂族烃基的示例包括甲基、乙基、丙基和乙烯基。从环境稳定性和储存稳定性的观点来看,R优选是甲基。
生产有机硅聚合物的方法的示例是溶胶-凝胶法。溶胶-凝胶法包括水解和缩合聚合用作起始材料的液体原料以制备溶胶,然后胶凝所得的溶胶,并用于合成玻璃、陶瓷、有机-无机杂化物和纳米复合材料。根据本方法,可在低温下由液相生产各种形式(诸如表面层、纤维、散装材料和微粒)的功能材料。
存在于调色剂颗粒的表面层中的有机硅聚合物可以通过硅化合物(特别是烷氧基硅烷)的水解和缩合聚合产生。
通过在调色剂颗粒中形成包含有机硅聚合物的表面层来提高环境稳定性,并因此可获得在长期使用中不会发生调色剂性能劣化并且具有优异的储存稳定性的调色剂。
此外,由于溶胶-凝胶法从液体开始并通过凝胶化液体来形成材料,因此可形成各种精细结构和形状。特别地,当在水性介质中形成调色剂颗粒时,由于亲水基团(诸如硅烷醇基)提供的亲水特性,更容易使有机硅化合物沉淀在调色剂颗粒的表面上。可通过调节反应温度、反应时间、反应溶剂、pH、有机硅化合物的类型和量等来调节上述精细结构和形状。
调色剂颗粒的表面层中的有机硅聚合物可以是具有由下式(Z)表示的结构的有机硅化合物的缩聚产物。
[化学式1]
在式(Z)中,R
由R
由上述式(Z)表示的化合物的示例如下。
三官能甲基硅烷,诸如甲基三甲氧基硅烷、甲基三乙氧基硅烷、甲基二乙氧基甲氧基硅烷、甲基乙氧基二甲氧基硅烷、甲基三氯硅烷、甲基甲氧基二氯硅烷、甲基乙氧基二氯硅烷、甲基二甲氧基氯硅烷、甲基甲氧基乙氧基氯硅烷、甲基二乙氧基氯硅烷、甲基三乙酰氧基硅烷、甲基二乙酰氧基甲氧基硅烷、甲基二乙酰氧基乙氧基硅烷、甲基乙酰氧基二甲氧基硅烷、甲基乙酰氧基甲氧基乙氧基硅烷、甲基乙酰氧基二乙氧基硅烷、甲基三羟基硅烷、甲基甲氧基二羟基硅烷、甲基乙氧基二羟基硅烷、甲基二甲氧基羟基硅烷、甲基乙氧基甲氧基羟基硅烷和甲基二乙氧基羟基硅烷。
三官能硅烷,诸如乙基三甲氧基硅烷、乙基三乙氧基硅烷、乙基三氯硅烷、乙基三乙酰氧基硅烷、乙基三羟基硅烷、丙基三甲氧基硅烷、丙基三乙氧基硅烷、丙基三氯硅烷、丙基三乙酰氧基硅烷、丙基三羟基硅烷、丁基三甲氧基硅烷、丁基三乙氧基硅烷、丁基三氯硅烷、丁基三乙酰氧基硅烷、丁基三羟基硅烷、己基三甲氧基硅烷、己基三乙氧基硅烷、己基三氯硅烷、己基三乙酰氧基硅烷和己基三羟基硅烷。
三官能苯基硅烷,诸如苯基三甲氧基硅烷、苯基三乙氧基硅烷、苯基三氯硅烷、苯基三乙酰氧基硅烷和苯基三羟基硅烷。
只要不影响本实施例的效果,可以使用通过使用以下材料结合具有由式(Z)表示的结构的有机硅化合物获得的有机硅聚合物。一个分子中具有四个活性基的有机硅化合物(四官能硅烷),一个分子中具有两个活性基的有机硅化合物(双官能硅烷),以及一个分子中具有一个活性基的有机硅化合物(单官能硅烷)。其示例如下。
三官能乙烯基硅烷,诸如二甲基二乙氧基硅烷、四乙氧基硅烷、六甲基二硅氮烷、3-氨基丙基三甲氧基硅烷、3-氨基丙基三乙氧基硅烷、3-(2-氨基乙基)氨基丙基三甲氧基硅烷、3-(2-氨基乙基)氨基丙基三乙氧基硅烷、乙烯基三异氰酸酯基硅烷、乙烯基三甲氧基硅烷、乙烯基三乙氧基硅烷、乙烯基二乙氧基甲氧基硅烷、乙烯基乙氧基二甲氧基硅烷、乙烯基乙氧基二羟基硅烷、乙烯基二甲氧基羟基硅烷、乙烯基乙氧基甲氧基羟基硅烷和乙烯基二乙氧基羟基硅烷。
调色剂颗粒中的有机硅聚合物含量可以按质量计是0.5%或更高且10.5%或更低。
当有机硅聚合物含量按质量计为0.5%或更高时,可进一步降低表面层的表面自由能,提高流动性,并且可抑制组分污染和起雾。当含量按质量计为10.5%或更低时,可抑制充电。可通过用于形成有机硅聚合物的有机硅化合物的类型和量、以及制备调色剂颗粒的方法、反应温度、反应时间、反应溶剂和有机硅聚合物形成过程中的pH来控制有机硅聚合物的含量。
包含有机硅聚合物的表面层可以与调色剂核心颗粒接触而没有任何间隙。以这种方式,可抑制相对于调色剂颗粒的表面层存在于内侧上的树脂组分、脱模剂等渗出,并且可获得具有优异的储存稳定性、环境稳定性和显影耐久性的调色剂。除了上述有机硅聚合物之外,表面层还可以包含树脂,诸如苯乙烯-丙烯酸共聚物树脂、聚酯树脂或聚氨酯树脂、各种添加剂等。
[粘合剂树脂]
调色剂颗粒包含粘合剂树脂。粘合剂树脂可以是任何已知的粘合剂树脂。粘合剂树脂优选为乙烯基树脂或聚酯树脂等。乙烯基树脂、聚酯树脂和其它粘合剂树脂的示例包括下列树脂和聚合物。
苯乙烯和取代苯乙烯的均聚物,诸如聚苯乙烯和聚乙烯基甲苯;苯乙烯共聚物,诸如苯乙烯-丙烯共聚物、苯乙烯-乙烯基甲苯共聚物、苯乙烯-乙烯基萘共聚物、苯乙烯-丙烯酸甲酯共聚物、苯乙烯-丙烯酸乙酯共聚物、苯乙烯-丙烯酸丁酯共聚物、苯乙烯-丙烯酸辛酯共聚物、苯乙烯-丙烯酸二甲氨基乙酯共聚物、苯乙烯-甲基丙烯酸甲酯共聚物、苯乙烯-甲基丙烯酸乙酯共聚物、苯乙烯-甲基丙烯酸丁酯共聚物、苯乙烯-甲基丙烯酸二甲氨基乙酯共聚物、苯乙烯-乙烯基甲醚共聚物、苯乙烯-乙烯基乙醚共聚物、苯乙烯-乙烯基甲基酮共聚物、苯乙烯-丁二烯共聚物、苯乙烯-异戊二烯共聚物、苯乙烯-马来酸共聚物和苯乙烯-马来酸酯共聚物;和聚甲基丙烯酸甲酯、聚甲基丙烯酸丁酯、聚乙酸乙烯酯、聚乙烯、聚丙烯、聚乙烯醇缩丁醛、硅树脂、聚酰胺树脂、环氧树脂、聚丙烯酸树脂、松香、改性松香、萜烯树脂、酚醛树脂、脂族或脂环族烃树脂和芳香族石油树脂。可以单独使用或组合使用这些粘合剂树脂。
从可充电性的观点来看,粘合剂树脂可以包含羧基,并且可以是通过使用包含羧基的可聚合单体生产的树脂。其示例包括:丙烯酸;α-烷基不饱和羧酸的衍生物和β-烷基不饱和羧酸的衍生物,诸如甲基丙烯酸、α-乙基丙烯酸和巴豆酸;不饱和二羧酸,诸如富马酸、马来酸、柠康酸和衣康酸;和不饱和二羧酸单酯衍生物,诸如琥珀酸单丙烯酰氧基乙基酯、琥珀酸单丙烯酰氧基乙烯酯、邻苯二甲酸单丙烯酰氧基乙基酯和邻苯二甲酸单甲基丙烯酰氧基乙基酯。
