近红外发光碳点的规模化合成方法及其应用
文献发布时间:2023-06-19 13:27:45
技术领域
本发明涉及近红外发光碳点的制备方法,尤其是一种规模化高产率的近红外发光碳点的制备方法,以及将碳点应用于碱性含氮有机物的检测。
背景技术
碳点作为一种零维碳基纳米材料近年来受到了人们的广泛关注,由于其原材料来源广泛,成本低、合成条件简单,以及优异的光学性质和良好的生物相容性被广泛应用于光催化、成像、载药、传感和光电器件等领域。然而现阶段有三个主要问题阻碍了碳点的进一步发展。1)碳点的大规模制备困难。碳点的规模化制备是其广泛应用的基础,因此开发碳点大规模制备必不可少。2)碳点分离纯化手段复杂。分离纯化是影响碳点性能的直接因素,不纯净的碳点无法让人放心使用,因此分离纯化对于制备碳点必不可少。现阶段碳点的分离纯化主要用透析和色谱柱,这两种方法繁琐耗时,无法扩大规模。3)荧光调节困难。碳点的大部分应用对其荧光有特定的要求,因此方便的荧光调节方法对于拓展碳点的应用至关重要。碱性含氮有机物作为主要水体污染物之一以及多种药物和化学物质的主要成分,对其灵敏检测异常重要。
发明内容
本发明提出一种高产率合成并简单快速的分离、提纯近红外发光碳点的方法以及方便的调节其荧光的手段。
近红外发光碳点的规模化合成方法,具体步骤如下:
步骤1,配制酸溶液,酸的体积浓度≥1%;
步骤2,将邻苯二胺加入酸溶液中,待充分溶解后加入过氧化物;均按质量比,酸:邻苯二胺=1:1-50:1,邻苯二胺:过氧化物=5:1-50:1;
步骤3,将步骤2的混合溶液进行微波加热;加热功率为150~800W,加热时间为5-60分钟;
步骤4,将步骤3得到的混合反应液置于密封的不锈钢水热反应釜中加热反应;反应的温度为100~250℃,反应时间为1~36小时;
步骤5,将步骤4中反应后的溶液调节pH为7~12使其去质子化沉淀;过滤后用水洗涤,得到黑色固体为近红外发光碳点固体。
所述步骤1中的酸为硫酸、盐酸、磷酸、硝酸、高锰酸钾、高氯酸、硒酸、氢溴酸、氢碘酸或氯酸中一种或任意几种的组合。
所述步骤2中的过氧化物为叔丁基过氧化氢、过氧苯甲酸、过氧化苯甲酰、甲乙酮过氧化物、过氧化新癸酸、过辛酸叔丁酯、过苯甲酸叔戊酯、过苯甲酸叔丁酯、异丙苯基过氧酯、过氧二碳酸酯、过氧缩酮、间氯过氧苯甲酸或叔丁基过氧缩酮中一种或任意几种的组合。
所述的黑色近红外发光碳点置于烘箱进行干燥烘干,干燥的温度为50~90℃。
制备过程中,使用酸能够活化邻苯二胺中的氨基,使用氧化物能增加聚合度,微波处理提供反应所需大量能量使邻苯二胺完成初步聚合。
将上述制备方法合成的碳点用于检测碱性含氮有机物,检测方法有四种,分别是溶液传感法、试纸法、吸收光谱法和荧光光谱法。
所述的碱性含氮有机物包括丁胺、麻黄碱、精胺、苯胺、秋水仙碱、槟榔碱、吡咯、吗啡、咪唑、吡啶、嘌呤、喹啉、吲哚、赖氨酸、四环素。
本发明的方法,具有以下效果:1)通过微波-水热法实现近红外发光碳点的千克水平制备。通过该方法合成的碳点的产量高于90%,成本低至0.4元/克。制备的碳点在540nm的最佳激发光下,在602nm处具有最佳的发射峰值,并且在 653nm处具有一个肩峰。 2)通过使用去质子化实现了碳点的纯化,得到的碳点的纯度高于95%。这种纯度可以完全满足碳点的研究和应用。 3)通过质子化和去质子化的过程,可以控制碳点的荧光。质子化可将碳点的最佳发射波长从602nm红移到653nm,导致51nm的红色偏移。去质子化则将碳点的荧光强度增加了11倍,荧光量子产率增加了3倍。4)该碳点可用于碱性含氮有机物的检测。
