一种促进神经元再生的靶标蛋白及其应用
文献发布时间:2023-06-19 12:18:04
技术领域
本发明属于生物医药领域,涉及一种促进神经元再生的靶标蛋白及其应用。
背景技术
有研究表明LRP1蛋白可调节Tau蛋白的摄取和扩散,Tau蛋白在阿尔茨海默症的研究中为目前最流行的假说之一,但其无法预测LRP1是否对神经元的再生有促进作用,且LRP1基因对AD神经元再生功能的研究目前尚未阐明。目前绝大部分神经退行性疾病最直观的病症表现为神经元功能损伤,LRP1是否可作为神经元再生的关键靶点?
发明内容
本发明的目的是针对现有技术的上述不足,提供抑制LRP1基因表达的物质在制备治疗神经退行性疾病或治疗阿尔兹海默症的药物中的应用。
本发明的另一目的是提供一种用于LRP1基因编辑的RNP复合物。
本发明的又一目的是提供RNP复合物的应用。
本发明的目的可通过以下技术方案实现:
抑制LRP1基因表达的物质在制备治疗神经退行性疾病或治疗阿尔兹海默症的药物中的应用。LRP1基因的genbank:
作为本发明的一种优选,所述的抑制LRP1基因表达的物质在制备促进神经元再生、提高Tau蛋白和Aβ
作为本发明的一种优选,所述的抑制LRP1基因表达的物质选自用于LRP1基因敲除的CRISPR/Cas9基因编辑系统、针对LRP1的特异性反义寡核苷酸、siRNA/shRNA、锌指核酸酶、转录激活因子样效应核酸酶、小分子抑制剂、腺病毒、慢病毒或小分子化合物。
作为本发明的进一步优选,所述的用于LRP1基因敲除的CRISPR/Cas9基因编辑系统包括靶向LRP1基因的crRNA、TracrRNA和Cas9蛋白;所述的靶向LRP1基因的crRNA选自crRNA1、crRNA2或crRNA3中的任意一种或多种;所述的crRNA1序列如SEQ ID NO.4所示,所述的crRNA2序列如SEQ ID NO.5所示,所述的crRNA3序列如SEQ ID NO.6所示。
作为本发明的进一步优选,所述的靶向LRP1基因的crRNA选自crRNA1、crRNA2和crRNA3的组合。
一种RNP复合物,包括靶向LRP1基因的crRNA、TracrRNA和Cas9蛋白;所述的靶向LRP1基因的crRNA选自crRNA1、crRNA2或crRNA3中的任意一种或多种;所述的crRNA1序列如SEQ ID NO.4所示,所述的crRNA2序列如SEQ ID NO.5所示,所述的crRNA3序列如SEQ IDNO.6所示。
作为本发明的一种优选,所述的靶向LRP1基因的crRNA选自crRNA1、crRNA2和crRNA3的组合。
本发明所述的RNP复合物在制备提高Tau蛋白和Aβ
本发明所述的RNP复合物在制备促进神经元再生药物中的应用。
LRP1基因作为治疗靶点在筛选治疗神经退行性疾病或治疗阿尔兹海默症的药物中的应用。
有益效果:
本发明前期利用CRISPR/Cas 9基因编辑技术在SH-SY5Y细胞中成功敲除ADAM10构建了更为全面反应AD的细胞模型,研究结果发现敲除ADAM10后,Aβ
LRP1蛋白是否能够成为神经退行性疾病的靶点目前尚无报道,在上述AD细胞模型的基础上,本发明首次发现LRP1蛋白在ADAM10 KO细胞中显著性升高,且LRP1与Tau的相互作用显著性增强。在ADAM10 KO细胞中使用LRP1进行基因治疗我们发现,LRP1蛋白被敲低后,Aβ42与Tau蛋白均可以显著性降低的同时,炎症小体NLRP3蛋白显著性下调,神经元状态显著性改善。综上所述,LRP1蛋白的下调可以改善神经元的状态,同时针对LRP1蛋白合成小分子化合物以及生物大分子可能是一个新型的治疗神经退行性疾病的靶点。针对此靶点合成的抑制该靶基因表达的药物能够更为全面的治疗神经退行性疾病和/或AD疾病。
