一种鉴定黄芩药材和黄芩饮片的方法
文献发布时间:2023-06-19 12:05:39
技术领域
本发明关于中药鉴定技术领域,尤其涉及一种鉴定黄芩药材和黄芩饮片的方法。
背景技术
黄芩为唇形科植物黄芩(Scutellaria baicalensis Georgi)的干燥根,具有清热燥湿,止血安胎的功效。临床研究发现黄芩具有抗肿瘤、抗炎、神经保护等功能,其主要的特征化学成分为黄芩苷、黄芩素、汉黄芩苷、汉黄芩素等黄酮类成分。2020版《中华人民共和国药典》黄芩饮片的炮制过程为“除去杂质,置沸水中煮10分钟,取出,闷透,切薄片,干燥;或蒸半小时,取出,切薄片,干燥”。黄芩饮片的炮制主要是达到杀酶保苷的作用,保证黄芩饮片的质量稳定。目前市场上销售的黄芩饮片存在没有进行高温杀酶处理或者杀酶处理不完全的黄芩饮片,炮制不规范的饮片在储藏过程中有效成分易发生转化,导致其质量下降。
2020版《中华人民共和国药典》中对于黄芩饮片的要求为黄芩苷含量不少于8%,但该要求并不能有效鉴别黄芩药材与黄芩饮片,并且从外观分析也很难有效地区别黄芩药材和黄芩饮片。为保证黄芩饮片的有效性以及质量稳定性,需要构建一种分析方法用于鉴别黄芩药材和黄芩饮片。
发明内容
针对以上技术问题,本发明提供一种鉴定黄芩药材和黄芩饮片的方法,该方法能够有效鉴别黄芩药材和黄芩饮片,能够用作保证黄芩饮片的有效性以及质量稳定性的监控方法。
为达到上述发明目的,本发明实施例采用了如下的技术方案:
一种鉴定黄芩药材和黄芩饮片的方法,具体包括以下步骤:
(1)称取质量为M1的待测黄芩,用体积为V1的低浓度乙醇提取获得第一供试品浓液,测定所述第一供试品溶液中的A成分和B成分的含量;
称取质量为M2的待测黄芩,用体积为V2的高浓度乙醇提取获得第二供试品浓液,测定所述第二供试品溶液中的A成分和B成分的含量;
其中,M1:V1=M2:V2;所述高浓度乙醇为体积百分比浓度≥50%的乙醇水溶液,所述低浓度乙醇为体积百分比浓度<50%的乙醇水溶液,且所述高浓度乙醇与所述低浓度乙醇的体积百分比浓度之差≥25%;
所述A成分为黄芩苷,所述B成分为黄芩素;或
所述A成分为汉黄芩苷,所述B成分为汉黄芩素;
(3)计算所述第一供试品溶液测得的A成分与所述第二供试品溶液测得的A成分的含量比值,记为a;计算所述第一供试品溶液测得的B成分与所述第二供试品溶液测得的B成分的含量比值,记为b;
在0.9≤a≤1.1,且0.5≤b≤1.1的情况下确定所述待测黄芩为黄芩饮片,否则所述待测黄芩为黄芩药材。
发明人经过广泛的研究发现,采用不同浓度的乙醇对未经蒸制的黄芩药材进行提取时,获得的提取液中黄芩苷和汉黄芩苷的含量会随着提取溶剂中乙醇浓度降低而降低,黄芩素和汉黄芩素的含量会随着提取溶剂中乙醇浓度降低而升高(如图1、2所示),且乙醇浓度差异越大,黄芩苷和黄芩素或汉黄芩苷和汉黄芩素的变化越明显,尤其是当乙醇浓度<50%时,这种变化趋势更加明显,当乙醇浓度≥50%时各指标成分含量基本不变。而采用不同浓度乙醇对蒸制的黄芩饮片进行提取时,获得的提取液中黄芩苷、黄芩素、汉黄芩苷、汉黄芩素的含量基本稳定(如图3、4所示)。