聚酯树脂可以是通过羧酸组分和醇组分的缩聚获得的聚酯树脂,其示例描述如下。羧酸组分的示例包括对苯二甲酸、间苯二甲酸、邻苯二甲酸、富马酸、马来酸、环己烷二羧酸和偏苯三酸。醇组分的示例包括双酚A、氢化双酚、双酚A环氧乙烷加成物、双酚A环氧丙烷加成物、甘油、三羟甲基丙烷和季戊四醇。
聚酯树脂可以是具有脲基团的聚酯树脂。聚酯树脂的末端羧基可以是未封端的。
粘合剂树脂可以具有可聚合的官能团,以便解决在高温下发生的调色剂粘度的变化。可聚合的官能团的示例包括乙烯基、异氰酸酯基、环氧基、氨基、羧基和羟基。
[交联剂]
为了控制粘合剂树脂的分子量,可以在可聚合单体的聚合过程中加入交联剂。
交联剂的示例包括:乙二醇二甲基丙烯酸酯、乙二醇二丙烯酸酯、二乙二醇二甲基丙烯酸酯、二乙二醇二丙烯酸酯、三乙二醇二甲基丙烯酸酯、三乙二醇二丙烯酸酯、新戊二醇二甲基丙烯酸酯、新戊二醇二丙烯酸酯、二乙烯基苯、双(4-丙烯酰氧基聚乙氧基苯基)丙烷、乙二醇二丙烯酸酯、1,3-丁二醇二丙烯酸酯、1,4-丁二醇二丙烯酸酯、1,5-戊二醇二丙烯酸酯、1,6-己二醇二丙烯酸酯、新戊二醇二丙烯酸酯、二乙二醇二丙烯酸酯、三乙二醇二丙烯酸酯、四乙二醇二丙烯酸酯、聚乙二醇#200、#400和#600的二丙烯酸酯、二丙二醇二丙烯酸酯、聚丙二醇二丙烯酸酯、聚酯型二丙烯酸酯(MANDA by Nippon Kayaku)和其中丙烯酸酯由甲基丙烯酸酯取代的前述任何一种化合物。
相对于100质量份的可聚合单体,加入的交联剂的量可以是0.001质量份或更多且15.000质量份或更少。
[脱模剂]
调色剂颗粒可以包含脱模剂。可用于调色剂颗粒的脱模剂的示例包括:石油蜡及其衍生物,诸如石蜡、微晶蜡和凡士林;褐煤蜡及其衍生物;通过费-托法得到的烃蜡及其衍生物;聚烯烃蜡及其衍生物,诸如聚乙烯和聚丙烯;天然蜡及其衍生物,诸如巴西棕榈蜡和小烛树蜡;高级脂肪醇;脂肪酸,诸如硬脂酸和棕榈酸及其酰胺、酯和酮;氢化蓖麻油及其衍生物;植物蜡;动物蜡和硅树脂。应注意,衍生物包括氧化物、含乙烯基单体的嵌段共聚物和接枝改性产物。
相对于100.0质量份的粘合剂树脂或可聚合单体,脱模剂的含量可以为5.0质量份或更多且20.0质量份或更少。
[着色剂]
调色剂颗粒包含着色剂。着色剂可是任何已知的着色剂,并且其示例如下。
黑色颜料的示例包括炭黑、苯胺黑、非磁性铁氧体、磁铁矿和通过使用下述黄色着色剂、红色着色剂和蓝色着色剂调色成黑色的颜料。这些着色剂可单独使用,也可作为两种或两种以上混合使用,或者可以固溶体形式使用。
其它颜色的着色剂的示例如下。黄色颜料的示例包括缩合的偶氮化合物,诸如氧化铁黄、那不勒斯黄、萘酚黄S、汉莎黄G、汉莎黄10G、联苯胺黄G、联苯胺黄GR、喹啉黄湖、永久黄NCG和柠檬黄湖;异吲哚酮化合物;蒽醌化合物;偶氮金属络合物;次甲基化合物和烯丙基酰胺化合物。具体示例如下。
C.I.颜料黄12、13、14、15、17、62、74、83、93、94、95、109、110、111、128、129、147、155、168和180。
橙色颜料的示例如下。
永久橙GTR、吡唑啉酮橙、火神橙、联苯胺橙G、阴丹士林艳橙RK和阴丹士林艳橙GK。
红色颜料的示例包括缩合的偶氮化合物,诸如孟加拉红、永久红4R、立索尔红、吡唑啉酮红、色淀红钙盐、湖红C、湖红D、亮胭脂红6B、亮胭脂红3B、曙红湖、罗丹明湖B和茜素湖;二酮吡咯并吡咯化合物;蒽醌化合物;喹吖啶酮化合物;碱性染料湖化合物;萘酚化合物;苯并咪唑酮化合物;硫靛化合物和苝化合物。具体示例如下。
C.I.颜料红2、3、5、6、7、23、48:2、48:3、48:4、57:1、81:1、122、144、146、166、169、177、184、185、202、206、220、221和254。
蓝色颜料的示例包括铜酞菁化合物及其衍生物,诸如碱性蓝湖、维多利亚蓝湖、酞菁蓝、无金属酞菁蓝、酞菁蓝部分氯化物、坚牢天蓝和阴丹士林蓝BG;蒽醌化合物和碱性染料湖化合物。具体示例如下。
C.I.颜料蓝1、7、15、15:1、15:2、15:3、15:4、60、62和66。
紫色颜料的示例包括坚牢紫B和甲基紫湖。
绿色颜料的示例包括颜料绿B、孔雀石绿湖和最终黄绿色G。白色颜料的示例包括锌白、氧化钛、锑白和硫化锌。
如果需要,着色剂可以用不阻碍聚合的材料进行表面处理。
相对于100.0质量份的粘合剂树脂或可聚合单体,着色剂的含量可为3.0质量份或更多且15.0质量份或更少。
[用于制造调色剂颗粒的方法]
已知的方法可以用于生产调色剂颗粒,并且示例包括捏合和粉碎方法以及湿法。从均匀粒径和形状可控性的观点来看,可使用湿法。湿法的示例包括悬浮聚合方法、溶解和悬浮方法、乳化聚合和聚集方法以及乳化和聚集方法。
这里,描述悬浮聚合方法。首先,通过使用分散器(诸如球磨机或超声波分散器)均匀溶解或分散用于生成粘合剂树脂的可聚合单体、着色剂和其它添加剂(如果需要)来制备可聚合单体组合物(可聚合单体组合物制备步骤)。在此阶段,如果需要,可以适当地加入多官能单体、链转移剂、用作脱模剂的蜡、电荷控制剂和增塑剂等。悬浮聚合方法中使用的可聚合单体的示例是以下基于乙烯基的可聚合单体。
苯乙烯;苯乙烯衍生物,诸如α-甲基苯乙烯、β-甲基苯乙烯、邻甲基苯乙烯、间-甲基苯乙烯、对甲基苯乙烯、2,4-二甲基苯乙烯、对正丁基苯乙烯、对叔丁基苯乙烯、对正己基苯乙烯、对正辛基苯乙烯、对正壬基苯乙烯、对正癸基苯乙烯、对正十二烷基苯乙烯、对甲氧基苯乙烯和对苯基苯乙烯;基于丙烯酸的可聚合单体,诸如丙烯酸甲酯、丙烯酸乙酯、丙烯酸正丙酯、丙烯酸异丙酯、丙烯酸正丁酯、丙烯酸异丁酯、丙烯酸叔丁酯、丙烯酸正戊酯、丙烯酸正己酯、丙烯酸2-乙基己酯、丙烯酸正辛酯、丙烯酸正壬酯、丙烯酸环己酯、丙烯酸苄酯、磷酸二甲酯丙烯酸乙酯、磷酸二乙酯丙烯酸乙酯、磷酸二丁酯丙烯酸乙酯和丙烯酸2-苯甲酰氧基乙酯;基于甲基丙烯酸酯的可聚合单体,诸如甲基丙烯酸甲酯、甲基丙烯酸乙酯、甲基丙烯酸正丙酯、甲基丙烯酸异丙酯、甲基丙烯酸正丁酯、甲基丙烯酸异丁酯、甲基丙烯酸叔丁酯、甲基丙烯酸正戊酯、甲基丙烯酸正己酯、甲基丙烯酸2-乙基己酯、甲基丙烯酸正辛酯、甲基丙烯酸正壬酯、磷酸二乙酯甲基丙烯酸乙酯和磷酸二丁酯甲基丙烯酸乙酯;亚甲基脂肪族一元羧酸酯;乙烯基酯,诸如乙酸乙烯酯、丙酸乙烯酯、苯甲酸乙烯酯、丁酸乙烯酯和甲酸乙烯酯;乙烯基醚,诸如乙烯基甲基醚、乙烯基乙基醚和乙烯基异丁基醚;和乙烯基甲基酮、乙烯基己基酮和乙烯基异丙基酮。