附图说明
图1 Sanger测序验证基因敲除
图A为CrRNA1,CrRNA2,CrRNA3在ADAM10基因基因序列中的位置示意图;图B为基因突变点测序图谱。与空白组Control KO相比,ADAM10敲除细胞株发生点突变。测序结果显示,已初步筛选到敲除ADAM10基因的细胞扩增株。对这些细胞株进行培养扩增,冻存做后续实验研究。
图2敲除ADAM10基因后靶标蛋白的变化
对敲除组的ADAM10在蛋白水平进行验证,如图A所示,与空白组control KO相比,ADAM10KO组ADAM10蛋白的表达极显著性降低,与Sanger测序结果相吻合。数据进一步表明运用CRISPR/Cas 9基因编辑技术在SH-SY5Y细胞中敲除ADAM10基因成功。可进行后续实验研究。
Aβ和细胞内高磷酸化Tau蛋白的累积是如今诊断AD的主要神经病理学标准,同时在临床上AD患者血浆中Aβ42/Aβ40比率可作为AD的预筛查指标。
因此我们对Aβ;Tau;S-199Tau;S-214Tau进行验证,如图B-E所示,与空白组control KO相比,ADAM10 KO组Aβ蛋白的表达显著性升高,Tau蛋白,S-199Tau蛋白,S-214Tau蛋白的表达均显著性上调。如图F-I所示,取细胞上清对Aβ42,Aβ40,p-Tau和Tau进行检测。数据表明,与空白组control KO相比,ADAM10 KO组Aβ40蛋白水平无显著性变化,而Aβ42蛋白水平的表达显著性上调,同时Aβ42/Aβ40,pTau/Tau的比值显著性升高。
以上数据表明Aβ与高磷酸化Tau蛋白在ADAM10基因敲除组均显著性高表达。目前数据初步表明,敲除ADAM10基因的细胞可作为AD细胞模型。
图3敲除ADAM10基因后炎症炎症小体NLRP3的变化
NLRP3炎性小体的失调与多种神经退行性疾病的进展有关。有研究表明NLRP3蛋白抑制剂可能会作为未来治疗AD的靶标。与Control KO组比,ADAM10 KO组NLRP3显著性增加,激活了炎症小体的表达。
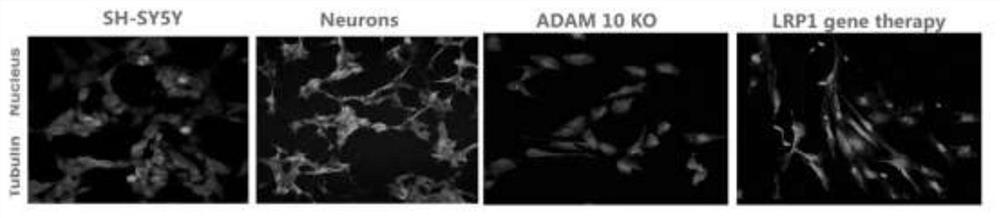
图4敲除ADAM10基因后神经元状态的变化
观察在SH-SY5Y细胞中敲除ADAM10基因后神经元细胞的生长速度,分化以及细胞密度的变化。与未进行分化处理的正常SH-SY5Y细胞相比,在经过神经细胞分化处理后,正常SH-SY5Y细胞神经元长度显著性变长,细胞间的连接更紧密。而敲除ADAM10基因后再经过相同的细胞分化处理,神经元长度显著性变短,细胞有皱缩趋势,细胞间的连接显著性变少,神经细胞间传递信息能力显著性减弱。
图5 LRP1的结构区域
其中CrRNA1、CrRNA2、CrRNA3分别为敲除LRP1基因的CrRNA序列。
图6 CrRNA1示意图
其中CrRNA1:GCUGCAAGUCUUAGGGGCUA(SEQ ID ON.4);Cutsite:57,138,459;Exon:Exon 2;On Target Score:0.536.
图7 crRNA2示意图
其中crRNA2:CUUGGGGCUGCAAGUCUUAG(SEQ ID ON.5);Cutsite:57,138,465;Exon:Exon 2;On Target Score:0.489.