对于黄芩药材和黄芩饮片,虽然提取获得的供试品溶液中4个指标成分含量变化趋势不同,但是提取溶剂中醇浓度差异较小时,也无法准确区别两种变化趋势,即无法确认待测黄芩是否为黄芩饮片。并且如果高浓度醇和低浓度醇体积百分比均>50%,也很难区分这两种变化趋势,即很难确定待测黄芩是否为黄芩饮片。所以本发明限定了高浓度乙醇的体积百分比浓度≥50%,高浓度和低浓度乙醇的体积百分比浓度的差值≥25%。结合上述规律,并且考虑到鉴别过程中各环节存在的误差,发明人将a的取值范围确定为0.9~1.1,b的取值范围确定为0.5~1.1。
本发明的方法用不同浓度的乙醇溶液对待测黄芩粉末进行提取,再对提取液中黄芩苷与黄芩素或汉黄芩苷与汉黄芩素的含量变化进行判断,能够有效鉴别待测黄芩为黄芩药材还是黄芩饮片。
本发明的一种更为优选实施方式中,所述的高浓度乙醇的体积百分比浓度为70%。
本发明的一种更为优选实施方式中,所述的低浓度乙醇的体积百分比浓度为25%。
本发明的一种更为优选实施方式中,所述待测黄芩为粉末状,以利于充分提取。
本发明的一种更为优选实施方式中,所述提取为超声提取,以保持黄芩药材中酶的活性。
本发明的一种更为优选实施方式中,步骤(1)中采用超高效液相色谱法测定所述第一供试品溶液和第二供试品溶液中所述A成分和B成分的含量。
可选地,所述超高效液相色谱法的预设色谱条件包括:
色谱柱:十八烷基硅烷键合硅胶的固定相;
流动相:A相为0.2%甲酸水溶液,B相为甲醇;梯度洗脱,所述梯度洗脱具体为:B相的体积百分比在0~10分钟由40%匀速增加到63%。
可选地,所述超高效液相色谱法的预设色谱条件还包括:柱温:35~45℃;进样量:1.5~2.5μL;检测波长:280nm;流速:0.25~0.35mL/分钟。进一步优选的条件为:柱温:40℃;进样量:2μL;检测波长:280nm;流速:0.3mL/分钟。
附图说明
下面将结合附图及实施例对本发明作进一步说明,附图中:
图1为本发明实施例2中用不同浓度的乙醇提取黄芩药材时提取液中黄芩苷、汉黄芩苷、黄芩素、汉黄芩素的含量曲线图;
图2为本发明实施例2中用不同浓度的乙醇提取黄芩药材时提取液中黄芩苷、汉黄芩苷、黄芩素、汉黄芩素的含量柱状图;
图3为本发明实施例2中用不同浓度的乙醇提取黄芩饮片时提取液中黄芩苷、汉黄芩苷、黄芩素、汉黄芩素的含量曲线图;
图4为本发明实施例2中用不同浓度的乙醇提取黄芩饮片时提取液中黄芩苷、汉黄芩苷、黄芩素、汉黄芩素的含量柱状图;
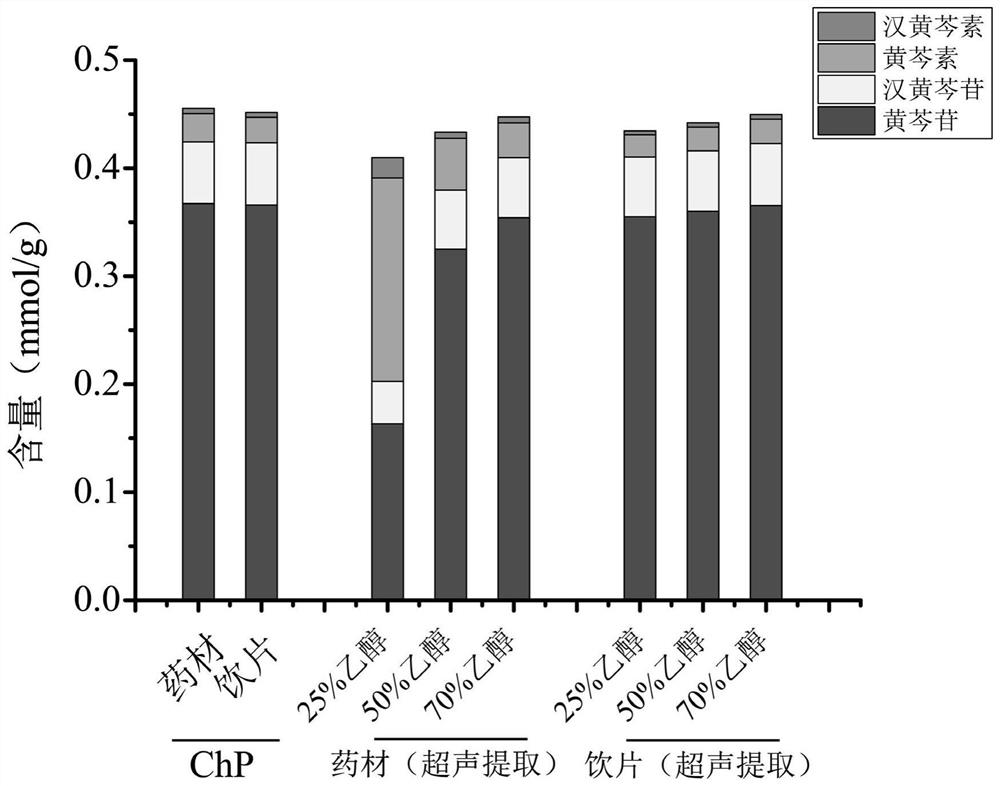
图5为本发明实施例2中采用药典法以及用不同浓度乙醇超声提取黄芩药材和黄芩饮片时供试品溶液中黄芩苷、汉黄芩苷、黄芩素、汉黄芩素的含量图。
具体实施方式
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图及实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
实施例1
