HLA基因扩增引物、试剂盒、测序文库构建方法及测序方法
文献发布时间:2023-06-19 13:48:08
技术领域
本发明涉及核酸测序技术领域,具体涉及HLA基因扩增引物、试剂盒、测序文库构建方法及测序方法。
背景技术
人类白细胞抗原HLA(Human Leukocyte Antigen)分子提呈抗原给T淋巴细胞识别,在适应性免疫应答中起着极其重要的作用。HLA是一个由一系列紧密连锁的基因座位组成的具有高度多态性的复合体,是编码人类主要组织相容性复合体(MHC)的基因簇,HLA定位于第6染色体短臂上。该系统是目前所知人体最复杂的多态系统。HLA是具有高度多态性的同种异体抗原,其化学本质为一类糖蛋白,由一条α重链(被糖基化的)和一条β轻链非共价结合而成。其肽链的氨基端向外(约占整个分子的3/4),羧基端穿入细胞质,中间疏水部分在胞膜中。HLA按其分布和功能分为Ⅰ类抗原和Ⅱ类抗原。HLA-I类分子为内源性抗原的递呈分子;HLA-Ⅱ类分子为外源性抗原的递呈分子。HLAⅠ类抗原的特异性取决于α重链,由HLA-A、B、C位点编码;其β轻链是β2-微球蛋白,编码基因在第15染色体。HLAⅡ类抗原受控于HLA-D区(包含5个亚区),由其中的A基因和B基因分别为α重链和β轻链编码,抗原多态性取决于β轻链。以上各基因均系多态性位点(复等位),且共显性。如果把MHC作为一个整体来看待,其多态性则更为突出。保守地估计,至少存在1300个不同的单体型,相应地约有17×10的七次方个基因型。HLA基因同时又是人类重要的遗传结构,在法医鉴定、器官移植配型等领域应用广泛。众所周知,高度的多态性是HLA基因一个非常重要的特点。以往对人类白抗原基因多态性的研究主要是针对编码抗原结合肤的外显子(HLA-A,-B基因的第2-4外显子,HLA-DQB1、-DRB1基因的第2外显子)。众多研究者对这些外显子的分型技术、多态位点的探测、疾病相关性以及进化分析研究做了大量工作。然而,越来越多的研究表明,HLA基因非编码区的多态位点及其功能也不容忽视。
早些年人类白细胞抗原(Human leukocyte antigen,HLA)分型鉴定是以血清学的方式进行,其缺点包含:会出现假阴性或假阳性结果,降低准确率;高特异性HLA抗血清来源有限,单克隆抗血清的交叉反应难易克服;血清学分型需要7-10ml全血制备有活力的淋巴细胞甚至有活力的B细胞,细胞采集与保存困难。随着科技的进步,开始产生了一些HLA的DNA分型方式,以下几种为目前市面上常见的HLA基因分型技术:序列特异性寡核酸法(Sequence-Specific Oligonucleotides Probes;SSO);序列特异性聚合酶连锁反应(Sequence-Specific Primers;SSP);核酸测序技术(Sequencing-Based Typing;SBT);次世代测序技术(Next Generation Sequencing;NGS)。SSO虽然具备实验简单与实验周期短的优势,但此技术并无法检测新的等位基因,只能针对已知的分型序列进行区分,而为了能有效分型,需要使用大量的探针也是其缺点之一。SSP也具备了实验简单与实验周期短的优势,但也同样具备无法检测新的等位基因的问题,并且无法分辨伪基因。SBT具备检测新的等位基因优势,但无法区分两条等位基因,并且产生模棱两可的结果。NGS的技术也有检测新的等位基因优势,虽然分型结果能达到4分位,但也因读长短的问题,组装非常依赖数据计算,造成会引入模棱两可的结果。虽然已经有很多HLA分型技术,但由于其极高的多态性,以现有的技术还是有问题无法解决,例如大量引入模棱两可的结果。