图8 crRNA3示意图
其中crRNA3:CUCUGCAGGCAAACUGCUUG(SEQ ID ON.6)Cutsite:57,138,481;Exon:Exon 2;On Target Score:0.465.
图9敲低LRP1蛋白后靶标蛋白的变化
与Control KO组比,ADAM10 KO组NLRP3显著性增加,激活了炎症小体的表达。同时,Tau蛋白和LRP1蛋白显著增加。使用CRISPR/Cas 9基因编辑技术在敲除模型细胞中通过敲低LRP1蛋白来观察后续指标。与ADAM10 KO组比,敲低LRP1蛋白后,LRP1蛋白的表达显著降低。同时Tau蛋白的表达显著降低,NLRP3炎症小体蛋白显著降低。
图10敲低LRP1蛋白后Aβ
ELISA测定Aβ
图11敲低LRP1蛋白后神经元的变化
与未进行分化处理的正常SH-SY5Y细胞相比,在经过神经细胞分化处理后,正常SH-SY5Y细胞神经元长度显著性变长,细胞间的连接更紧密。而敲除ADAM10基因后再经过相同的细胞分化处理,神经元长度显著性变短,细胞有皱缩趋势,细胞间的连接显著性变少,神经细胞间传递信息能力显著性减弱。在模型AD细胞中敲低LRP1蛋白的表达后,神经元状态显著回调。
具体实施方式
本发明通过下面的实施例进行详细的解释,但并不意味着本发明仅限于此。
实施例1 AD细胞模型构建
(1)构建CRISPR/CAS9基因编辑方法
以下过程均在无菌操作台中全程无菌的条件下操作:
①构建针对ADAM10基因的RNP复合物:分别取0.33μl的crRNA
②将RNP复合物电转导入待敲除基因的SH-SY5Y细胞中:预热含15%FBS培养基,取密度为80%的细胞,弃去培养基。使用适量的PBS溶液清缓的清洗细胞,弃去溶液后,加入适量的胰蛋白酶于37℃下消化细胞,等数分钟80%细胞脱壁后,使用含15%FBS的培养基终止消化,1100rpm离心10min弃去上清。用10ml PBS溶液重悬细胞,充分混匀细胞后在取10μl细胞溶液计数。取250,000个细胞在1100rpm离心10min,弃去PBS上清,使用15.5μl的P3溶液重悬细胞。加入4.5μl的RNP复合物,得到20μl体系的细胞溶液。将20μl体系的细胞溶液加入16孔的电转孔板中,将孔板放入Lonza-4D电转仪中,选择SH-SY5Y程序进行电转。
(2)构建单细胞克隆技术方法
将电转好的细胞逐级稀释到1个细胞/10μl,加入96孔板中,补足200μl含15%FBS的培养基。稳定12小时等待细胞贴壁后,在显微镜下观察并筛选记录出96孔板中含一个细胞的孔。筛选后,继续等待这些单细胞生长扩增,待单细胞在96孔板中扩增到细胞密度为80%后将细胞转移至12孔板中,待单细胞在12孔板中扩增到细胞密度为80%后将细胞转移至6孔板中,待细胞密度为80%后,取一部分细胞进行Sanger测序,筛选敲除成功的单细胞系,剩下的细胞继续培养,每隔48小时换细胞培养液一次。
(3)Sanger测序
①细胞DNA样本的提取
取细胞弃去培养基,加入适量的PBS溶液润洗细胞后弃去溶液,加入适量的Trpsin消化细胞数分钟后加入含15%FBS培养基终止消化,1100rpm离心10minPBS溶液充分重悬后取少量细胞计数,取500,000个细胞严格按照。基因组DNA小量抽提试剂盒(离心柱式;D00D0063)的说明书操作提取细胞DNA。
②聚合酶链式反应(PCR)
如表1所示,目的基因ADAM10的引物设计如下:
表1目的基因ADAM10的引物
③反应体系如下
表2 PCR反应体系
④PCR反应条件
表3 PCR反应程序
⑤PCR产物纯化
根据PCR产物电泳结果切割所需的DNA目的条带,纯化步骤严格按照说明书SK8131胶回收操作。
⑥Sanger测序阶段
测序反应体系如下表所示:
表4 Sanger测序反应体系
反应条件:
表5 Sanger测序反应条件