接下来,将可聚合单体组合物放入预先制备的水性介质中,并通过使用高剪切力搅拌器或分散器将由可聚合单体组合物形成的液滴形成为所需尺寸的调色剂颗粒(颗粒形成步骤)。
颗粒形成步骤中的水性介质可以包含分散稳定剂以便控制调色剂颗粒的粒径,获得尖锐的粒度分布,并抑制生产过程中调色剂颗粒的结合。分散稳定剂通常大致分类为通过空间位阻展现排斥力的聚合物和通过静电排斥力稳定分散体的微溶于水的无机化合物。微溶于水的无机化合物的微粒溶解在酸或碱中,并因此可在聚合后通过用酸或碱洗涤而容易地溶解和除去。
用作分散稳定剂的微溶于水的无机化合物可以包含镁、钙、钡、锌、铝或磷。用作分散稳定剂的微溶于水的无机化合物更优选包含镁、钙、铝或磷。具体示例如下。
磷酸镁、磷酸三钙、磷酸铝、磷酸锌、碳酸镁、碳酸钙、氢氧化镁、氢氧化钙、氢氧化铝、硅酸钙、硫酸钙、硫酸钡和羟基磷灰石。上述分散稳定剂可以与有机化合物结合使用,诸如聚乙烯醇、明胶、甲基纤维素、甲基羟丙基纤维素、乙基纤维素、羧甲基纤维素钠盐或淀粉。相对于100质量份的可聚合单体,可以使用0.01质量份或更多且2.00质量份或更少的这些分散稳定剂。
相对于100质量份的可聚合单体,可以组合使用0.001质量份或更多且0.1质量份或更少的表面活性剂,以使这些分散稳定剂更细。表面活性剂的示例包括市售的非离子、阴离子和阳离子表面活性剂。其具体示例包括十二烷基硫酸钠、十四烷基硫酸钠、十五烷基硫酸钠、辛基硫酸钠、油酸钠、月桂酸钠、硬脂酸钾和油酸钙。
在颗粒形成步骤之后或期间,温度可以设定为50℃或更高且90℃或更低,并且聚合包含在可聚合单体组合物中的可聚合单体以获得调色剂颗粒分散液(聚合步骤)。
在聚合步骤中,可以搅拌容器内部,使得温度分布变得均匀。当要加入聚合引发剂时,加入的时刻和时长可以是任意的。为了获得所需的分子量分布,可以在聚合反应的后期提高温度。此外,为了将未反应的可聚合单体、副产物等除至系统外部,可以在反应的后期或反应完成后将水性介质部分蒸馏掉。蒸馏可以在常压或减压下进行。
悬浮聚合方法中使用的聚合引发剂通常是油溶性引发剂。其示例如下。
偶氮化合物,诸如2,2'-偶氮二异丁腈,2,2'-偶氮二-2,4-二甲基戊腈,1,1'-偶氮二(环己烷-1-腈)和2,2'-偶氮二-4-甲氧基-2,4-二甲基戊腈;和基于过氧化物的引发剂,诸如乙酰环己基磺酰过氧化物、二异丙基过氧碳酸酯、过氧化癸酰、过氧化月桂酰、过氧化硬脂酰、过氧化丙酰、过氧化乙酰、叔丁基过氧-2-乙基己酸酯、过氧化苯甲酰、叔丁基过氧异丁酸酯、过氧化环己酮、过氧化甲乙酮、过氧化二枯基、氢过氧化叔丁基、过氧化二叔丁基、过氧化新戊酸叔丁酯和氢过氧化枯烯。
如果需要,水溶性引发剂可组合用作聚合引发剂,并且其示例如下。过硫酸铵、过硫酸钾、2,2'-偶氮双(N,N’-二甲基异丁酯脒)盐酸盐、2,2’-偶氮双(2-脒基丙烷)盐酸盐、偶氮二(异丁脒)盐酸盐、2,2’-偶氮二异丁腈磺酸钠、硫酸亚铁和过氧化氢。
可单独使用或组合使用这些聚合引发剂。为了控制可聚合单体的聚合度,可以附加地使用链转移剂和阻聚剂等。
从获得高精度、高分辨率图像的观点来看,调色剂颗粒的重量平均粒径可以是3.0μm或更大且10.0μm或更小。可通过孔径形式的电阻法来测量调色剂的重量平均粒径。例如,Coulter计数器Multisizer3(贝克曼库尔特公司生产)可用于测量。然后将如此获得的调色剂颗粒分散液送至过滤步骤,以进行调色剂颗粒和水性介质的固液分离。
可通过典型的过滤方法进行用于从获得的调色剂颗粒分散液中获得调色剂颗粒的固液分离,随后,调色剂颗粒可以再次形成浆液或者可以用漂洗水漂洗以从调色剂颗粒表面去除残留的杂质。彻底洗涤后,再次进行固液分离以获得调色剂饼。然后,通过已知的干燥方法干燥调色剂饼,并且如果需要,通过分类分离粒径在特定范围之外的颗粒群来获得调色剂颗粒。可以再次使用粒径在特定范围之外的颗粒群以提高最终产率。
如上所述,当通过在水性介质中形成调色剂颗粒来形成包含有机硅聚合物的表面层时,可通过在水性介质中进行聚合步骤和其它适当步骤的同时加入有机硅化合物的水解液来形成表面层。聚合后的调色剂颗粒的分散液可以用作核心颗粒分散液,并且可以向其中加入有机硅化合物的水解液以形成表面层。当不使用水性介质时,诸如当采用捏合和粉碎方法时,可将获得的调色剂颗粒分散在水性介质中以制备核心颗粒分散液,并且可以向其中加入有机硅化合物的水解液以形成表面层。
[用于测量调色剂物理性质的方法]
[分离用于核磁共振(NMR)测量的调色剂颗粒的四氢呋喃(THF)不溶物部分的方法]
可按如下获得调色剂颗粒的四氢呋喃(THF)不溶物部分。
首先,称取10.0克调色剂颗粒,放入圆筒形过滤器(Toyo Roshi Kaisha有限公司生产的No.86R)中,并置于索氏提取器中。使用200毫升THF作为溶剂进行提取20小时,并将圆柱形过滤器中的残留物在40℃下真空干燥数小时。所得产物用作用于NMR测量的调色剂颗粒的THF不溶物部分。
当调色剂颗粒的表面已经用外部添加剂等处理时,通过以下方法除去外部添加剂以获得调色剂颗粒。