发明内容
基于此,有必要针对传统HLA基因分型方法引入模棱两可的结果问题,提供一种能够提高结果准确性且工艺简化的HLA基因扩增引物、试剂盒、测序文库构建方法及测序方法。
本发明的第一目的在于提供一种HLA基因扩增引物,包括以下9组组引物组中的任意一组种或任意多组种组成的混合物:
第一引物组:SEQ ID NO:1和SEQ ID NO:2;
第二引物组:SEQ ID NO:3和SEQ ID NO:4;
第三引物组:SEQ ID NO:5和SEQ ID NO:6;
第四引物组:SEQ ID NO:7和SEQ ID NO:8;
第五引物组:SEQ ID NO:9和SEQ ID NO:10;
第六引物组:SEQ ID NO:11和SEQ ID NO:12;
第七引物组:SEQ ID NO:12和SEQ ID NO:13;
第八引物组:SEQ ID NO:14和SEQ ID NO:15;
第九引物组:SEQ ID NO:16、SEQ ID NO:17和SEQ ID NO:18。
本发明的第二目的在于提供一种用于HLA基因测序的试剂盒,其包括所述的HLA基因扩增引物。
本发明的第三目的在于提供一种HLA基因测序文库的构建方法,包括以下步骤:
采用所述的HLA基因扩增引物对受试样品进行扩增得到扩增产物;
使所述扩增产物与接头发生连接反应构建得到文库;
对所述文库进行纯化。
本发明的第四目的在于提供一种HLA基因测序方法,其包括:采用所述的HLA基因测序文库的构建方法构建HLA基因测序文库,然后对所述文库进行测序。
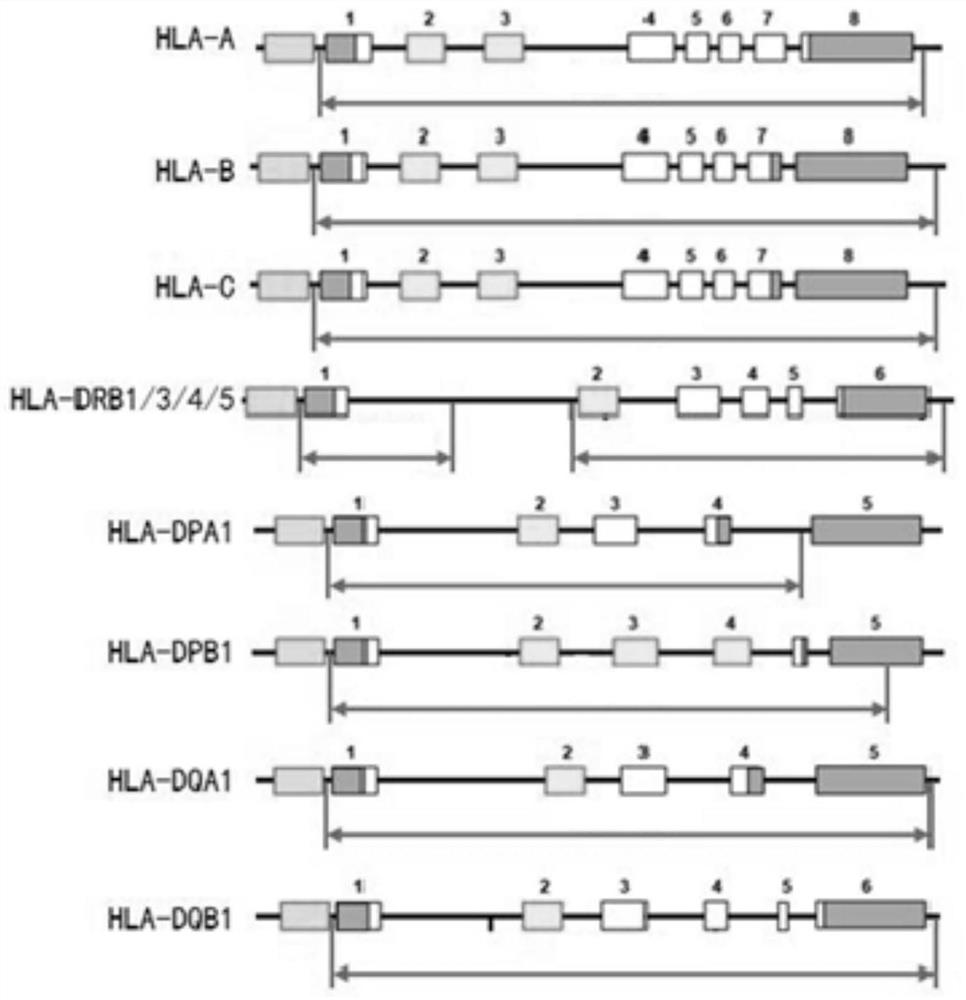
本发明提出9组用于HLA基因的扩增引物组,9组引物组分别针对HLA基因不同的靶序列。此9组用于HLA基因的扩增引物组既可以单独进行PCR扩增得到扩增基因。并且,此9组引物组中的任意组可以组合进行单管多重PCR扩增,9组扩增引物组可单管多重PCR扩增得到HLA 11个基因完整区域(包含:DPB1、DQA1、DQB1、DPA1、DRB1/3/4/5、HLA-A、HLA-B、HLA-C),使实验过程更简便(目前二代测序实现11个基因的扩增最少需要3管分别扩增,实验繁琐),简化工艺流程。
利用本发明的引物组扩增产物制备文库,可应用于基于Pacific Biosciences(PacBio)的三代单分子测序技术检测新的等位基因,并且具备长读长与高准确度的优势,解决了SBT与NGS无法做phasing的问题,在分辨率上可以达到普遍6分位,并且无模棱两可的结果。本发明优于其他技术除了三代单分子测序技术本身的优势之外,利用三代单分子测序技术长读长的优势,本发明可实现在引物的设计上增加扩增区域,提高扩增区域的覆盖度。传统的引物设计仅针对外显子区域,本发明的引物组不仅可扩增得到外显子区域信息,还可提供更为全面的内含子信息。PacBio测序技术和本发明引物组的设计相结合,能有效的提供完整的HLA信息,为未来进一步研究HLA基因的功能提供了一套完整的解决方案,得到全面的HLA基因信息,提高分型的灵敏度和准确度。
附图说明
图1为本发明一实施例的引物扩增部分样本的电泳结果图;
图2为本发明另一实施例的引物扩增部分样本的电泳结果图;