将PCR反应板从PCR仪上取下后每孔加4μl EDTA Mix(0.5mol/L)EDTA,3mol/L醋酸钠,灭菌去离子水比例混合和60μl 95%乙醇,在冰水浴中反应30min。
将溶液于4000g离心20min,轻轻将板倒置,600rmp/min甩干后每孔加150μl 70%乙醇,4000g离心5min。将板倒置600rmp/min甩干,于超净台上晾5min,加10μl HidiFormamide,漩涡器上振荡1min。
将反应板放在PCR仪上96℃,3min迅速将反应板取下放入冰浴冷却,等待上样。
将96孔板置于ABI 3730XL测序仪电泳分析。测序电泳仪操作按照ABI Prism 3730使用手册严格执行操作。
与空白组Control KO相比,ADAM10敲除细胞株发生点突变。测序结果(图1)显示,已初步筛选到敲除ADAM10基因的细胞扩增株。对这些细胞株进行培养扩增,冻存做后续实验研究。
实施例2
2.1 AD细胞模型中ADAM10靶标蛋白和炎症炎症小体NLRP3的测定
通过Western对敲除组的ADAM10在蛋白水平进行验证。如图2A所示,与空白组control KO相比,ADAM10 KO组ADAM10蛋白的表达极显著性降低,与Sanger测序结果相吻合。数据进一步表明运用CRISPR/Cas 9基因编辑技术在SH-SY5Y细胞中敲除ADAM10基因成功。可进行后续实验研究。
通过Western对敲除组的炎症炎症小体NLRP3在蛋白水平进行验证。如图3所示,与Control KO组比,ADAM10 KO组NLRP3显著性增加,激活了炎症小体的表达。
2.2 AD细胞模型中Aβ与Tau蛋白的测定
Aβ和细胞内高磷酸化Tau蛋白的累积是如今诊断AD的主要神经病理学标准,同时在临床上AD患者血浆中Aβ42/Aβ40比率可作为AD的预筛查指标。因此我们对Aβ;Tau;S-199Tau;S-214Tau进行验证:
取细胞弃去培养基,加入适量的PBS溶液润洗细胞后弃去溶液,加入适量的Trpsin消化细胞数分钟后加入含15%FBS培养基终止消化,1100rpm离心10min。PBS溶液充分重悬后取少量细胞计数,取2,000,000个细胞严格按照ELISA试剂盒说明书操作步骤严格进行,每个样本的总蛋白由BCA试剂盒测定,矫正总蛋白后,测定Aβ40;Aβ42;p-Tau及Tau的含量后进行数据统计。如图2B-E所示,与空白组control KO相比,ADAM10 KO组Aβ蛋白的表达显著性升高,Tau蛋白,S-199Tau蛋白,S-214Tau蛋白的表达均显著性上调。如图2F-I所示,取细胞上清对Aβ42,Aβ40,p-Tau和Tau进行检测。数据表明,与空白组control KO相比,ADAM10KO组Aβ40蛋白水平无显著性变化,而Aβ42蛋白水平的表达显著性上调,同时Aβ42/Aβ40,pTau/Tau的比值显著性升高。
以上数据表明Aβ与高磷酸化Tau蛋白在ADAM10基因敲除组均显著性高表达。目前数据初步表明,敲除ADAM10基因的细胞可作为AD细胞模型。
2.3 AD细胞模型中神经元状态变化
(1)细胞神经元分化A培养
将SH-SY5Y细胞依次培养在如下表6所示的不同类型的培养基中,首先将细胞在Growth Media培养基中培养使其细胞密度达到70%。随后将培养基配方更换为Differentiation media 1培养7-10天,在此期间,每48小时更换培养液一次。将细胞按照1:1的比例传代于新鲜的培养基Differentiation media 2中培养4-7天,在此期间,每48小时更换培养液一次。将培养基换成Differentiation media 3培养7-10天,在此期间,每48小时更换培养液一次。此阶段细胞分化为神经元用于后续的检测与分析。
表6 SH-SY5Y细胞神经分化的培养基组成成分
(2)共聚焦显微镜观察神经元的生长