本发明实施例提供了用超高效液相色谱法检测黄芩苷、汉黄芩苷、黄芩素、汉黄芩素的检测方法以及标准曲线。
超高效液相色谱法的色谱条件为:
仪器:Waters Acquity HPLC-H Class plus(美国沃特世公司),PDA检测器;
色谱柱:
柱温:40℃;
进样量:2μL;
检测波长:280nm;
流速:0.3mL/分钟;
流动相:A相为0.2%甲酸水溶液,B相为甲醇;梯度洗脱,洗脱程序见表1。
表1流动相梯度洗脱条件
标准曲线的绘制:
取黄芩苷10.01mg、汉黄芩苷10.11mg、黄芩素10.11mg、汉黄芩素10.01mg,精密称定,分别置于10mL容量瓶中,用甲醇溶解(黄芩苷、汉黄芩苷采用少量的二甲基亚砜助溶)并定容至刻度线,摇匀,分别获得浓度为1.001mg/mL黄芩苷对照品储备液、1.011mg/mL汉黄芩苷对照品储备液、1.011mg/mL黄芩素对照品储备液、1.001mg/mL汉黄芩素对照品储备液,备用。
分别精密量取上述汉黄芩苷、黄芩素、汉黄芩素对照品储备液各1mL,分别置于10mL、5mL、10mL容量瓶中,用甲醇定容至刻度线,摇匀,获得浓度为0.1011mg/mL汉黄芩苷对照品溶液、0.2022mg/mL黄芩素对照品溶液、0.1001mg/mL汉黄芩素对照品溶液,备用。
分别取上述黄芩苷对照品储备液2.5mL、汉黄芩苷对照品溶液4mL、黄芩素对照品溶液4mL、汉黄芩素对照品溶液1mL于25mL容量瓶中,用甲醇定容至刻度线,摇匀获得混合对照品储备液,混合对照品储备液中,黄芩苷的浓度为100.1μg/mL、汉黄芩苷的浓度为16.18μg/mL、黄芩素的浓度为32.35μg/mL、汉黄芩素的浓度为4.004μg/mL。
用50%甲醇将上述混合对照品储备液逐级稀释2、4、8、16、32、64、128倍,得到一系列不同浓度的混合对照品溶液。
采用超高效液相色谱法测定系列的混合对照品溶液,得到相应的超高效液相色谱图,分别以黄芩苷、汉黄芩苷、黄芩素、汉黄芩素的浓度为横坐标,相应的超高效液相色谱图的峰面积作为纵坐标,绘制得到4条标准曲线,各标准曲线的线性回归方程、线性范围,r
表2黄芩苷、汉黄芩苷、黄芩素、汉黄芩素的线性回归方程以及r
实施例2
本发明实施例提供了待测黄芩提取方法和乙醇浓度的筛选过程。
(a)黄芩药材粉末以及黄芩饮片粉末的制备
将黄芩药材粉碎,过65目筛,即得黄芩药材粉末。
将同批次黄芩药材蒸制30分钟,干燥后粉碎,过65目筛,即得黄芩饮片粉末。
(b)采用2020版《中华人民共和国药典》中记录的黄芩提取方法制备上述黄芩药材及黄芩饮片的供试品溶液。其制备过程具体如下:
取上述黄芩药材粉末0.3g,加入体积百分比浓度为70%乙醇40mL,加热回流3小时,放冷,将溶液倒入100mL量瓶中,用少量的体积百分比浓度为70%乙醇分次洗涤容器和残渣,洗液倒入同一量瓶中,加体积百分比浓度为70%乙醇至刻度线,摇匀,12700rpm离心10分钟,精密量取上清液1mL,置10mL量瓶中,加甲醇定容至刻度,摇匀,取1mL,用水1:1稀释即得黄芩药材供试品溶液。
同法制备黄芩饮片供试品溶液。
