一种化学交联透明质酸水凝胶及其制备方法与应用
文献发布时间:2023-06-19 12:18:04
技术领域
本发明涉及一种化学交联透明质酸水凝胶及其制备方法与应用,属于材料技术领域。
背景技术
由于先天或后天的原因造成人体组织和器官的缺损、畸形等一系列问题,再加上医美行业的需求,研究者和医疗器械企业一直在探索一种安全、方便且可靠的填充物,用以整形和促进组织修复,在20世纪40年代起,由于材料科学的进一步发展,许多植入材料被用作组织填充剂应用于人体组织填充,在过去的三十年中,通过组织填充的手段治疗患者的方式使用频率越来越高,美国涉及软组织填充的手术数量从2011年160万例增加到2015年的240 万例。组织填充是通过扩充体积来实现组织填充,近几十年来,多种高粘度液体或聚合物颗粒悬浮液组成的真皮填充剂被应用于皱纹和痤疮疤痕下填充。目前已上市组织填充剂主要以胶原和透明质酸为主。
透明质酸是一种高分子长链黏多糖,广泛分布于人体各部位,具有良好的亲水性、黏弹性、润滑性和生物相容性,并且已经在医学领域得到广泛的应用。天然透明质酸存在稳定性差、对透明质酸酶及自由基敏感、体内保留时间短,常用的方法是使用交联剂(二乙烯基砜 (DVS)或者丁二醇二缩水甘油醚(BDDE))将其交联,通过交联度来调控其体内保留时间。
目前,已经报道的文献中,交联过程均是在碱性环境下,优选pH 11-13。此方法交联效率低,反应回收率低(凝胶转化率),必然使得产品的注射频次增加、产品价格昂贵。因此,寻找一种高效交联水凝胶及其制备方法是解决目前组织填充剂领域材料需求和成本降低的关键。
为了解决上述问题,本发明提供了一种高效交联透明质酸、制备方法及其应用。基本构思是透明质酸是长链多糖在溴化锂溶液中具有更高的自由度,暴露更多的反应基团。化学交联反应在溴化锂溶液中反应可以明显提高反应均匀性、交联效率和产品回收率。
发明内容
为解决上述问题,本发明提供了一种化学交联透明质酸水凝胶及其制备方法。利用透明质酸是长链多糖在溴化锂溶液中具有更高的自由度,暴露更多的反应基团,提高反应交联效率和产品回收率。
本发明的第一个目的是提供一种化学交联透明质酸水凝胶,所述的透明质酸水凝胶是由透明质酸或其盐在溴化锂溶液中,通过化学交联剂交联得到。
进一步地,所述的化学交联剂为二缩水甘油醚、二乙烯基砜、1,2,7,8-二环氧辛烷、1,3-二环氧丁烷和三偏磷酸钠中的一种或两种以上。
进一步地,所述的透明质酸的分子量为400k-3000kDa。
进一步地,所述的透明质酸的分子量优选为500k-2500kDa,进一步优选为1000k-2200 kDa。
本发明的第二个目的是提供所述的化学交联透明质酸水凝胶的制备方法,包括如下步骤:
S1、将透明质酸或其盐溶于溴化锂溶液,得到透明质酸溴化锂溶液,并将化学交联剂加入到透明质酸溴化锂溶液中,或预先加入到溴化锂溶液中;
S2、进行交联反应,得到所述的化学交联透明质酸水凝胶。
进一步地,在S2步骤中,还包括反应后去除溴化锂、未反应的化学交联剂和未反应的透明质酸的步骤。
进一步地,所述的透明质酸或其盐的质量分数为1mg/mL~300mg/mL。
进一步地,所述的透明质酸或其盐的质量分数优选为10mg/mL~250mg/mL,进一步优选为20mg/mL~200mg/mL。
进一步地,所述的透明质酸或其盐与所述的化学交联剂的质量体积比为1g: 0.2-1000μL。
进一步地,所述的透明质酸或其盐与所述的化学交联剂的质量体积比优选为1g:2-800μL,进一步优选为1g:5-500μL。