向100毫升离子交换水中加入160克蔗糖(由Kishida Chemical有限公司生产),并在热水浴中溶解,以制备蔗糖重溶液。向离心分离管(体积:50毫升)中放入31克蔗糖重溶液和6毫升Contaminon N(由Wako Pure Chemical公司生产,用于洗涤精密测量仪器的中性洗涤剂的10%水溶液(按质量计),洗涤剂的pH为7并含有非离子表面活性剂、阴离子表面活性剂和有机助洗剂),并制备分散液。向该分散液中加入1.0克调色剂,并用抹刀或类似物将调色剂团块松开。
将离心分离管用振荡器以350spm(每分钟冲程数)振荡20分钟。振荡后,将液体放入用于摆动转子的玻璃管(体积:50毫升)中,并用离心分离器(Kokusan有限公司生产的H-9R)以3500rpm的速度分离30分钟。结果,调色剂颗粒和分离的外部添加剂彼此分离。在目测确认调色剂被从水溶液中充分分离后,用抹刀或类似物对已经分离形成顶层的调色剂进行取样。取样的调色剂通过真空过滤器被过滤,并在干燥器中被干燥1小时或更长时间,以获得调色剂颗粒。多次执行该过程以确保所需的数量。
[用于确认由式(1)表示的亚结构的方法]
使用以下方法来确认调色剂颗粒中包含的有机硅聚合物中由式(1)表示的亚结构。
通过
仪器:JEOL RESONANCE公司生产的JNM-ECX500II
样本试管:直径3.2毫米
样品:用于NMR测量的调色剂颗粒的四氢呋喃不溶物部分,150毫克
测量温度:室温
脉冲模式:CP/MAS
测量核频率:123.25MHz(
标准物质:金刚烷(外标法:29.5ppm)
样本旋转速率:20kHz
接触时间:2ms
延迟时间:2s
运行次数:1024
本方法用于基于是否存在源自键合到硅原子的基团的信号来识别由式(1)中的R表示的烃基,基团例如是甲基(Si-CH
<用于计算调色剂颗粒中包含的有机硅聚合物中归因于由式(1)表示的结构的峰面积比的方法>
在以下条件下进行调色剂颗粒的THF不溶物部分的
[
仪器:JEOL RESONANCE公司生产的JNM-ECX500II
样本试管:直径3.2毫米
样品:用于NMR测量的调色剂颗粒的四氢呋喃不溶物部分,150毫克
测量温度:室温
脉冲模式:CP/MAS
测量核频率:97.38MHz(
标准物质:DSS(外标法:1.534ppm)
样本旋转速率:10kHz
接触时间:10ms
延迟时间:2s
运行次数:2000至8000
在上述测量之后,通过曲线拟合,将调色剂颗粒的四氢呋喃不溶物部分中具有不同取代基和键合基团的多个硅烷组分的峰分离成下述X1结构、X2结构、X3结构和X4结构,并计算每个结构的峰面积。
X1结构:(Ri)(Rj)(Rk)SiO
X2结构:(Rg)(Rh)Si(O
X3结构:RmSi(O
X4结构:Si(O
[化学式2]
X1结构
X2结构
X3结构
X4结构
在式(2)、(3)和(4)中,Ri、Rj、Rk、Rg、Rh和Rm各自独立地表示有机基团,诸如具有1至6个碳原子的烃基、卤素原子、羟基、乙酰氧基或键合到硅上的烷氧基。
在本实施例中,在通过调色剂颗粒的THF不溶物部分的
当需要进一步详细确认由式(1)表示的亚结构时,除了上述
<通过用透射电子显微镜(TEM)对调色剂颗粒进行横截面观察来测量其中含有有机硅聚合物的表面层的厚度为2.5纳米或更小的部分的比率的方法>
在本实施例中,通过以下方法进行调色剂颗粒的横截面观察。
观察调色剂颗粒横截面的具体方法包括将调色剂颗粒完全分散在室温可固化的环氧树脂中,并在40℃的环境中固化树脂2天。用配有金刚石刀片的切片机从获得的固化产物上切下样品的薄带。在放大10,000至100,000倍的量级下,用透射电子显微镜(TEM)(由JEOL有限公司生产的JEM-2800)观察该样品中调色剂颗粒的横截面。
由于粘合剂树脂和表面层材料之间的原子量存在差异,并且其中原子量大的部分以较浅的阴影出现,因此可进行识别。为了增强材料之间的对比度,采用四氧化钌染色法或四氧化锇染色法。
由上述TEM图像中的调色剂颗粒的横截面确定的在测量中使用的每个物品的等效圆直径Dtem在由下述方法确定的调色剂颗粒的重量平均粒径D4的±10%范围内。
如上所述,在200kV的加速电压下,通过使用由JEOL有限公司生产的JEM-2800获得调色剂颗粒横截面的暗场图像。接下来,通过三窗口法,使用由Gatan公司生产的EELS检测器GIF Quantam获得映射图像,以确认表面层。
对于等效圆直径Dtem在调色剂颗粒的重量平均粒径D4的±10%范围内的每个调色剂颗粒,相对于调色剂颗粒横截面的长轴L和通过长轴L的中心并且垂直于长轴L的轴L90之间的交点,调色剂颗粒横截面被等分为16个部分。用An(n=1至32)分别表示均从上述中心延伸到表面层的各分割轴,用RAn表示各分割轴的长度,以及用FRAn表示表面层的厚度。
计算其上含有机硅聚合物的表面层的厚度为2.5纳米或更小的分割轴的数量,并确定这些轴相对于32个分割轴的比率。测量十个调色剂颗粒,并计算每个调色剂颗粒的平均值。
<根据从透射电子显微镜(TEM)图像获得的调色剂颗粒的横截面确定的等效圆直径Dtem>
通过以下方法确定根据从TEM图像获得的调色剂颗粒的横截面确定的等效圆直径Dtem。首先,对于一个调色剂颗粒,由以下公式确定根据从TEM图像获得的该调色剂颗粒的横截面确定的等效圆直径Dtem。
根据从TEM图像获得的调色剂颗粒的横截面获得的等效圆直径(Dtem)=(RA1+RA2+RA3+RA4+RA5+RA6+RA7+RA8+RA9+RA10+RA11+RA12+RA13+RA14+RA15+RA16+RA17+RA18+RA19+RA20+RA21+RA22+RA23+RA24+RA25+RA26+RA27+RA28+RA29+RA30+RA31+RA32)/16
确定10个调色剂颗粒的等效圆直径并取平均值,以确定由调色剂颗粒的横截面确定的等效圆直径(Dtem)。
[其中含有机硅聚合物的表面层厚度为2.5纳米或更小的部分的比率]