(c)超声法制备黄芩药材及黄芩饮片的供试品溶液
取上述黄芩药材粉末0.3g,于100mL容量瓶中,加入体积百分比浓度为70%乙醇40mL,常温超声提取(600w,35kHZ)30分钟,放冷,加体积百分比浓度为70%乙醇至刻度线,摇匀,12700rpm离心10分钟,精密量取上清液1mL,置10mL容量瓶中,加甲醇定容至刻度,摇匀,取1mL,用水1:1稀释即得黄芩药材供试品溶液。同法制备黄芩饮片供试品溶液。
将上述步骤提取用的体积百分比浓度为70%乙醇,变更为体积百分比浓度为50%、25%乙醇,重复上述提取操作。进行常温超声提取时,用相应溶剂定容、离心后,精密量取上清液1mL,置10mL容量瓶中,立即加甲醇定容至刻度,然后依法处理,分别获得不同浓度乙醇提取的黄芩药材供试品溶液以及黄芩饮片供试品溶液。
(d)测定各供试品溶液中四个指标成分黄芩苷、汉黄芩苷、黄芩素、汉黄芩素的浓度。
对上述的黄芩供试品溶液,分别采用实施例1的超高效液相色谱法测定供试品溶液中4个指标成分,得到超高效液相色谱图,分别从色谱图中读取4个指标成分的峰面积带入实施例1中相应的回归方程中,得到各供试品溶液中各指标成分的浓度C1(μg/mL)。需要说明的是,精密称定是指称取质量应准确所取质量的千分之一。考虑到在称量过程中的差异,为了使结果对比更加精确,按照下式将上述浓度C1换算成含量C2(mmol/g)。
C2=(C1×2×10×100×10
其中,M为C1所对应的指标成分的分子量,m为C1所对应供试品溶液制备时精密称定的对应黄芩药材粉末或黄芩饮片粉末的质量。
以超声提取制备供试品溶液使用的乙醇浓度为横坐标轴,各指标成分含量C2为纵坐标轴,得到图1、3,并可进一步得到柱状图,即图2和图4。由图1~4可知在黄芩药材的供试品溶液中,黄芩苷、汉黄芩苷含量随提取溶剂中乙醇的浓度降低而降低,黄芩素、汉黄芩素的含量随提取溶剂中乙醇的浓度降低而升高;而黄芩饮片供试品溶液中的4个指标成分基本不随提取溶剂中乙醇的浓度变化而变化。从图5可以发现用体积百分比浓度为70%乙醇超声提取和采用2020版《中华人民共和国药典》法对黄芩饮片和黄芩药材进行提取,得到的测定结果基本一致。由于超声提取操作简单,且保留了黄芩药材中酶的活性,因此在实际操作中提取方法优选超声提取。
实施例3
本发明实施例提供了一种鉴定黄芩药材和黄芩饮片的方法的建立:
样品:
YC-1:黄芩药材;
YP-1:由YC-1蒸制30分钟后自然干燥,得到的黄芩饮片;
YC-2:黄芩药材;
YP-2:由YC-2蒸制30分钟后自然干燥,得到的黄芩饮片;
YC-3:黄芩药材;
YP-3:由YC-3蒸制30分钟后自然干燥,得到的黄芩饮片;
分别将6个样品粉碎过65目筛,得到6个样品的粉末。
鉴定黄芩药材和黄芩饮片的方法的建立步骤:
(1)称取0.3g待测黄芩粉末YC-1,于100mL容量瓶中,加入体积百分比浓度为70%乙醇40mL,超声(600w,35kHZ)30分钟,放冷,加体积百分比浓度为70%乙醇至刻度线,摇匀,12700rpm离心10分钟,精密量取上清液1mL,置10mL容量瓶中,加甲醇至刻度,摇匀,取1mL,用水1:1稀释即得YC-1的供试品溶液,命名为70-YC-1。