将分化的SH-SY5Y神经细胞使用4%PFA在室温下固定20min。弃去溶液后使用0.1%PBS-T溶液轻缓的润洗细胞3次,每次2min。固定后,将细胞放置在5%NGS-T封闭溶液中于室温孵育2小时。在4℃下加入一抗anti-tubulin III primary antibody(#ab179513,Abcam plc.),一抗稀释浓度为1:1000,孵育过夜。去除一抗后使用0.1%PBS-T溶液轻缓的润洗细胞3次。在室温下加入二抗Alexa Fluor 488(ab150113,Abcam),二抗稀释浓度为1:2000,于室温下孵育1h。去除二抗后,将细胞固定在载玻片中,在24h内用过共聚焦显微镜对样品进行成像检测。
使用Zeiss880共聚焦显微镜采集所有神经元分化图片。Laser power设置为4%;gain设置为650;Digital offset设置为350;Acquiring speed设置1.03s;Z-stack images设置为5μm;平均每张片子拍2次。
共聚焦显微镜采集的图像使用Image J插件Neuron J进行分析。选定每张图片中大于3个区域进行分析,随后测量轴突长度,并用Neuron J测量坐标之间的距离。
如图4所示,与未经过神经细胞分化的正常的SH-SY5Y细胞相比,在经过神经细胞分化处理后,正常的SH-SY5Y细胞神经元长度显著性变长,细胞间的连接更紧密。而敲除ADAM10基因后再经过相同的细胞分化处理,神经元长度显著性变短,细胞有皱缩趋势,细胞间的连接显著性变少,神经细胞间传递信息能力显著性减弱。
实施例3构建LRP1的基因敲除细胞系
以下过程均在无菌操作台中全程无菌的条件下操作:
①构建RNP复合物:分别取0.33μl的crRNA1(SEQ ID ON.4,100μM),0.33μl的crRNA2(100μM),0.33μl的crRNA3(SEQ ID ON.5,100μM)与1μl Alt-R.CRISPR-Cas9tracrRNA(SEQ ID ON.6,100μM)充分混合均匀后使用无核酸酶的缓冲液稀释到浓度为20-80μM,得到RNA混合物。将RNA混合物在95℃下加热5min,在室温中冷却10min,得到退火RNA混合物。取退火RNA混合物2.9μl与Cas 9蛋白1μl按照1.2:1-2:1的比例充分混合后放置室温10min。加入0.6μl电转加强液后在室温中反应5min,得到RNP复合物。
②将RNP复合物电转导入待敲除基因的SH-SY5Y细胞中:预热含15%FBS培养基,取密度为80%的细胞,弃去培养基。使用适量的PBS溶液清缓的清洗细胞,弃去溶液后,加入适量的胰蛋白酶于37℃下消化细胞,等数分钟80%细胞脱壁后,使用含15%FBS的培养基终止消化,1100rpm离心10min弃去上清。用10ml PBS溶液重悬细胞,充分混匀细胞后在取10μl细胞溶液计数。取250,000个细胞在1100rpm离心10min,弃去PBS上清,使用15.5μl的P3溶液重悬细胞。加入4.5μl的RNP复合物,得到20μl体系的细胞溶液。将20μl体系的细胞溶液加入16孔的电转孔板中,将孔板放入Lonza-4D电转仪中,选择SH-SY5Y程序进行电转。
此部分实验研究内容在实施例4得到的AD模型细胞中运用基因编辑技术敲低LRP1蛋白构建针对LRP1蛋白的基因治疗体系。
实施例6
①细胞总蛋白的提取:
弃去培养液后加入2ml预冷PBS进行润洗细胞,弃去PBS洗液。此操作重复2次。将培养瓶置于冰上,细胞计数后取1000,000个细胞,加入100μl含PMSF的裂解液,充分混合后于冰上裂解30min,隔段时间要摇动细胞瓶使细胞充分裂解。裂解后的细胞快速使用刮棒将细胞刮下,使用移液枪吸取细胞碎片和裂解液转移至1.5ml离心管中。于4℃下13000g离心20min。取上清,留取5μl对蛋白含量进行测定。取适量的上清以4:1体积加入上样缓冲液,于沸水煮沸10min。样品放于-20℃保存。