其中含有机硅聚合物的表面层的厚度(FRAn)为2.5纳米或更小的部分的比率={(其中含有机硅聚合物的表面层的厚度(FRAn)为2.5纳米或更小的轴的数量)/32}×100
对10个调色剂颗粒进行此计算,并且将其中含有机硅聚合物的表面层的厚度(FRAn)为2.5纳米或更小的部分的比率进行平均,并且将结果用作其中含有机硅聚合物的表面层的厚度(FRAn)为2.5纳米或更小的部分的比率。
[调色剂颗粒中有机硅聚合物含量的测量]
通过使用波长色散X射线荧光光谱仪“Axios”(由PANalytical生产)和封装的专用软件“SuperQ ver.4.0F”(由PANalytical生产)来测量有机硅聚合物的含量,该软件用于设置测量条件并分析测量数据。Rh用作X射线管阳极,测量环境为真空,测量直径(准直器掩膜直径)为27mm,以及测量时间为10秒。当要测量轻元素时,使用比例计数器(PC)进行检测,以及当要测量重元素时,使用闪烁计数器(SC)进行检测。
测量样品是通过以下方法形成的粒料:将4g调色剂颗粒放置在特殊铝环中以用于压制,将颗粒整平,并通过使用片剂压缩机“BRE-32”(由Maekawa Testing Machine MFG有限公司生产)在20MPa下对颗粒加压60秒,从而形成厚度为2mm以及直径为39mm的颗粒。
相对于不含有机硅聚合物的100质量份调色剂颗粒,加入0.5质量份二氧化硅(SiO
对于样品中的每个样品,如上所述使用片剂压缩机制备校准曲线样品的粒料,并且用PET作为分散晶体测量在109.08°的衍射角(2θ)处观察到的Si-Kα辐射的计数率(单位:cps)。在这个过程中,X射线发生器的加速电压和电流值分别为24千伏和100毫安。通过在纵轴上绘制所得的X光计数率以及在横轴上绘制每个校准曲线样品中添加的SiO
[用于测量有机硅聚合物的附着率的方法]
向100毫升离子交换水中加入160克蔗糖(由Kishida Chemical有限公司生产),蔗糖在热水浴中溶解,以制备蔗糖重溶液。向离心分离管(体积:50毫升)中放入31克蔗糖重溶液和6毫升Contaminon N(由Wako Pure Chemical公司生产,用于洗涤精密测量仪器的中性洗涤剂的10%水溶液(按质量计),洗涤剂的pH为7并含有非离子表面活性剂、阴离子表面活性剂和有机助洗剂),并制备分散液。向该分散液中加入1.0克调色剂,并用抹刀或类似物将调色剂团块松开。
将离心分离管用振荡器以350spm(每分钟冲程数)振荡20分钟。振荡后,将液体放入用于摆动转子的玻璃管(体积:50毫升)中,并用离心分离器(Kokusan有限公司生产的H-9R)以3500rpm的速度分离30分钟。在目测确认调色剂被从水溶液中充分分离后,用抹刀或类似物对已经分离形成顶层的调色剂进行取样。含有调色剂的取样水溶液通过真空过滤器被过滤,并在干燥器中干燥1小时或更长时间。用抹刀将干燥的产物粉碎,并用X射线荧光测量Si含量。附着率(%)是根据洗涤的调色剂的目标元素量与初始调色剂的目标元素量的比率来计算的。
各元素的X射线荧光按照JIS K 0119-1969进行测量,并且具体细节如下。
作为测量仪器,使用波长色散X射线荧光光谱仪“Axios”(由PANalytical生产)和封装的专用软件“SuperQ ver.4.0F”(由PANalytical生产),该软件用于设置测量条件并分析测量数据。Rh用作X射线管阳极,测量环境为真空,测量直径(准直器掩膜直径)为10mm,以及测量时间为10秒。当要测量轻元素时,使用比例计数器(PC)进行检测,以及当要测量重元素时,使用闪烁计数器(SC)进行检测。
测量样品是厚度为约2毫米的粒料,其通过将约1克初始调色剂和水洗调色剂放入直径为10毫米的特殊铝环中进行压制,将调色剂整平,并通过使用片剂压缩机在20MPa下对调色剂加压60秒而形成。将“BRE-32”(由Maekawa Testing Machine MFG有限公司生产)用作片剂压缩机。
在上述条件下进行测量,并且基于所获得的X射线峰值位置来识别元素。由计数率(单位:cps)计算得出每种元素的浓度,计数率是每单位时间X射线光子数。
如下确定关于量化调色剂中元素(例如,硅含量)的方法。首先,向100质量份调色剂颗粒中加入0.5质量份二氧化硅(SiO
对于样品中的每个样品,如上所述使用片剂压缩机制备校准曲线样品的粒料,并且用PET作为分散晶体测量在109.08°的衍射角(2θ)处观察到的Si-Kα辐射的计数率(单位:cps)。在这个过程中,X射线发生器的加速电压和电流值分别为24千伏和100毫安。通过在纵轴上绘制所得的X光计数率以及在横轴上绘制每个校准曲线样品中添加的SiO
[示例]
现在将通过以下示例详细描述本发明,这些示例不限制本发明。除非另有说明,示例和对比示例中每种材料的“份”和“%”是基于质量的。
[示例1]
[水性介质1的制备步骤]
向反应器中的1000.0份离子交换水中加入14.0份磷酸钠(十二水合物)(由RASAIndustries有限公司生产),并在氮气吹扫下将温度保持在65℃1.0小时。
在用T.K.Homomixer(由Tokushu Kika Kogyo有限公司生产)以12000rpm搅拌混合物的同时,将氯化钙水溶液立即加入到混合物中以制备含有分散稳定剂的水性介质,该氯化钙水溶液是通过将9.2份氯化钙(二水合物)溶解到10.0份离子交换水中制备的。向水性介质中加入10%(按质量计)的盐酸以将pH调节至5.0,结果获得水性介质1。
[水解用于表面层的有机硅化合物的步骤]
向配备有搅拌器和温度计的反应器中,称量60.0份离子交换水,并通过使用10%(按质量计)的盐酸将pH调节至3.0。在搅拌混合物的同时,将温度调节至70℃。随后,向其中加入40.0份用作表面层的有机硅化合物的甲基三乙氧基硅烷,随后搅拌2小时或更长时间以进行水解。当油和水不再分离并形成一层时,目测确认水解完成,随后进行冷却。结果,获得了用于表面层的有机硅化合物的水解液。
