取代吡啶羧酸、其制备方法及其组合物
文献发布时间:2023-06-19 12:14:58
相关申请的交叉引用
本申请要求于2018年11月16日提交的印度专利申请号为201841043166的专利申请的优先权。
技术领域
本发明涉及新的取代吡啶羧酸衍生物及其制备方法。这些化合物及其药学上可接受的盐和酯可用于治疗或控制各种代谢性疾病。本发明还涉及含有此类化合物的药物组合物及其与至少一种治疗剂的结合。
背景技术
吡啶衍生物在治疗各种疾病方面有着广泛的治疗应用,并正在探索其各种新的活性。
美国专利3,655,679公开了具有抗炎、止痛特性的芳基吡啶羧酸及其衍生物。
欧洲专利申请EP0109027A1公开了2-烷氧基-5-(吡啶基)吡啶衍生物及其作为强心剂的用途。
US2004/0081672A1公开文本涉及对皮肤有益的有机酸的烟酰胺、烟酸和烟酸酯衍生物在协同治疗或预防皮肤局部疾病中的应用,例如痤疮、酒渣鼻、皮肤皱纹、老年斑、口腔溃疡、萎缩纹、粉刺和皮肤发红。
W02005/075464Al公开了用于治疗大麻素2受体介导的疼痛的吡啶衍生物。美国专利6,656,957公开了具有调节谷氨酸信号传递活性的卤素取代的吡啶衍生物。
尽管有多种选择,但仍未满足开发最合适的取代吡啶羧酸衍生物及其组合物的药物化合物的需要。因此有必要开发具有多种治疗活性的新的吡啶衍生物。本发明者在药物和药物化学领域中使用简单且经济的方法通过改变活性开发了具有良好收率和纯度的特定新的吡啶衍生物。
发明内容
本发明涉及取代的吡啶羧酸衍生物化合物,用于管理心血管疾病、炎症疾病、糖尿病、癌症、营养失调和皮肤病症以及其他代谢性疾病。
具体而言,本发明的目的是提供通式(I)的化合物或其药学上可接受的盐、异构体、酯、前药或溶剂化物:
其中,Ar选自以下给出的化合物:
并且,R
R
R
并且其中R
本发明的另一个目的是提供一种制备式(I)化合物的简单且经济的方法。
本发明的另一个目的是提供一种药物组合物,其包含药学上有效量的式(I)化合物或其药学上可接受的盐、异构体、酯、前药或溶剂化物以及药学上可接受的载体或赋形剂。
本发明的另一个目的是提供一种结合组合物,其包含药学上有效量的式(I)化合物或其药学上可接受的盐、异构体、酯、前药或溶剂化物以及药学上可接受的与另一种治疗剂组合的载体或赋形剂。
附图说明
图1示出了式(I)化合物的
图2示出了式(I)化合物的
图3示出了式(I)化合物的FT-IR谱图,特别是INL3001115。
图4示出了式(I)化合物的质谱图,特别是INL3001115。
图5示出了式(I)化合物的
图6示出了式(I)化合物的
图7示出了式(I)化合物的FT-IR谱图,特别是IINL3001136。
图8示出了式(I)化合物的质谱图,特别是INL3001136。
图9示出了式(I)化合物的
图10示出了式(I)化合物的
图11示出了式(I)化合物的FT-IR谱图,特别是INL3001119。
图12示出了式(I)化合物的质谱图,特别是INL3001119。
图13示出了式(I)化合物的