②制备分离胶与浓缩胶
分离胶(15%):超纯水2.3mL;30%丙烯酰胺5.0mL;1.5M Tris-HCl(pH 8.8)2.5mL;10%SDS 0.1mL;10%APS(过硫酸铵)0.1mL;TEMED 4μl。
分离胶(8%):超纯水4.6mL;30%丙烯酰胺2.7mL;1.5M Tris-HCl(pH 8.8)2.5mL;10%SDS 0.1mL;10%APS(过硫酸铵)0.1mL;TEMED 8μl。
分离胶(6%):超纯水5.3mL;30%丙烯酰胺2.0mL;1.5M Tris-HCl(pH 8.8)2.5mL;10%SDS 0.1mL;10%APS(过硫酸铵)0.1mL;TEMED 8μl。
浓缩胶(5%):超纯水2.7mL;30%丙烯酰胺0.4mL;1M Tris-HCl溶液(pH 6.8)0.5mL;10%SDS 40μl;10%APS 40μl;TEMED 4μl。
过硫酸铵与TEMED为促凝剂,加入溶液后要立即混匀后灌胶。灌好分离胶后,轻缓的加入超纯水,加超纯水时移动移液枪,保证水在同一水平线上。静置直到看到分离胶与水之间出现较明显的界限。等待分离胶凝固后,倾斜倒掉水,沥干水后,再灌入浓缩胶,插上梳齿,等待浓缩胶凝固后上样。
③蛋白样品上样
沿着垂直方向缓慢的拔出梳齿。使用50μl微量上样器取适量的样本,上样总蛋白一般为20μg,将上样器缓慢插入梳齿最底部,以适当的速度打入蛋白样品。
④电泳
第一阶段:75V,40~60min直至看到目的蛋白在分离胶与浓缩胶分界处出现一条线。
第二阶段:115V,60~90min直至目的蛋白迁移于距分离胶底部约1/3处。
⑤转膜
提前剪裁好与胶条大小匹配的PVDF膜,放入甲醇中浸泡活化15-20s后转移到超纯水中放置2min,将PVDF膜放入转膜缓冲液中浸泡10min。取出转膜滤纸放入转膜缓冲液中浸泡10min。等待电泳结束后切割所需分子量范围内的胶条。转膜装置从上至下的顺序依次为阴极碳板,转膜滤纸,胶,PVDF膜,滤纸。按照顺利放好,精确对齐,每一步均要保证无气泡存在。接通电源,1.5mm的胶恒流0.3A转膜时间2h,1mm的胶恒流0.18A转膜时间1.5h。将转膜槽放置在冰浴中防止转膜过热。
⑥封闭
转膜阶段结束后,断开电源取出PVDF膜。于5%的脱脂奶粉中4℃封闭过夜,或37℃封闭1h。
⑦孵育抗体
a.使用5%脱脂牛奶或者5%BSA溶液按照抗体说明书以适当的比例稀释一抗,孵育4℃过夜后于37℃条件下孵育1h。
b.弃去一抗,使用TBST溶液洗膜30min,每5min换一次液。
c.使用5%脱脂牛奶或者5%BSA溶液按照抗体说明书以适当的比例稀释二抗,于37℃条件下孵育1h。
d.弃去二抗,使用TBST溶液洗膜30min,每5min换一次液。
⑧凝胶图像分析
依据膜的面积将每条膜均匀的加入100~200ul的显影液,将条带放入凝胶成像仪中避光显色。对条带进行扫描,拍照。凝胶图象处理系统处理并分析条带的分子量与净光密度值。曝光时间依据条带的曝光难易程度做适当的调整,最起始的曝光时间为5~20s,总曝光时间根据每个条带做适当调整。
如图9所示,与空白组Control KO相比,ADAM10 KO组LRP1蛋白显著升高,LRP1基因治疗组LRP1蛋白的表达显著回调。运用CRISPR/Cas9基因编辑技术敲低LRP1蛋白成功,可进行后续实验测定。如图9所示,与Control KO组比,ADAM10 KO组Tau蛋白,NLRP3炎症小体蛋白显著上调,敲低LRP1蛋白后,Tau蛋白与NLRP3炎症小体蛋白显著回调,结果表明,LRP1蛋白的下调能回调炎症因子的水平,并降低Tau蛋白的表达。
实施例7敲低LRP1蛋白后Aβ
取细胞弃去培养基,加入适量的PBS溶液润洗细胞后弃去溶液,加入适量的Trpsin消化细胞数分钟后加入含15%FBS培养基终止消化,1100rpm离心10min。PBS溶液充分重悬后取少量细胞计数,取2,000,000个细胞严格按照ELISA试剂盒说明书操作步骤严格进行,每个样本的总蛋白由BCA试剂盒测定,矫正总蛋白后,测定Aβ