[制备可聚合单体组合物的步骤]
苯乙烯:50.0份
炭黑(Orion Engineered Carbon生产的Nipex 35):7.0份
将上述材料置于磨碎机(由Mitsui Miike Chemical Engineering Machinery有限公司生产)中,并用直径1.7毫米的氧化锆珠以220rpm分散5.0小时,从而制备颜料分散液。将下列物质加入到上述颜料分散液中。
苯乙烯:20.0份
丙烯酸正丁酯:30.0份
交联剂(二乙烯基苯):0.3份
饱和聚酯树脂:5.0份(环氧丙烷改性的双酚A(2摩尔加合物)和对苯二甲酸(摩尔比为10:12)之间的缩聚产物,玻璃转变温度Tg=68℃,重量平均分子量Mw=10000,分子量分布Mw/Mn=5.12)
费托蜡(熔点:78℃):7.0份
将所得混合物保持在65℃的温度下,并通过使用T.K.Homomixer(由Tokushu KikaKogyo有限公司生产)以500rpm均匀地溶解和分散,从而制备可聚合单体组合物。
[颗粒形成步骤]
在将水性介质1的温度保持在70℃并且将T.K.Homomixer的旋转速率保持在12000rpm的同时,将可聚合单体组合物加入到水性介质1中,并向其中加入9.0份用作聚合引发剂的叔丁基过氧新戊酸酯。在用搅拌器保持12000rpm的情况下,形成颗粒10分钟。
[聚合步骤]
在颗粒形成步骤后,用螺旋桨搅拌叶片代替搅拌器,并且在混合物以150rpm的速度被搅拌时,通过将温度保持在70℃进行聚合反应5.0小时,并且通过将温度升高到85℃并在该温度下加热2.0小时进行聚合反应以获得核心颗粒。将含有核心颗粒的浆液的温度降至55℃,并测量pH。pH值为5.0。在55℃下继续搅拌的同时,通过加入20.0份用于表面层的有机硅化合物的水解液,开始形成调色剂的表面层。在将混合物在相同条件下保持30分钟后,通过使用氢氧化钠水溶液将所得浆液的pH调节至9.0以终止缩合,并将浆液在此条件下保持300分钟以形成表面层。
[洗涤和干燥步骤]
聚合步骤完成后,冷却调色剂颗粒的浆液,向调色剂颗粒的浆液中加入盐酸以将pH调节至1.5或更低,并将所得混合物搅拌1小时并静置。然后,用真空过滤器进行固液分离以获得调色剂饼。用离子交换水将调色剂饼再次形成为浆液以再次制备分散液,并且通过上述过滤器对分散液进行固液分离。重复浆液的制备和固液分离,直到滤液的电导率为5.0μS/cm或更低,然后进行最后一次固液分离以获得调色剂饼。
在气流干燥器Flashjet Drier(由Seishin Enterprise有限公司生产)中干燥获得的调色剂饼,并通过使用利用了康达效应的多区分级机除去粗颗粒。结果,获得调色剂颗粒1。调节干燥条件,使得吹风温度为90℃,干燥器出口温度为40℃,并且根据调色剂饼中的水含量调节调色剂饼进料速度,使得出口温度不偏离40℃。
在调色剂颗粒1的横截面TEM观察中进行硅映射以确认硅原子存在于表面层中,并确认其上含有有机硅聚合物的调色剂颗粒的表面层的厚度为2.5纳米或更小的分割轴的数量的比率为20.0%或更小。在随后的示例中也进行相同的硅映射以确认硅原子存在于表面层中,并确认其上表面层的厚度为2.5纳米或更小的分割轴的数量的比率为20.0%或更小。在本示例中,所获得的调色剂颗粒1直接用作调色剂1,而无需外部添加。
评估调色剂1的方法如下。
[马氏硬度的测量]
硬度是关于物理物体表面或表面附近的机械性能中的一种,并在异物导致物理物体变形或损坏时表示物理物体抵抗变形或损坏的能力。硬度有多种测量方法和定义。例如,根据待测区域的大小选择测量方法。例如,当待测量的区域是10微米或更大时,采用维氏方法。当待测量的区域是10微米或更小时,采用纳米压痕法。当待测量的区域是1微米或更小时,使用原子力显微镜(AFM)方法。硬度的定义的示例包括作为压痕硬度的布氏硬度和维氏硬度、作为刮擦硬度的马氏硬度和作为回弹硬度的肖氏硬度。视情况地使用这些定义。
由于典型的粒径为3微米至10微米,纳米压痕法可以用于测量调色剂。根据发明人进行的研究,表明耐刮擦性的马氏硬度适合于定义提供本发明效果的硬度。这大概是因为刮擦硬度可指示调色剂抵抗被显影单元内的硬物质(诸如金属和外部添加剂)刮擦的强度。
通过纳米压痕法测量调色剂的马氏硬度的方法包括通过在ISO 14577-1中描述的压痕试验程序使用符合ISO 14577-1的市售设备获得载荷-位移曲线,并从载荷-位移曲线计算硬度。在本发明中,将纳米压痕测试仪“ENT-1100b”(由ELIONIX公司生产)用作符合ISO标准的设备。该设备的“ENT1100操作手册”中描述了测量方法。具体的测量方法如下。
通过使用附接的温度控制器,防护罩内的测量环境保持在30.0℃。保持大气温度恒定对于减少由热膨胀、漂移等引起的测量数据的变化是有效的。温度设定为30.0℃,这是其中调色剂被摩擦的显影设备附近的估计温度。将附接到设备的标准样品台用作样品台。施加调色剂后,吹入弱空气以分散调色剂,并将样品台装载到设备上。将样品台在该处保持至少1小时后,开始测量。
将附接到设备上的具有20微米方形尖端的平压头(带有金刚石尖端的钛压头)用作测量压头。如果使用尖头压头来测量小球体、其上附着有外部添加剂的物体以及具有表面不规则部的物体(诸如调色剂),测量精度会明显受到影响。因此,使用了平压头。用于测试的最大载荷设定为2.0×10
选择在由附接到设备的显微镜拍摄的测量图像(视场尺寸:160微米宽,并且120微米长)中与其它颗粒分离的调色剂颗粒作为待测量的颗粒。为了使位移量的误差最小化,选择那些粒径(D)在数均粒径(D1)的±0.5微米范围内的颗粒(“D1-0.5微米”等于或小于D,并且D等于或小于“D1+0.5微米”)。通过使用与该设备一起封装的软件测量调色剂颗粒的长轴和短轴,并通过[(长轴长度+短轴长度)/2]来确定直径D(微米),从而确定待测量的颗粒的粒径。通过下述方法,使用Coulter计数器Multisizer 3(贝克曼库尔特公司生产)测量数均粒径。