进一步地,所述的溴化锂浓度为0.1-10M。
进一步地,所述的溴化锂浓度优选为1-10M,进一步优选2-9.8M。
进一步地,所述的交联反应是在反应温度-80~80℃条件下反应3分钟~72小时。
本发明的第三个目的是提供所述的化学交联透明质酸水凝胶的应用,所述的应用包括在作为软组织、硬组织或者腔体的填充或修复材料中的应用。
本发明的第四个目的是提供一种水凝胶组合物,包括由所述的化学交联透明质酸水凝胶。
进一步地,所述的化学交联透明质酸水凝胶中还包含保湿剂、生物活性剂、美容活性剂、细胞附着剂、细胞、细胞外基质、药物中的一种或多种组合。
其中保湿剂包括:甘油、聚谷氨酸等。
其中生物活性剂包括:细胞生长因子、肽、拟肽、抗体、核酸和多糖等。
其中细胞包括:干细胞、成体细胞、成骨细胞、成软骨细胞、成脂肪细胞等。
其中美容活性剂包括:抗衰老剂、抗自由基剂、抗氧化剂、水合剂、美白剂、着色剂、防晒剂、肌肉松弛剂等。
本发明的有益效果是:
本发明采用溴化锂交联替代传统碱性反应条件,可以降低交联剂用量,加快交联反应速度,提高产品回收率(凝胶转化率),与传统制备方法相比,所制备的注射用交联透明质酸凝胶产品具有更高的交联效率及产品回收率(凝胶转换率),明显延长凝胶体内的维持时间。
附图说明:
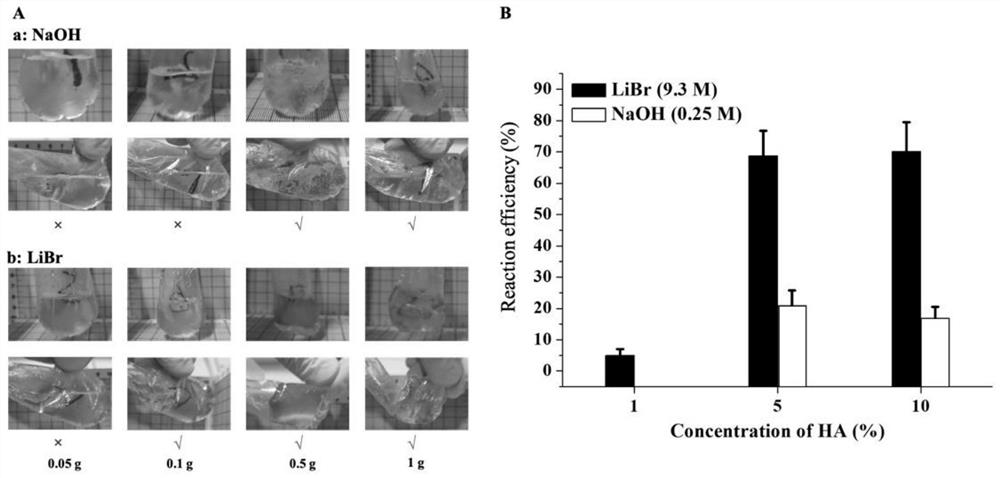
图1为透明质酸在碱性体系中(0.25M氢氧化钠)和溴化锂体系中反应情况及反应效率。 A:透明质酸在氢氧化钠和溴化锂体系中反应情况;B:透明质酸在氢氧化钠和溴化锂体系中的反应效率。
图2为不同反应体系制备化学交联透明质酸修饰度。
图3为不同反应体系制备化学交联透明质酸体外降解情况。
具体实施方式
下面结合具体实施例对本发明作进一步说明,以使本领域的技术人员可以更好地理解本发明并能予以实施,但所举实施例不作为对本发明的限定。
实施例1:不通过反应体系制备交联透明质酸
为了验证本发明适用的范围及其反应条件,发明人对透明质酸或其盐分子量、反应质量分数、交联剂种类、交联剂添加量,反应温度和反应时间进行了测试。
透明质酸或其盐分子量:400kDa、500kDa、700kDa、900kDa、1000kDa、1300kDa、1400kDa、 1500kDa、1600kDa、1800kDa、2000kDa、2100kDa、2200kDa、2400kDa、2500kDa、2700kDa、 2800kDa、3000kDa。
反应质量分数:1mg/mL、2mg/mL、5mg/mL、10mg/mL、15mg/mL、20mg/mL、30 mg/mL、50mg/mL、75mg/mL、90mg/mL、100mg/mL、130mg/mL、140mg/mL、160mg/mL、 180mg/mL、200mg/mL、210mg/mL、230mg/mL、250mg/mL、290mg/mL、300mg/mL
交联剂种类:二缩水甘油醚、二乙烯基砜、1,2,7,8-二环氧辛烷、1,3-二环氧丁烷、三偏磷酸钠。
交联剂添加量:1g:0.2μL、1g:0.40μL、1g:0.7μL、1g:1μL、1g:4μL、1g:7μL、 1g:8μL、1g:10μL、1g:30μL、1g:60μL、1g:80μL、1g:100μL、1g:300μL、1g: 600μL、1g:900μL、1g:1000μL
反应温度:-80、-20、4、25、37、45、60、80