图14示出了式(I)化合物的
图15示出了式(I)化合物的FT-IR谱图,特别是INL3001117。
图16示出了式(I)化合物的质谱图,特别是INL3001117。
图17示出了式(I)化合物的FT-IR谱图,特别是INL3001101。
图18示出了式(I)化合物的质谱图,特别是INL3001101。
图19示出了INL3001117的合成方案。
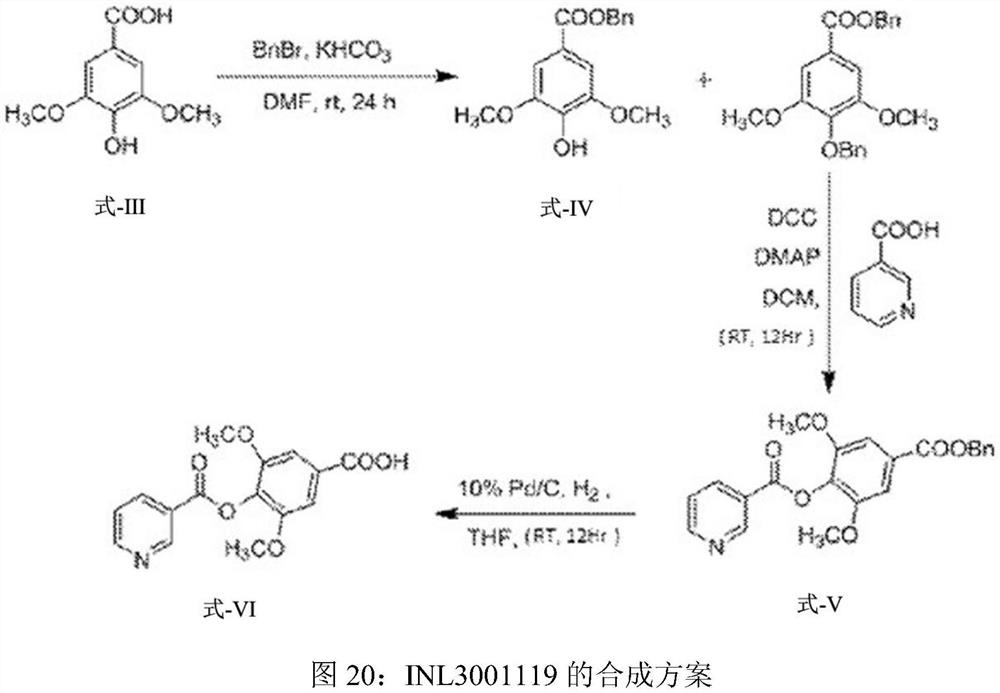
图20示出了INL3001119的合成方案。
具体实施方式
以下段落将详细说明本发明的各种实施例。为免发生歧义,明确规定这些段落(或其部分)中任何一段中单独描述的任何特定特征可与剩余段落(或其部分)中一段或多段中描述的一个或多个其他特征相结合。换言之,明确的意图是下面在每一段(或其部分)中单独描述的特征代表本发明的重要方面,这些方面可以单独地实施,和与本说明书中其他地方描述的本发明的其他重要方面相结合作为一个整体,包括示例和附图。本领域技术人员将理解,本发明扩展到这些特征的这类结合,并且为了简洁起见,这里没有详细叙述这些特征。
本发明涉及通式(I)的取代的吡啶衍生物化合物或其药学上可接受的盐、异构体、酯、前药或溶剂化物:
其中,Ar选自下述化合物:
并且
R
R
R
并且其中R
在另一优选实施例中,本发明涉及用于制备式(I)化合物、其分离和表征的方法。
在另一优选实施例中,本发明提供一种结合组合物,其包含药学上有效量的式(I)化合物或其药学上可接受的盐、异构体、酯、前药或溶剂化物以及药学上可接受的载体或赋形剂与另一治疗剂的组合。
在另一优选实施例中,本发明提供用于治疗胆碱能受体介导的疾病的药物组合物,其中所述组合物包含药学上有效量的式(I)化合物或其药学上可接受的盐、异构体、酯、前药或溶剂化物和药学上可接受的载体或赋形剂。
在另一实施例中,本发明化合物用于管理高脂血症和高胆固醇血症及相关的心血管疾病、糖尿病、营养失调、炎症、增生性疾病、皮肤病症和其他代谢性疾病。
在一些实施例中,本发明的组合物为固体、液体或半固体剂型的形式,用于口服、局部、直肠、静脉内、肌肉内给药。
合适固体剂型的非限制性实例包括片剂(例如悬浮片、咬合悬浮片剂、快速分散片、咀嚼片、融化片、泡腾片、双层片等)、囊片、胶囊剂(例如填充有固体和/或液体的软或硬明胶胶囊)、粉末剂(例如包装粉末,可分配的粉末或泡腾粉末)、锭剂、小袋、扁囊剂、糖锭、丸剂、颗粒剂、微粒、胶囊微粒、粉末气雾剂或任何其他合理适于口服给药的固体剂型。
合适的液体剂型的非限制性示例包括溶液、悬浮液、酏剂、糖浆、液体气雾剂等。
合适的半固体剂型的非限制性示例包括软膏、凝胶、乳液和乳膏等。
在另一实施例中,根据本发明的化合物可转化为各种给药形式。这可以通过与惰性、无毒、药学上合适的赋形剂混合用一种本身已知的方式来实现。这些赋形剂包括崩解剂、粘合剂、载体、溶剂、乳化剂和分散剂或润湿剂、表面活性剂、润滑剂、助流剂、合成和天然聚合物、稳定剂、染料、味道和/或气味校正剂。
在本发明中,以下术语具有下面详述的含义。