图3为本发明另一实施例的引物扩增部分样本的电泳结果图;
图4为本发明另一实施例的引物扩增部分样本的电泳结果图;
图5为本发明另一实施例的引物扩增部分样本的电泳结果图;
图6为本发明另一实施例的引物扩增部分样本的电泳结果图;
图7为本发明另一实施例的引物扩增部分样本的电泳结果图;
图8为本发明另一实施例的引物扩增部分样本的电泳结果图;
图9为本发明另一实施例的引物扩增部分样本的电泳结果图;
图10为本发明一实施例的引物扩增区域图;
图11为本发明一实施例的文库片段大小检测结果图。
具体实施方式
为了便于理解本发明,下面将参照相关附图对本发明进行更全面的描述。附图中给出了本发明的较佳实施例。但是,本发明可以以许多不同的形式来实现,并不限于本文所描述的实施例。相反地,提供这些实施例的目的是使对本发明的公开内容的理解更加透彻全面。
除非另有定义,本文所使用的所有的技术和科学术语与属于本发明的技术领域的技术人员通常理解的含义相同。本文中在本发明的说明书中所使用的术语只是为了描述具体的实施例的目的,不是旨在于限制本发明。本文所使用的术语“和/或”包括一个或多个相关的所列项目的任意的和所有的组合。
gDNA:基因组DNA,是指有机体在单倍体状态下的全部DNA。
如本文中所使用的,术语“靶核酸序列”、“靶核酸”和“靶序列”是指待检测的目标核酸序列。在本申请中,术语“靶核酸序列”、“靶核酸”和“靶序列”具有相同的含义,并且可互换使用。如本文中所使用的,术语“靶向序列”和“靶特异性序列”是指,在允许核酸杂交、退火或扩增的条件下,能够与靶核酸序列选择性/特异性杂交或退火的序列,其包含与靶核酸序列互补的序列。在本申请中,术语“靶向序列”和“靶特异性序列”具有相同的含义,并且可互换使用。易于理解的是,靶向序列或靶特异性序列对于靶核酸序列是特异性的。换言之,在允许核酸杂交、退火或扩增的条件下,靶向序列或靶特异性序列仅与特定的靶核酸序列杂交或退火,而不与其他的核酸序列杂交或退火。
“F”和“R”分别表示上游和下游,引申为上游引物和下游引物。
如本文中所使用的,术语“上游”用于描述两条核酸序列(或两个核酸分子)的相对位置关系,并且具有本领域技术人员通常理解的含义。例如,表述“一条核酸序列位于另一条核酸序列的上游”意指,当以5'至3'方向排列时,与后者相比,前者位于更靠前的位置(即,更接近5'端的位置)。如本文中所使用的,术语“下游”具有与“上游”相反的含义。
如本文中所使用的,术语“PCR反应”具有本领域技术人员通常理解的含义,其是指使用核酸聚合酶和引物来扩增靶核酸的反应(聚合酶链式反应)。
如本文中所使用的,术语“杂交”和“退火”意指,互补的单链核酸分子形成双链核酸的过程。在本申请中,“杂交”和“退火”具有相同的含义,并且可互换使用。通常,完全互补或实质上互补的两条核酸序列可发生杂交或退火。两条核酸序列发生杂交或退火所需要的互补性取决于所使用的杂交条件,特别是温度。
第一方面,本发明实施例提供一种HLA基因扩增引物,包括以下9组组引物组中的任意一组种或任意多组种组成的混合物:
第一引物组:SEQ ID NO:1和SEQ ID NO:2;第二引物组:SEQ ID NO:3和SEQ IDNO:4;第三引物组:SEQ ID NO:5和SEQ ID NO:6;第四引物组:SEQ ID NO:7和SEQ ID NO:8;第五引物组:SEQ ID NO:9和SEQ ID NO:10;第六引物组:SEQ ID NO:11和SEQ ID NO:12;第七引物组:SEQ ID NO:12和SEQ ID NO:13;第八引物组:SEQ ID NO:14和SEQ ID NO:15;第九引物组:SEQ ID NO:16、SEQ ID NO:17和SEQ ID NO:18。
在一些实施方式中,HLA扩增引物包括以上9组种引物组中的任意两组种、三组种、四组种、五组种、六组种、七组种、八组种或九组种组成的混合物。
本发明提出9组用于HLA基因的扩增引物组,针对HLA基因不同的靶序列。此9组用于HLA基因的扩增引物组既可以单独进行PCR扩增得到扩增基因。并且,此9组引物组中的任意组可以组合进行单管多重PCR扩增,9组扩增引物组可单管多重PCR扩增得到HLA 11个基因完整区域(包含:DPB1、DQA1、DQB1、DPA1、DRB1/3/4/5、HLA-A、HLA-B、HLA-C),使实验过程更简便(目前二代测序实现11个基因的扩增最少需要3管分别扩增,实验繁琐),简化工艺流程。
在多重PCR反应中,因为同时需要扩增多对引物,并且扩增序列长短不一,各片段的扩增之间难免会存在干扰。因此,调整引物组的配比以及各引物组中的引物的配比对于实现高质量的多重扩增影响重大。
在一些实施方式中,各引物组中的引物的摩尔比关系为:
SEQ ID NO:1与SEQ ID NO:2为1:(0.9~1.1);SEQ ID NO:3与SEQ ID NO:4为1:(0.9~1.1);SEQ ID NO:5与SEQ ID NO:6为1:(0.9~1.1);SEQ ID NO:7与SEQ ID NO:8为1:(0.9~1.1);SEQ ID NO:9与SEQ ID NO:10为1:(0.9~1.1);SEQ ID NO:11与SEQ ID NO:12为1:(0.9~1.1);SEQ ID NO:12与SEQ ID NO:13为1:(0.9~1.1);SEQ ID NO:14与SEQID NO:15为1:(0.9~1.1);SEQ ID NO:16与SEQ ID NO:17与SEQ ID NO:18为(1.9~2.1):(0.9~1.1):(0.9~1.1)。
在一些实施方式中,各引物组的引物的摩尔数之和的比值为第一引物组:第二引物组:第三引物组:第四引物组:第五引物组:第六引物组:第七引物组:第八引物组:第九引物组=(8.9~9.1):(5.4~5.6):(21~23):(1.4~1.6):(1.8~2):(0.9~1.1):(2~2.2):(0.9~1.1):(2.4~2.6)。
在一些实施方式中,各引物组的中的引物摩尔数之和分别为:
各引物组中的引物的配比可以为上述实施例的配比关系。各引物组的摩尔数之和可以为上述摩尔数之和的相同倍数,仅需要满足相应的比值关系即可。
针对PacBio三代测序平台,研究一种简单快速的HLA扩增的建库测序方法。利用三代测序的优势,结合特殊的引物扩增手段,在达到一次多重扩增目的的同时,以克服传统测序方法带来的缺陷。
第二方面,本发明实施例提供一种用于HLA基因测序的试剂盒,包括上述任一实施方式所述的HLA基因扩增引物。该试剂盒适用于建库测序。
可选的,所述试剂盒还包括PCR扩增试剂、测序文库构建试剂及基因纯化试剂中的任意一种或多种。
在一些实施方式中,测序文库构建试剂包括接头、接头连接反应试剂及核酸外切酶中的任意一种或多种。