反应时间:3分钟、5分钟、10分钟、30分钟、45分钟、60分钟、1小时、3小时、5 小时、8小时、12小时、24小时、48小时、72小时。
对照组仅有反应体系为0.25M氢氧化钠溶液。
实验发现本发明样品组在修饰度测试中均优于对照组。说明本发明采用溴化锂反应体系使交联效率得到显著的提高。
发明人列举了将0.05g、0.1g、0.5g和1g透明质酸(分子量1400KDa)分别溶于10mL0.25M NaOH或者9.3M溴化锂中,迅速向反应体系中加入500μL 1,4-丁二醇二缩水甘油醚(BDDE),并搅拌均匀,置于60℃烘箱反应3小时,观察反应体系中是否有块状凝胶形成,收集块状凝胶,水洗、冻干称重,计算交联效率。
交联效率=W
其中,W
研究发现:使用二缩水甘油醚、二乙烯基砜、1,2,7,8-二环氧辛烷、1,3-二环氧丁烷和三偏磷酸钠作为交联剂均能得到较为理想的玻尿酸水凝胶。
在透明质酸的分子量为400k-3000kDa范围内,均能得到较为理想的玻尿酸水凝胶。在 500k-2500kDa范围内,玻尿酸水凝胶性能更加优异。在1000k-2200kDa范围内,玻尿酸水凝胶性能进一步优秀。
在透明质酸或其盐的质量分数为1mg/mL~300mg/mL范围内,均能得到较为理想的玻尿酸水凝胶。在10mg/mL~250mg/mL范围内,玻尿酸水凝胶性能更加优异。在20 mg/mL~200mg/mL范围内,玻尿酸水凝胶性能进一步优秀。
在透明质酸或其盐与所述的化学交联剂的质量体积比为1g:0.2-1000μL范围内,均能得到较为理想的玻尿酸水凝胶。在1g:2-800μL范围内,玻尿酸水凝胶性能更加优异。在1g:5-500μL范围内,玻尿酸水凝胶性能进一步优秀。
在溴化锂浓度为0.1-10M范围内,均能得到较为理想的玻尿酸水凝胶。在1-10M范围内,玻尿酸水凝胶性能更加优异。在2-9.8M范围内,玻尿酸水凝胶性能进一步优秀。
在反应温度-80~80℃范围内,均能得到较为理想的玻尿酸水凝胶。
在反应时间3分钟~72小时范围内,均能得到较为理想的玻尿酸水凝胶。
实施例2:透明质酸修饰度测试
实施例1中0.5g和1g组,所得到的交联透明质酸凝胶上的9处位置的样品,每个样品 1g左右,测定9个样品修饰度,并计算RSD值。修饰度测试条件:称取0.25g纯化后的实施例一的样品于5mL容量瓶中,用酶解液定容至5mL,42℃静置2h,灭活后静置备用。使用高效液相色谱测其修饰度,色谱柱:SuperdexTM 75,进样量:20μL,流速:0.5mL/min,柱温:35℃,流动相:20mmol/L,波长:232nm。在其他实验条件均一致的情况下,修饰度越高,说明交联效率越高。9个取样点的修饰度越接近,说明交联均匀性越高。通过图2 结果可以看出,使用本发明方法所得到的交联透明质酸凝胶上的9个取样的修饰度几乎完全一致,而对比例中修饰度差异则比较大,说明该本发明方法使得交联均匀性得到有效保证。对比碱性条件下反应所得样品可以得出,本发明所得样品的修饰度远远高于对比组,说明本发明采用微波反应方法使交联效率得到显著的提高。
实施例3:体外降解测试
取适量实施例一中0.5g和1g组,化学交联透明质酸凝胶(约含HA 8mg),置于西林瓶中,加入自制透明质酸凝胶酶(酶活60U/mL)4mL,涡旋混匀。42℃水浴振摇,每 10min取样50μL并适当稀释,在232nm下测吸光度,直至吸光度不再变化,认为降解完全,记录完全降解所用时间。测试结果见图3。实施例制备得到的注射用交联透明质酸凝胶在体外降解的时间相对较长,而对比例所制备得到的注射用交联透明质酸凝胶在体外降解的时间相对较短,说明采用本发明所述的方法制备得到的注射用交联透明质酸凝胶具有优异的抗酶解性能,从而说明了所得到的注射用交联透明质酸凝胶交联效率高。