术语“盐”必须理解为根据本发明使用的化合物的任何形式,其中所述化合物呈离子形式或带电并与反离子(阳离子或阴离子)偶联或呈溶液形式。这一定义还包括季铵盐和分子与其他分子和离子的络合物,特别是通过离子相互作用形成的络合物。该定义特别包括生理上可接受的盐;该术语必须理解为等同于“药理学上可接受的盐”或“药学上可接受的盐”。
本发明上下文中的术语“药学上可接受的盐”是指当以适当方式用于治疗时,尤其是在人类和/或哺乳动物中应用或使用时,在生理上可耐受的任何盐(通常意味着其无毒,尤其是由于反离子而无毒)。盐的非限制性示例包括本发明化合物的无毒的、无机的和有机的碱或酸加成盐。在许多情况下,本发明的化合物能够凭借氨基和/或羧基或与其类似的基团的存在而形成酸和/或碱盐。药学上可接受的酸加成盐可由无机酸和有机酸形成。可衍生盐的无机酸包括例如盐酸、氢溴酸、硫酸、硝酸、磷酸等。可衍生盐的有机酸包括例如乙酸、丙酸、乙醇酸、丙酮酸、草酸、马来酸、丙二酸、琥珀酸、富马酸、酒石酸、柠檬酸、苯甲酸、肉桂酸、扁桃酸、甲磺酸、乙磺酸、对甲苯磺酸、水杨酸等。药学上可接受的碱加成盐可用无机和有机碱形成。可衍生盐的无机碱包括例如钠、钾、锂、铵、钙、镁、铁、锌、铜、锰、铝等;尤其优选铵盐、钾盐、钠盐、钙盐和镁盐。可衍生盐的有机碱包括例如伯胺、仲胺及叔胺、取代胺(包括天然存在的取代胺)、环胺、碱性离子交换树脂等,特别是例如异丙胺、三甲胺、二乙胺、三乙胺、三丙胺和乙醇胺。本发明的药学上可接受的盐可通过常规化学方法由母体化合物、碱性或酸性部分合成。通常,可通过使这些化合物的游离酸形式与化学计量的适当碱(例如氢氧化钠、氢氧化钙、氢氧化镁或氢氧化钾、碳酸盐、碳酸氢盐等)反应,或通过使这些化合物的游离碱形式与化学计量的适当酸反应,来制备所述盐。这类反应通常在水或有机溶剂,或在两者的混合物中进行。通常,在可行的情况下,优选非水介质,如醚、乙酸乙酯、乙醇、异丙醇或乙腈。额外合适盐的列表可在Remington's Pharmaceutical Sciences,20th ed.,Mack Publishing Company,Easton,Pa.,(1985)中找到,其通过引用并入本文中。
根据本发明的术语“溶剂化物”应理解为意指根据本发明的化合物的任何形式,其中所述化合物通过非共价键结合到另一分子(通常为极性溶剂),尤其包括水合物和醇化物。优选溶剂化物是水合物。
如本文所使用的术语“药学上可接受的载体”是指一种或多种相容的固体或液体填料稀释剂或所有溶剂、分散介质、包衣、表面活性剂、抗氧化剂、防腐剂、等渗剂、吸收延迟剂、盐、防腐剂、药物、药物稳定剂、粘合剂、赋形剂、崩解剂、润滑剂、甜味剂、调味剂、染料等本领域普通技术人员已知的类似材料及其组合(例如,参考Remington's PharmaceuticalSciences,18th Ed.Mack Printing Company,1990,第1289-1329页,其通过引用并入本文)。除非任何常规载体与活性成分不相容,否则考虑将其用于治疗或药物组合物中。