通过选择粒径D(微米)满足上述条件的一百个调色剂颗粒来进行测量。测量的输入条件如下。
测试模式:加载-卸载测试
测试加载:20.000mgf(=2.0×10
分隔数:1000步
步间隔:10毫秒
当通过选择分析菜单“数据分析(ISO)”进行测量时,测量后通过与设备一起封装的软件对马氏硬度进行分析并输出。该测量是在一百个调色剂颗粒上进行的,并且将算术平均值用作本发明中的马氏硬度。
[用于测量附着率的方法]
通过[用于测量调色剂物理性质的方法]中描述的方法进行测量。
[打印输出的评估]
使用了CANON KABUSHIKI KAISHA生产的市售激光束打印机LBP7600C的改进型号。改进包括修改用于评估的设备的主体和与其一起使用的软件,使得显影辊的旋转速度是原始周向速度的1.8倍。具体地,就周向速度而言,改进前显影辊的转速为200毫米/秒,而改进后的转速为360毫米/秒。
将40克调色剂装入LBP7600C的调色剂盒中。调色剂盒在常温、正常湿度(NN)(25℃/50%RH)的环境中放置24小时。24小时后,调色剂盒在相同的环境中附接到LBP7600C。
在4000张横向取向的A4纸上打印出打印率为35.0%的图像之后,在NN环境中评估电荷上升。也在初始阶段评估了电荷上升。
[显影条纹的评估]
将半色调(调色剂涂布量:0.2mg/cm
[评估标准]
A:在显影辊或图像上没有观察到沿纸张排出方向延伸的竖直条纹。
B:在显影辊的两侧均观察到沿周向方向延伸的5条或更少的细条纹。或者,在图像上观察到沿纸张排出方向延伸的模糊竖直条纹。
B:在显影辊的两侧均观察到沿周向方向延伸的6条或更多且20条或更少的细条纹。或者,在图像上观察到5条或更少的细条纹。
D:在显影辊上观察到21条或更多的条纹。或者,在图像上观察到1条或多条明显的条纹或6条或更多的细条纹。
[重影评估]
将由3厘米宽的实心图像竖直线和3厘米宽的空白竖直线交替构成的图像连续印刷在10张片材上,然后将半色调图像印刷在一张片材上。视觉评估残留在图像上的先前图像的历史。调节半色调图像的图像密度,使得用Macbeth密度计(由Macbeth生产)和SPI过滤器通过反射密度测量确定的反射密度为0.4。
A:未发生重影。
B:先前图像的历史在某些部分隐约可见。
C:先前图像的历史在某些部分是可见的。
D:先前图像的历史在所有部分都是可见的。
[清洁性能评估]
将调色剂涂布量为0.2mg/cm
B:未发现清洁失败的图像,但发现充电辊污染。
C:在半色调图像上观察到轻微的清洁失败。
D:在半色调图像上发现明显的清洁失败。
[电荷上升的评估]
在10张片材上输出了实心图像。在打印出第10张片材的过程中,强行关闭机器,并在通过管制刮刀后立即测量显影辊上的调色剂电荷量。通过使用图6的透视图中所示的法拉第笼来测量显影辊上的电荷量。内部(图中的右侧)被置于减压状态,以便抽吸显影辊上的调色剂,并且通过安装调色剂过滤器33捕获调色剂。法拉第笼13还包括抽吸部分31和支架32。使用捕获的调色剂的质量M和用库仑计直接测量的电荷Q来计算每单位质量的电荷量Q/M[μC/g],并且将结果分级为如下调色剂电荷量(Q/M)。
A:小于-40μC/g
B:-40μC/g或更大,但小于-30μC/g
C:-30μC/g或更大,但小于-20μC/g
D:-20μC/g或更大
(示例2至示例12)
如表1所示,除了在“聚合步骤”中加入水解液的条件和加入后的保持时间被改变外,调色剂的制备与实施例1相同。通过使用盐酸和氢氧化钠水溶液调整浆液的pH。按实施例1的方法对得到的调色剂进行评估。评估结果见表2。
(示例13至示例18)
如表1所示,除了在“水解用于表面层的有机硅化合物的步骤”中使用的用于表面层的有机硅化合物被改变外,调色剂的制备与示例1相同。按示例1的方法对得到的调色剂进行评估。评估结果见表2。
(示例19至示例23)
如表1所示,除了在“聚合步骤”中加入水解液的条件被改变外,调色剂的制备与示例1相同。按示例1的方法对得到的调色剂进行评估。评估结果见表2。
(比较例1和比较例2)
如表1所示,除了在“聚合步骤”中加入水解液的条件和加入后的保持时间被改变外,调色剂的制备与示例1相同。按示例1的方法对得到的调色剂进行评估。评估结果见表2。
(比较例3)
未进行“水解用于表面层的有机硅化合物的步骤”。替代性地,在“制备可聚合单体组合物的步骤”中,加入8份仍为单体形式并用作用于表面层的有机硅化合物的甲基三乙氧基硅烷。
在“聚合步骤”中,在冷却至70℃后测量pH后,不加入水解液。在70℃下继续搅拌的同时,通过使用氢氧化钠水溶液将浆液的pH调节至9.0以终止缩合,并将浆液在此条件下保持300分钟以形成表面层。
除了上述变化之外,按照示例1制备调色剂。按示例1评估所获得的调色剂。评估结果见表2。
(比较例4)
将比较例3中“制备可聚合单体组合物的步骤”中加入的甲基三乙氧基硅烷的量改为15份。
除了上述变化之外,按照比较例3制备调色剂。按示例1评估所获得的调色剂。评估结果见表2。
(比较例5)
将比较例3中“制备可聚合单体组合物的步骤”中加入的甲基三乙氧基硅烷的量改为30份。
除了上述变化之外,按照比较例3制备调色剂。按示例1评估所获得的调色剂。评估结果见表2。
(比较例6)
(粘合剂树脂1的生产示例)
对苯二甲酸:25.0%(摩尔百分比)
己二酸:13.0%(摩尔百分比)
偏苯三酸:8.0%(摩尔百分比)
环氧丙烷改性双酚A(2.5摩尔加合物):33.0%(摩尔百分比)
环氧乙烷改性双酚A(2.5摩尔加合物):21.0%(摩尔百分比)