将上述步骤提取用的体积百分比浓度为70%乙醇,变更为体积百分比浓度为50%、25%乙醇,重复上述提取操作,特别要注意的是样品常温超声提取,用相应溶剂定容、离心后,精密量取上清液1mL,置10mL容量瓶中,立即加甲醇定容至刻度,然后依法处理,分别获得不同浓度乙醇YC-1的供试品溶液。
采用实施例1的超高效液相色谱法对70-YC-1供试品溶液进行测定,得到70-YC-1的供试品溶液色谱图,从色谱图中读取四个指标成分的峰面积带入实施例1中相应的线性回归方程中得到70-YC-1的溶液中黄芩苷、汉黄芩苷、黄芩素、汉黄芩素的浓度分别命名为,C1-黄芩苷、C1-汉黄芩苷、C1-黄芩素、C1-汉黄芩素,由于考虑到精密称定时粉末质量的差异按照前述的浓度C1和含量C2的换算公式,将浓度C1换算成含量C2,由于不同样品的含水量有差异,为了使样品的含量更加精确,将C2换算成C3,换算公式如下:
C3=C2/(1-W)
其中W为对应样品的含水量(%)。
重复上述步骤,得到用体积百分比浓度为50%、25%乙醇的提取溶剂得到的样品中黄芩苷、汉黄芩苷、黄芩素、汉黄芩素的含量。
以相同的方法得到6个样品用不同体积百分比浓度乙醇获得的黄芩苷、汉黄芩苷、黄芩素、汉黄芩素的含量。以第一供试品溶液测得的黄芩苷与第二供试品溶液测得的黄芩苷含量的比值为a1,第一供试品溶液测得的黄芩素与第二供试品溶液测得的黄芩素含量的比值为b1,第一供试品溶液测得的汉黄芩苷与第二供试品溶液测得的汉黄芩苷含量的比值为a2,第一供试品溶液测得的汉黄芩素与第二供试品溶液测得的汉黄芩素含量的比值为b2。
以体积百分比浓度为25%的乙醇提取得到的供试品溶液为第一供试品溶液,体积百分比浓度为70%的乙醇提取得到的供试品溶液为第二供试品溶液,测定结果见表3、4。
表3不同批次黄芩饮片各指标成分在第一供试品溶液(25%乙醇)与第二供试品溶液(70%乙醇)中测定的含量的比值
表4不同批次黄芩药材各指标成分在第一供试品溶液(25%乙醇)与第二供试品溶液(70%乙醇)中测定的含量的比值
以体积百分比浓度为50%的乙醇提取得到的供试品溶液为第一供试品溶液,体积百分比浓度为70%的乙醇提取得到的供试品溶液为第二供试品溶液,测定结果见表5、6。
表5不同批次黄芩饮片各指标成分在第一供试品溶液(50%乙醇)与第二供试品溶液(70%乙醇)中测定的含量的比值
表6不同批次黄芩药材各指标成分在第一供试品溶液(50%乙醇)与第二供试品溶液(70%乙醇)中测定的含量的比值
以体积百分比浓度为25%的乙醇提取得到的供试品溶液为第一供试品溶液,体积百分比浓度为50%的乙醇提取得到的供试品溶液为第二供试品溶液,测定结果见表7、8。
表7不同批次黄芩饮片各指标成分在第一供试品溶液(25%乙醇)与第二供试品溶液(50%乙醇)中测定的含量的比值
表8不同批次黄芩药材各指标成分在第一供试品溶液(25%乙醇)与第二供试品溶液(50%乙醇)中测定的含量的比值