作为式(I)化合物的前药的任何化合物也在本发明的范围内。术语“前药”以其最广义使用,并且包括在体内转化为本发明化合物的那些衍生物。前药的示例包括但不限于式(I)化合物的衍生物及代谢物。优选地,具有羧基官能团的化合物的前药是羧酸的低级烷基酯。通过酯化分子上存在的任何羧酸部分,方便地形成羧酸酯。前药通常可以使用众所周知的方法来制备,例如“Medicinal Chemistry and Drug Discovery 6th ed.(DonaldJ.Abraham ed.,2001,Wiley)以及“Design and Applications of Prodrugs”(H.Bundgaard ed.,1985,Harwood Academic Publishers)中描述的那些方法。
本发明化合物的术语“治疗有效量”是指将引起受试者的生物或医学反应、或改善症状、减缓或延迟疾病进展、或预防疾病等的本发明化合物的量。
图1-18通过对新的取代的吡啶衍生物化合物的质谱、FT-IR和NMR谱的分析简要表征了通式(I)的一些非限制性的取代的吡啶衍生物化合物或其药学上可接受的盐、异构体、酯、前药或溶剂化物:
即INL3001115、INL3001136、INL3001119、INL3001117和INL3001101。
图19-20为制备化合物INL3001117和INL3001119的示意合成工艺。
在本发明的另一实施例中,本发明涉及一种制备式(I)化合物的方法,其中该方法步骤通过降低总体工艺成本避免了传统合成工艺中使用的多个纯化和提取步骤,并且还避免了多次溶剂洗涤。
在又一实施例中,在制备式(I)化合物的工艺步骤中获得的残余物可在有或没有任何附加纯化步骤的情况下用于下一步骤。
在另一实施例中,该方法包括各种非限制性反应组分,例如氯化剂、有机和无机溶剂、杂环芳香化合物、极性和非极性溶剂、结晶剂或其混合物。
如下所述的制备方法旨在帮助理解本发明,并且无意也不应理解为以任何方式限制本发明的范围。本领域技术人员将容易认识到在不改变本发明范围的情况下可以进行各种修改和变化。这样的修改和变型包含在本发明的范围内,并且这些实施例决不以任何方式限制本发明的范围。
实施例:
实施例1:式(I)的取代的吡啶羧酸衍生物的合成过程:
·将吡啶羧酸放入烧瓶中,并向其中添加合适的氯化剂。
·然后在较高温度下将所得混合物搅拌足够的时间。
·然后从反应混合物中去除过量的氯化剂,并将所得残留物在纯化或未纯化的情况下用于下一个反应步骤。
·此后,在溶剂中制备上述残余物的溶液,向其中加入杂环芳族有机化合物,然后加入酚衍生物的溶液。
·将所得混合物在室温以上搅拌足够的时间。
·监测反应完成情况,这通过TLC(薄层色谱)或任何其他合适的已知分析技术进行确认。
·然后将反应混合物冷却,并用极性溶剂淬灭。
·在旋转蒸发器上使用芳烃将溶剂完全去除。
·此后,将残留物溶解在乙醇或任何其他有机溶剂中,并向其中添加硅胶,然后干燥,得到干燥的浆液。
·然后将浆液加载到色谱柱上,并使用合适的溶剂洗脱,以获得具有高收率的吡啶衍生物。