向四颈烧瓶中加入总计100份上述酸组分和醇组分,以及0.02份用作酯化催化剂的2-乙基己酸锡。将减压设备、水分离设备、氮气引入设备、温度计和搅拌器附接到烧瓶上,并在氮气环境中将温度升高至230℃以进行反应。反应结束后,将产物从反应器中取出、冷却并粉碎,得到粘合剂树脂1。
(粘合剂树脂2的生产示例)
除了如下改变单体组合物比率和反应温度之外,按照粘合剂树脂1制备粘合剂树脂2。
对苯二甲酸:50.0%(摩尔百分比)
偏苯三酸:3.0%(摩尔百分比)
环氧丙烷改性双酚A(2.5摩尔加合物):47.0%(摩尔百分比)
反应温度:190℃
(对比调色剂6的生产示例)
粘合剂树脂1:70.0份
粘合剂树脂2:30.0份
磁性氧化铁颗粒:90.0份
(数均粒径:0.14μm,Hc=11.5kA/m,σs=84.0Am
费托蜡(熔点:105℃):2.0份
电荷控制剂1(结构式如下):2.0份
电荷控制剂1
[化学式3]
式中,tBu代表叔丁基。
在Henschel混合机中预混合上述材料,并在具有螺杆区和三个捏合区的双螺杆捏合机挤出机中进行熔融捏合。在此过程中,将靠近供应端口的第一捏合区中的加热温度设定为110℃,将第二捏合区中的加热温度设定为130℃,将第三捏合区中的加热温度设定为150℃,将桨叶旋转速度设定为200rpm,并冷却在这些条件下熔融捏合得到的捏合产物。在锤磨机中粗粉碎冷却后的产物后,粗粉碎的产物通过细粉碎机用射流进行粉碎。通过多区分级机对得到的细粉碎的粉末进行分级,该多区分级机利用了康达效应。结果,获得重量平均粒径为7.0微米的调色剂颗粒。
从外部向100份调色剂颗粒中加入1.0份疏水性二氧化硅细粉末(BET:140m
(比较例7)
制备日本专利特开No.2015-45860中的示例中描述的磁性调色剂颗粒1。粘合剂中存在用作填料的磁性材料,并表面被热处理。按示例1评估所获得的调色剂。评估结果见表2。
[表1]
[表2]
[调色剂的效果]
如表中所示,通过将马氏硬度调节到200[MPa]或更高且1100[MPa]或更低,与常规调色剂相比,显影部分中调色剂的耐磨性显著提高,并且与相关技术相比,可以抑制由打印引起的调色剂摩擦起电的变化。结果,可抑制具有劣化可充电性的积聚的劣化调色剂的量的增加。此外,可抑制补充雾化的发生,并且由于出现较少的显影条纹和重影,图像得到改善。
当调色剂摩擦起电的变化减少并且补充雾化被抑制时,停机时间和诸如测量显影电流的复杂操作不再是必要的。此外,如在前述实施例中,即使当成像装置具有其中显影设备内部的旧调色剂在补充期间快速与补充的新调色剂接触的结构时,也不会发生补充雾化,并且可显著减少调色剂补充所需的停机时间。
这些表还表明,当马氏硬度低于200MPa时,不能令人满意地获得本发明的效果。
[外部添加剂]
可以在没有任何外部添加剂的情况下将调色剂颗粒用作调色剂;然而,为了进一步提高流动性、可充电性和清洁性能等,可以向调色剂颗粒中加入流化剂、清洁助剂和其它外部添加剂,并且所得混合物可以用作调色剂。
外部添加剂的示例包括:无机氧化物细颗粒(诸如二氧化硅细颗粒、氧化铝细颗粒和氧化钛细颗粒),以及无机硬脂酸盐化合物细颗粒(诸如硬脂酸铝细颗粒和硬脂酸锌细颗粒)。其它示例包括无机钛酸盐化合物细颗粒,诸如钛酸锶和钛酸锌。这些可以单独使用或以两者或更多者的组合使用。
相对于100质量份调色剂颗粒,添加的这些外部添加剂的总量优选为0.05质量份或更多且5质量份或更少,并且更优选为0.1质量份或更多且3质量份或更少。可以组合使用各种外部添加剂。
调色剂可以在调色剂颗粒的表面上具有带正电荷的颗粒。带正电荷的颗粒的数均粒径优选为0.10微米或更大且1.00微米或更小,以及更优选为0.20微米或更大且0.80微米或更小。
当提供此种带正电荷的颗粒时,转移效率在整个耐久性和使用过程中是极好的。具有此种粒径的带正电荷的颗粒可在调色剂颗粒的表面上滚动,并且当颗粒在感光鼓和转印带之间摩擦时促进调色剂的负电荷。推测因此,抑制了由施加转移偏压引起的正电荷。本发明的调色剂的特征在于具有坚硬的表面,并且因此带正电荷的颗粒不容易附着或嵌入调色剂颗粒表面中。因此,可保持高转移效率。带正电荷的颗粒的示例包括水滑石、氧化钛和三聚氰胺树脂。其中,水滑石是特别优选的。
调色剂可以在调色剂颗粒的表面上具有氮化硼。氮化硼可以通过任何方法提供给调色剂颗粒表面,例如,氮化硼可以从外部添加到调色剂颗粒表面。已经发现的是,只要调色剂的马氏硬度在本发明的范围内,氮化硼可均匀地并以高附着率存在于调色剂颗粒表面上,并且附着率在整个耐久性和使用过程中的劣化很小。
[变型]
当调色剂瓶12附接到开口34时,调色剂瓶12可以布置成不向上突出并且位于设备外壳的外侧,并且可以布置成容纳在装置的内部,尽管与各个实施例中描述的情况相比,这具有增大装置尺寸的缺点。在这种情况下,也可通过更简单的系统补充显影剂,该系统涉及通过利用调色剂自身的重量将调色剂从调色剂瓶12移动到显影剂容纳室37。此外,通过使用第三实施例中描述的调色剂获得了协同效果,因为即使当每次调色剂用完时通过使用新的调色剂瓶12继续调色剂补充时,也可进一步抑制补充雾化和积聚的劣化调色剂的量的增加。应注意的是,尽管新的调色剂瓶被用来补充,但是不更换诸如感光鼓1和显影设备3的其它处理单元。
根据上述描述,可以提供一种通过更简单的结构实现显影剂补充的机构和一种实现更用户友好的显影剂补充的机构。
虽然已经参考示例性实施例描述了本发明,但是应当理解的是,本发明不限于所公开的示例性实施例。所附权利要求的范围应被赋予最广泛的解释,以便包含所有此类修改和等效结构和功能。
本申请要求2018年11月14日提交的日本专利申请No.2018-213995的权益,其全部内容在此以引用的方式并入本文中。
- 广角成像光学系统以及使用该广角成像光学系统的广角成像装置、监视成像装置、车载成像装置以及投影装置
- 磁共振成像装置、磁共振成像装置用的磁场调整件、磁共振成像方法及磁共振成像装置的磁场调整方法