本发明的方法中,步骤1的酸性环境有利于提高产品的产率,步骤2中加入过氧化物,氧化环境有利于提高产品产率,步骤3中微波前处理有利于最终产品产率。步骤5中,去质子化和大量水洗涤,有利于提高最终产品的纯度。
附图说明
图1千克级碳点合成图片。
图2为碳点溶液荧光光谱图;
图中碳点在400-600 nm的光激发下,在550-850nm波长范围内发射荧光。
图3为碳点溶液的紫外-可见光吸收光谱;
图中碳点对200-700 nm波长范围内的光有吸收。
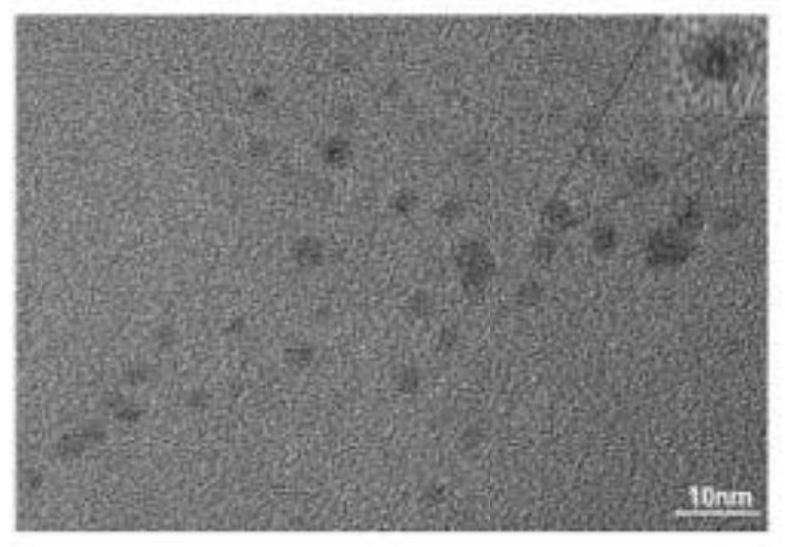
图4为碳点TEM图谱;
通过透射电子显微镜可以直接观察到碳点的形貌与形状。
图5为碳点粒径分布图。
图6为碳点红外图谱;
图中,500-3500 cm
图7为碳点XPS图谱;
图a显示碳点含有的元素;
图b显示碳原子种类;
图c显示了氧原子种类;
图d显示氮原子种类。
图8为实施例1溶液法检测吡啶图;
图显示当吡啶浓度高于10
图9为实施例1试纸法检测吡啶图;
将吡啶滴到试纸上,浓度高于10
图10实施例1紫外-可见光吸收光谱法检测吡啶图;
图a为随吡啶浓度增加碳点吸收光谱变化图;
图b为波长626nm处吸收强度与吡啶浓度的关系曲线图。
图11为实施例1荧光光谱法检测吡啶图;
图a为随吡啶浓度增加碳点荧光光谱变化;
图b为波长606nm处荧光强度与吡啶浓度关系曲线图。
图12为实施例2溶液法检测精胺图;
图显示当精胺浓度高于10
图13为实施例2试纸法检测精胺图;
将精胺滴到试纸上,浓度高于10
图14为实施例2紫外-可见光吸收光谱法检测精胺图;
图a为随精胺浓度增加碳点吸收光谱变化图;
图b为波长626nm处吸收强度与精胺浓度的关系曲线图。
图15为实施例2荧光光谱法检测精胺图;
图a为随精胺浓度增加碳点荧光光谱变化;
图b为波长606nm处荧光强度与精胺浓度关系曲线图。
图16为实施例3溶液法检测丁胺图;
图显示当丁胺浓度高于10
图17为实施例3试纸法检测丁胺图;
将丁胺滴到试纸上,浓度高于10
图18为实施例3紫外-可见光吸收光谱法检测丁胺图;
图a为随丁胺浓度增加碳点吸收光谱变化图;
图b为波长626nm处吸收强度与丁胺浓度的关系曲线图。
图19为实施例3荧光光谱法检测丁胺图;
图a为随丁胺浓度增加碳点荧光光谱变化;
图b为波长606nm处荧光强度与丁胺浓度关系曲线图。
具体实施方式
实施例1:近红外发光碳点的规模化合成方法,具体步骤如下:
(1)将98%浓硫酸250 mL放入烧杯中备用;
(2)取步骤(1)中备用的硫酸加入到4.65L水中,配制体积浓度为5%的硫酸溶液;
(3)将1.15kg邻苯二胺加入到步骤(2)的溶剂中,待邻苯二胺充分溶解后加入50g间氯过氧苯甲酸;
(4)将步骤(3)的混合溶液放入微波。炉中加热,微波炉功率设定为300W,加热时间为15分钟;
(5)将步骤(4)得到的混合反应液置于不锈钢水热反应釜中,在200℃下加热12小时;
(6)步骤(5)反应后的溶液调节pH=8,使其去质子化沉淀。过滤后用5L水洗涤。将得到的黑色固体置于烘箱干燥得到近红外发光碳点固体1.104kg,如图1。
使用本实施例制备的碳点,分别采用溶液传感法、试纸法、吸收光谱法和荧光光谱法对碱性含氮有机物吡啶进行检测。
溶液传感法检测步骤如下:
(a)将碳点合成实施例1中获得的碳点溶于甲醇中,配制1mg/mL的碳点溶液;
(b)将吡啶溶于甲醇中,分别配制浓度为10
(c)将5 uL步骤(a)中配制的碳点溶液分别加入到3 mL步骤(b)中配制的吡啶溶液中,观察颜色变化;当吡啶浓度高于10
试纸法检测步骤如下:
(1)将滤纸剪裁为1×5 cm之后在步骤(a)中获得的碳点溶液中浸泡5分钟,制备检测试纸;
(2)将10 uL步骤(b)中的吡啶溶液依次滴加到试纸上观察颜色变化;当吡啶浓度高于10
吸收光谱法检测步骤如下:
(1)将步骤(a)中配制的1 mg/mL的碳点溶液15 uL加入到3 mL不同浓度的吡啶中测试其紫外吸收光谱;不同浓度吡啶分别是0,2×10
(2)以光谱626 nm处吸收强度做标准曲线,用于吡啶浓度测定,如图10b;
(3)将1 mg/mL的碳点溶液15uL滴入到3mL未知浓度的吡啶中,根据步骤(2)得到的标准曲线,以检测出的吸收强度点对应被测吡啶溶液的浓度。
荧光光谱法检测步骤如下:
(1)将配制的1 mg/mL的碳点溶液15 uL加入3 mL不同浓度的吡啶中测试其荧光光谱;不同浓度吡啶分别是10
(2)以荧光光谱606 nm处荧光强度做标准曲线,用于吡啶浓度测定;如图11b;
(3)将1 mg/mL的碳点溶液15uL滴入到3mL未知浓度的吡啶中,根据步骤(2)得到的标准曲线,以检测出的荧光强度点对应被测吡啶溶液的浓度。
实施例2:近红外发光碳点的规模化合成方法,具体步骤如下:
(1)将35%盐酸500 mL放入烧杯中备用;
(2)取步骤(1)中备用的盐酸加入到2L水中,配制成体积浓度为7%的盐酸溶液;
(3)将1.15kg邻苯二胺加入到步骤(2)的溶液中,待邻苯二胺充分溶解后加入20g叔丁基过氧化氢;
(4)将步骤(3)的混合溶液放入微波炉中加热,微波炉功率设定为250W,加热时间为20分钟;
(5)将步骤(4)得到的混合反应液置于不锈钢反应釜中,在密封的反应器中230℃加热10小时反应;
(6)将步骤(5)反应后的溶液调节pH=7,使其去质子化沉淀。过滤后用5L水洗涤。将得到的黑色固体置于烘箱干燥得到近红外发光碳点固体1.04kg。
使用本实施例制备的碳点,分别采用溶液传感法、试纸法、吸收光谱法和荧光光谱法对碱性含氮有机物精胺进行检测。
溶液传感法检测步骤如下:
(a)将碳点合成实施例2中获得的碳点溶于甲醇中,配制1mg/mL的碳点溶液;
(b)将精胺溶于甲醇中,分别配制浓度为10
(c)将5 uL步骤(a)中配制的碳点溶液分别加入到3 mL步骤(b)中配制的精胺溶液中,观察颜色变化;当精胺浓度高于10
试纸法检测步骤如下:
(1)将滤纸剪裁为1×5 cm之后在步骤(a)获得的碳点溶液中浸泡5 min,制备检测试纸;
(2)将10 uL步骤(b)中的精胺溶液分别滴加到试纸上观察颜色变化;当精胺浓度高于10
吸收光谱法检测步骤如下:
(1)将步骤(a)中配制的1 mg/mL的碳点溶液15 uL加入到3 mL不同浓度的精胺溶液中测试其紫外吸收光谱;不同浓度精胺分别是0,2×10
(2)以光谱626 nm处吸收强度做标准曲线,用于精胺浓度测定,如图14b;
(3)将15uL的碳点溶液(1 mg/mL)滴入到3mL未知浓度的精胺中,根据步骤(2)得到的标准曲线,以检测出的吸收强度点对应被测精胺溶液的浓度。
荧光光谱法检测步骤如下:
(1)将配制的1 mg/mL的碳点溶液15 uL加入到3 mL不同浓度的精胺中测试其荧光光谱;不同浓度精胺分别是6×10
(2)以荧光光谱606 nm处荧光强度做标准曲线,用于精胺浓度测定;如图15b;
(3)将1 mg/mL的碳点溶液15uL滴入到3mL未知浓度的精胺中,根据步骤(2)得到的标准曲线,以检测出的荧光强度点对应被测精胺溶液的浓度。
实施例3:近红外发光碳点的规模化合成方法,具体步骤如下:
(1)将85%浓磷酸250 mL放入烧杯中备用;
(2)取步骤(1)中备用的磷酸加入到3.25L水中,配制成体积浓度为6%的磷酸溶液;
(3)将1.15kg邻苯二胺加入到步骤(2)的溶液中,待邻苯二胺充分溶解后加入20g间氯过氧苯甲酸;
(4)将步骤(3)的混合溶液放入微波炉中加热,微波炉功率设定为200W,加热时间为25分钟;
(5)将步骤(4)得到的混合反应液置于不锈钢反应釜中,在密封的反应器中180℃加热14小时反应;
(6)将步骤(5)反应后的溶液调节pH=9,使其去质子化沉淀。过滤后用5L水洗涤。将得到的黑色固体置于烘箱干燥得到近红外发光碳点固体1.112kg。
使用本实施例制备的碳点,分别采用溶液传感法、试纸法、吸收光谱法和荧光光谱法对碱性含氮有机物丁胺进行检测。
溶液传感法检测步骤如下:
(a)将碳点合成实施例3中获得的碳点溶于甲醇中,配制1mg/mL的碳点溶液;
(b)将丁胺溶于甲醇中,分别配制浓度为10
(c)将5 uL步骤(a)中配制的碳点溶液分别加入到3 mL步骤(b)中配制的丁胺中,观察颜色变化;当丁胺浓度高于10
试纸法检测步骤如下:
(1)将滤纸剪裁为1×5 cm之后在步骤(a)中获得的碳点溶液中浸泡5 min,制备检测试纸;
(2)将10 uL步骤(b)中的丁胺溶液分别滴加到试纸上观察颜色变化;当丁胺浓度高于10
吸收光谱法检测步骤如下:
(1)将配制的1 mg/mL的碳点溶液15 uL加入到3 mL不同浓度的丁胺中测试其紫外吸收光谱;不同浓度丁胺分别是0,2×10
(2)以光谱626 nm处吸收强度做标准曲线,用于丁胺浓度测定,如图18b;
(3) 将1 mg/mL的碳点溶液15 uL滴入到3mL未知浓度的丁胺中,根据步骤(2)得到的标准曲线,以检测出的吸收强度点对应被测丁胺溶液的浓度。
荧光光谱法检测步骤如下:
(1) 将配制的1 mg/mL的碳点溶液15 uL加入到3 mL不同浓度的丁胺中测试其荧光光谱;不同浓度丁胺分别是10
(2)以荧光光谱606 nm处荧光强度做标准曲线,用于丁胺浓度测定;如图19b;
(3)将1 mg/mL的碳点溶液15 uL加入到3mL未知浓度的丁胺中,根据步骤(2)得到的标准曲线,以检测出的荧光强度点对应被测丁胺溶液的浓度。
实施例4:近红外发光碳点的规模化合成方法,具体步骤如下:
(1)将98%浓硫酸100 mL放入烧杯中备用;
(2)取步骤(1)备用的硫酸中加入到4.80L水中,配制为体积浓度为2%的硫酸溶液;
(3)将1.15kg邻苯二胺加入到步骤(2)的溶液中,待邻苯二胺充分溶解后加入40g叔丁基过氧化氢;
(4)将步骤(3)的混合溶液放入微波炉中加热,微波炉功率设定为200W,加热时间为25分钟;
(5)将步骤(4)得到的混合反应液置于不锈钢反应釜中,在密封的反应器中180℃加热12小时反应;
(6)将步骤(5)反应后的溶液调节pH=7,使其去质子化沉淀。过滤后用5L水洗涤。将得到的黑色固体置于烘箱干燥得到近红外发光碳点固体1.022kg。