·此后,可以通过将分离的化合物溶解在有机溶剂中并加入良好的结晶溶剂进行结晶,然后冷却以得到晶体并将该晶体与母液分离并干燥。
实施例2:取代的吡啶羧酸衍生物的合成过程:
其中R
实施例3:甲酯基芳基烟酸盐的合成
其中,
R=OH,R
R=OH,R
R=H,R
R=OH,R
R=OH,R
R=OH,R
在将酚甲酯衍生物(1当量)、烟酸(1.5当量)、N,N’-二环己基碳二亚胺(DCC)(1.5当量)、4-二甲氨基吡啶(DMAP)(0.1当量)和二氯甲烷(10w/v)的混合物室温下搅拌6-12小时。采用薄层色谱法对反应过程进行了监测。反应完成后,将反应混合物过滤并将二氯甲烷层与饱和NaHCO
实施例3.1:4-(甲氧基羰基)烟酸苯酯的合成
使用与实施例3相同的合成方法制备4-(甲氧基羰基)烟酸苯酯,收率为85%。
表征(分析数据):收率:85%;Mp:127-128;
实施例3.2:2-甲氧基-4-(甲氧羰基)烟酸苯酯的合成
采用与实施例3相同的合成工艺制备2-甲氧基-4-(甲氧羰基)烟酸苯酯,收率为82%。
表征(分析数据):收率:82%;
实施例3.3:2,6-二甲氧基-4-(甲氧羰基)烟酸苯酯的合成
采用与实施例3相同的合成工艺制备2,6-二甲氧基-4-(甲氧羰基)烟酸苯酯,收率为80%。
表征(分析数据):收率:80%;Mp:173-175℃;
实施例3.4:4-(甲氧羰基)-1,2-亚苯二烟酸酯的合成
采用与实施例3相同的合成工艺制备4-(甲氧羰基)-1,2-亚苯二烟酸酯,收率为76%。
在上述反应中,使用2.2当量的烟酸代替1.5当量的烟酸。
表征(分析数据):收率:76%;Mp:159-161℃;
实施例4:2-羟基-4-(甲氧羰基)烟酸苯酯的合成
表征(分析数据):收率:75%;
在双颈圆底烧瓶中,将2-(苄氧基)-4-(甲氧基羰基)烟酸苯酯(0.5g,1当量)、四氢呋喃(THF)(10mL)和10%Pd/C(0.lg)进行混合。然后将烧瓶用隔膜盖上,施加氮气、真空并释放。之后,使用气球将烧瓶充满氢气。将悬浮液在室温搅拌4小时。通过薄层色谱法确认原料的完全转化。反应完成后,将Pd/C在硅藻土床上过滤,并用THF洗涤。最后,在真空下蒸发THF,并将粗产物在二氯甲烷中搅拌,得到白色固体状的脱苄基产物。
表征(分析数据):收率:55%;
实施例5:2-羟基-5-(甲氧羰基)烟酸苯酯的合成
步骤1:
4-((叔丁基二甲基甲硅烷基)氧基)-3-羟基苯甲酸酯(lg,1当量)、烟酸(0.65g,1.5当量)、N,N’-二环己基碳二亚胺(DCC)(1.1g,1.5当量)和4-二甲氨基吡啶(DMAP)(0.1g,0.1当量)在二氯甲烷(10mL)中,在室温下搅拌,直到TLC指示原料完全转化。然后过滤反应混合物,并用饱和NaHCO
表征(分析数据):
用乙酸(0.3mL)和1M的四丁基氟化铵(0.41g,2当量)溶液在低于5℃的温度下处理2-((叔丁基二甲基甲硅烷基)氧基)-5-(甲氧基羰基)烟酸苯酯(0.3g,1当量)(来自上述步骤1)在THF(10mL)中的混合物,直至TLC指示反应完成(30分钟)。然后将反应混合物倒入冰冷的水中,并用乙酸乙酯萃取(2×20mL)。合并的有机层经无水Na