实施例4:可注射凝胶组合物制备、生物安全性评价及动物皮下填充实验
将10g大分子量透明质酸(分子量1400KDa)溶于100mL 9.3M溴化锂中,迅速向反应体系中加入5mL 1,4-丁二醇二缩水甘油醚(BDDE),并搅拌均匀,置于60℃烘箱反应3 小时,将凝胶切成约1cm
取小分子量透明质酸(分子量80KDa)1.0g,加入到100mL 0.5%的甘油水(也可选用 0.9%生理盐水、磷酸盐、氨基酸等等渗体系)溶液中,搅拌至溶解,经5μm滤芯过滤,得到pH为6.0-8.0的非交联透明质酸钠凝胶。将透明质酸钠凝胶和非交联透明质酸钠凝胶以质量比为1:10混合,混合均匀后,在100℃下进行热处理60分钟,得到可注射透明质酸盐凝胶体系。并进行如下测试:
1、推挤力,按照YY/T 0962-2014整形手术用交联透明质酸钠凝胶中附录B测试方法进行检测。得本发明所制备的透明质酸组合物推挤力为5-30N。
2、渗透压,按照《中华人民共和国药典》(2015版)第四部0632渗透压摩尔浓度测定法中的冰点下降发进行测试。所述可注射凝胶体系的粒径为250-350mOsmol。
3、粒径,按照《中华人民共和国药典》(2015版)第四部0982粒度和粒度分布测定法中光色she发中的显色发测定。所述可注射凝胶体系的粒径为10-300μm。
4、旋转粘度,按照《中华人民共和国药典》(2015版)第四部0633粘度测定法第三方转转粘度计测定法测定。所述可注射凝胶体系的运动粘度为10-80mm
5、pH值,按照《中华人民共和国药典》(2015版)第四部0631pH值测定法进行测试。所述可注射凝胶体系的pH值为6.0-8.0。
6、重金属,按照《中华人民共和国药典》(2015版)第四部0821金属检测法中第二法进行试压。所述可注射凝胶体系的重金属残留≤10μg/g。
7、溶血实验,按照GB/T16886.4-2003规定的方法进行试验。所述可注射凝胶体系的≤ 5%。
8、细胞毒性实验,按照GB/T16886.5-2008规定的方法进行试验。所述可注射凝胶体系的≤I级。
9、皮肤致敏实验,按照GB/T16886.10-2005规定的方法进行试验。所述可注射凝胶体系的≤I级。
10、亚曼毒性实验,按照GB/T16886.11-2011规定的方法进行试验。所述可注射凝胶体系无亚曼毒性。
11、皮下植入实验。选250g左右健康大鼠30只,背部左侧注射本发明样品组,右侧注射对照组,注射量1mL。跟踪观察受试打住一周、两周、一个月、三个月、六个月效果。实验结果发现三个月有效数量样品组30只、对照组28只;六个月有效数量样品组28只、对照组15只。说明本发明方法制备的注射用交联透明质酸凝胶的安全性与传统交联方法均能满足注射美容填充手术的需求,而长期有效性结果显示,本发明方法制备的注射用交联透明质酸凝胶在体内的维持时间远远高于传统交联方式制备的产品,是一种非常理想的组织填充剂。
以上所述实施例仅是为充分说明本发明而所举的较佳的实施例,本发明的保护范围不限于此。本技术领域的技术人员在本发明基础上所作的等同替代或变换,均在本发明的保护范围之内。本发明的保护范围以权利要求书为准。
- 一种化学交联透明质酸水凝胶及其制备方法与应用
- 化学交联的透明质酸水凝胶纳米颗粒及其制备方法