如图10所示,与Control KO组比,模型组Aβ
实施例8
(1)细胞神经元分化培养
将SH-SY5Y细胞依次培养在如表6所示的不同类型的培养基中,首先将细胞在Growth Media培养基中培养使其细胞密度达到70%。随后将培养基配方更换为Differentiation media 1培养7-10天,在此期间,每48小时更换培养液一次。将细胞按照1:1的比例传代于新鲜的培养基Differentiation media 2中培养4-7天,在此期间,每48小时更换培养液一次。将培养基换成Differentiation media 3培养7-10天,在此期间,每48小时更换培养液一次。此阶段细胞分化为神经元用于后续的检测与分析。
(2)共聚焦显微镜观察神经元的生长
将分化的SH-SY5Y神经细胞使用4%PFA在室温下固定20min。弃去溶液后使用0.1%PBS-T溶液轻缓的润洗细胞3次,每次2min。固定后,将细胞放置在5%NGS-T封闭溶液中于室温孵育2小时。在4℃下加入一抗anti-tubulin III primary antibody(#ab179513,Abcam plc.),一抗稀释浓度为1:1000,孵育过夜。去除一抗后使用0.1%PBS-T溶液轻缓的润洗细胞3次。在室温下加入二抗Alexa Fluor 488(ab150113,Abcam),二抗稀释浓度为1:2000,于室温下孵育1h。去除二抗后,将细胞固定在载玻片中,在24h内用过共聚焦显微镜对样品进行成像检测。
使用Zeiss880共聚焦显微镜采集所有神经元分化图片。Laser power设置为4%;gain设置为650;Digital offset设置为350;Acquiring speed设置1.03s;Z-stack images设置为5μm;平均每张片子拍2次。
共聚焦显微镜采集的图像使用Image J插件Neuron J进行分析。选定每张图片中大于3个区域进行分析,随后测量轴突长度,并用Neuron J测量坐标之间的距离。
如图11所示,与模型组相比,LRP1蛋白敲低后神经元长度显著变长,神经元状态显著回调。
序列表
<110> 中国药科大学
<120> 一种促进神经元再生的靶标蛋白及其应用
<160> 6
<170> SIPOSequenceListing 1.0
<210> 1
<211> 20
<212> RNA
<213> 人工序列(Artificial Sequence)
<400> 1
uuuuuuuuua uaggucagua 20
<210> 2
<211> 20
<212> RNA
<213> 人工序列(Artificial Sequence)
<400> 2
uuuuuuuuau aggucaguau 20
<210> 3
<211> 20
<212> RNA
<213> 人工序列(Artificial Sequence)
<400> 3
aaauauauca gacauuauga 20
<210> 4
<211> 20
<212> RNA
<213> 人工序列(Artificial Sequence)
<400> 4
gcugcaaguc uuaggggcua 20
<210> 5
<211> 20
<212> RNA
<213> 人工序列(Artificial Sequence)
<400> 5
cuuggggcug caagucuuag 20
<210> 6
<211> 20
<212> RNA
<213> 人工序列(Artificial Sequence)
<400> 6
cucugcaggc aaacugcuug 20
- 一种促进神经元再生的靶标蛋白及其应用
- 一种MNK2蛋白激酶与穿膜肽的融合蛋白及其水凝胶与促进心肌再生的应用