表征(分析数据):
实施例6:3-羟基-5-(甲氧羰基)烟酸苯酯和5-(甲氧羰基)-l,3-亚苯二烟酸酯的混合物的合成:(合成过程与实施例3相同)
从混合物中分离出两种化合物,并且表征如下表1所示:
实施例7:4-((苄氧基)羰基)烟酸苯酯的合成
酚苄基酯衍生物(1当量)在二氯甲烷(10w/v)、带电烟酸(1.5当量)、DCC(1.5当量)和DMAP(0.1当量)的溶液。将反应混合物在室温下搅拌8-15小时。通过薄层色谱法观察了反应进程。反应完成后,将反应混合物过滤并将滤液(二氯甲烷层)转移至饱和NaHCO
表征(分析数据):收率:76%;
实施例8:4-((苄氧基)羰基)-2-甲氧基烟酸苯酯的合成
将与实施例7中相同的合成工艺用于制备4-((苄氧基)羰基)-2-甲氧基烟酸苯酯。
表征(分析数据):收率:82%;
实施例9:4-((苄氧基)羰基)-2,6-二甲氧基烟酸苯酯的合成
采用与实施例7中相同的合成工艺制备4-((苄氧基)羰基)-2,6-二甲氧基烟酸苯酯。
表征(分析数据):收率:80%;Mp:111-112℃;
实施例10:2-(苄氧基)-5-((苄氧基)羰基)烟酸苯酯的合成
采用与实施例7中相同的合成工艺制备2-(苄氧基)-5-((苄氧基)羰基)烟酸苯酯。
表征(分析数据):收率:76%;Mp:70-72℃;
实施例11:2-(苄氧基)-4-((苄氧基)羰基)烟酸苯酯的合成
采用与实施例7中相同的合成工艺制备2-(苄氧基)-4-((苄氧基)羰基)烟酸苯酯。
表征(分析数据):收率:82%;
实施例12:合成4-((苄氧基)羰基)-2-羟基烟酸苯酯和4-((苄氧基)羰基)-1,2-亚苯二烟酸酯的混合物。
采用与实施例7相同的合成工艺制备上述混合物。
从混合物中分离出两种化合物,其特征如下表2所示:
实施例13:合成3-((苄氧基)羰基)-5-羟基烟酸苯酯和5-((苄氧基)羰基)-l,3-亚苯二烟酸酯的混合物。
采用与实施例7相同的合成工艺制备上述混合物。
从混合物中分离出两种化合物,其特征如下表3所示:
实例14:2-(苄氧基)-5-((苄氧基)羰基)-3-羟基烟酸苯酯的合成:
采用与实施例7中相同的合成工艺制备2-(苄氧基)-5-((苄氧基)羰基)-3-羟基烟酸苯酯。
表征(分析数据):收率:56%;
实施例15:2,6-双(苄氧基)-4-((苄氧基)羰基)烟酸苯酯的合成:
使用与实施例7相同的合成方法制备2,6-双(苄氧基)-4-((苄氧基)羰基)烟酸苯酯。
表征(分析数据):收率:78%;
实施例16:4-((烯丙基氧基)羰基)烟酸苯酯的合成
采用与实施例7相同的合成方法制备4-((烯丙基氧基)羰基)烟酸苯酯,其中使用酚烯丙基酯衍生物代替酚苄基酯衍生物。
表征(分析数据):收率:75%;
- 取代吡啶羧酸、其制备方法及其组合物
- 6-(三取代的苯基)-4-氨基吡啶-2-羧酸类除草剂和解草酯类化合物的用于谷类作物的安全性组合物