由表3~8可知,采用黄芩药材或饮片各指标成分在第一供试品溶液(25%乙醇)与第二供试品溶液(70%乙醇)中测定的含量的比值作为判断依据是合理的,以0.9≤a1≤1.1,且0.5≤b1≤1.1或0.9≤a2≤1.1,且0.5≤b2≤1.1作为判断黄芩为黄芩饮片的指标是可行的。由表6可知在提取黄芩药材时,所用的乙醇体积百分比浓度都≥50%时,虽然可以判断出其为黄芩药材,但a1、a2的值接近于0.9,而b1、b2的值也接近于1.1,为了避免在鉴别过程中引起的误判,在使用高浓度乙醇制备黄芩的第二供试品溶液时,其高浓度乙醇体积百分比浓度需≥50%,而用低浓度乙醇制备黄芩的第一供试品溶液时,其低浓度乙醇体积百分比浓度需小于50%。而且当提取溶剂中醇浓度差异较小时,也无法区别两种变化趋势,即无法确认黄芩是否为黄芩饮片。所以本发明的实例中高浓度醇的体积百分比浓度≥50%,其低浓度乙醇体积百分比浓度需小于50%,高浓度和低浓度的醇的体积百分比浓度的差值≥25%,更为优选的高浓度乙醇的体积百分比浓度为70%,低浓度乙醇的体积百分比浓度为25%。
实施例4
本发明实施例提供了一种鉴定黄芩药材和黄芩饮片的方法,包括如下步骤:
(1)称取质量为M1的待测黄芩粉末,用体积为V1、体积百分比浓度为25%的低浓度乙醇超声提取获得第一供试品浓液,用超高效液相色谱法测定所述第一供试品溶液中的黄芩苷和黄芩素的含量;
称取质量为M2的待测黄芩粉末,用体积为V2、体积百分比浓度为70%的高浓度乙醇超声提取获得第二供试品浓液,用超高效液相色谱法测定所述第二供试品溶液中的黄芩苷和黄芩素的含量;
其中,M1:V1=M2:V2;
(2)计算所述第一供试品溶液测得的黄芩苷与所述第二供试品溶液测得的黄芩苷的含量比值,记为a;计算所述第一供试品溶液测得的黄芩素与所述第二供试品溶液测得的黄芩素的含量比值,记为b;
在0.9≤a≤1.1,且0.5≤b≤1.1的情况下,该待测黄芩粉末为黄芩饮片粉末,否则该待测黄芩粉末为黄芩药材粉末。
实施例5
本发明实施例提供了一种鉴定黄芩药材和黄芩饮片的方法,包括如下步骤:
(1)称取质量为M1的待测黄芩粉末,用体积为V1、体积百分比浓度为25%的低浓度乙醇超声提取获得第一供试品浓液,用超高效液相色谱法测定所述第一供试品溶液中的汉黄芩苷和汉黄芩素的含量;
称取质量为M2的待测黄芩粉末,用体积为V2、体积百分比浓度为70%的高浓度乙醇超声提取获得第二供试品浓液,用超高效液相色谱法测定所述第二供试品溶液中的汉黄芩苷和汉黄芩素的含量;
其中,M1:V1=M2:V2;
(2)计算所述第一供试品溶液测得的汉黄芩苷与所述第二供试品溶液测得的汉黄芩苷的含量比值,记为a;计算所述第一供试品溶液测得的汉黄芩素与所述第二供试品溶液测得的汉黄芩素的含量比值,记为b;
在0.9≤a≤1.1,且0.5≤b≤1.1的情况下,该待测黄芩粉末为黄芩饮片粉末,否则该待测黄芩粉末为黄芩药材粉末。
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
- 一种鉴定黄芩药材和黄芩饮片的方法
- 一种半枝莲药材或其配方颗粒中野黄芩苷和野黄芩素的含量测定方法