接头为发卡状的接头,通过在基因扩增片段两端分别连接发卡状的接头,使得基因扩增片段形成圆环状闭合结构。
在一些实施方式中,以3.5L体系为基准,所述接头连接反应试剂为以下反应试剂的混合物:dNTP 1mmol、dATP 10mmol、T4 DNA聚合酶7.5×10
第三方面,本发明实施例提供一种HLA基因测序文库的构建方法,包括以下步骤:
采用上述任一实施方式所述的HLA基因扩增引物中的任意一种引物组或任意多种引物组组成的混合物对受试样品进行扩增得到扩增产物;
使所述扩增产物与接头发生连接反应构建得到文库;
对所述文库进行纯化。
在一些实施方式中,所述扩增的反应条件为:93℃~95℃,2min~3min;98℃,10s,68℃,11min~13min,30个循环,第11个循环开始,每循环增加30s;68℃,10min。
针对要扩增的区域设计引物,因为同时需要扩增多对引物,并且扩增序列长短不一,透过调整引物配比和调整适合的反应环境,达到一管多重扩增。
在一些实施方式中,所述连接反应的条件为:37℃,20min~30min;16℃~25℃,20min~30min;65℃,10min。
文库的纯化的目的是为了去除文库构建过程引入的各反应溶剂和杂质。
第四方面,本发明实施例提供一种HLA基因测序方法,其包括:采用上述任一实施方式所述的HLA基因测序文库的构建方法构建HLA基因测序文库,然后对所述文库进行测序。
在一些实施方式中,所述测序基于PacBio测序平台进行。
Pacbio第三代测序基于边合成边测序的原理,以单分子实时测序系统(SingleMolecule Real Time,SMRT)芯片为载体进行测序反应。基本原理如下:聚合酶捕获文库DNA序列,锚定在零模波导孔底部;4种不同荧光标记的dNTP随机进入零模波导孔底部;荧光dNTP被激光照射,发出荧光,检测荧光;荧光dNTP与DNA模板的碱基匹配,在酶的作用下合成一个碱基;统计荧光信号存在时间长短,区分匹配碱基与游离碱基,获得DNA序列;酶反应过程中,一方面使链延伸,另一方面使dNTP上的荧光基团脱落;聚合反应持续进行,测序同时持续进行。
Pacbio第三代测序中,DNA分子被接上发卡状的接头,因此,构建的文库整个是圆环的分子,利于其周而复始的复制。并且,对于一个片段的重复测序,可以提高准确度,不会像illumina测序那样,因为同时测多个碱基而出现phasing和prephasing的情况,制造噪音限制读长。使用Pacific Biosciences测序平台时由于其技术特点,获得的原始数据无需要组装,即可获得完整的信息,避免了一、二代测序技术自身区分无法分姐妹染色体信息以及组装可能造成的错误信息等,将整个HLA分型技术提升到了新的层次。
在一些实施方式中,所述测序方法适用的受试样品来自纯DNA样本或其他样本形式,如全血、血浆等。
以下为具体实施例。
首先针对要扩增的区域设计引物,因为同时需要扩增多对引物,并且扩增序列长短不一,透过调整引物配比、寻找适合的试剂、调整适合的反应环境,达到一管多重扩增。又基于三代单分子测序技术长读长的特性,使用扩增产物直接进行Pacific Biosciences三代建库,搭配Pacific Biosciences测序平台得到所扩增的HLA分型资讯。整个流程分为五个部份,依序为扩增、建库、纯化、混库、上机。
实施例1
一.扩增
使用从Coriell购买的88个人类gDNA标准品样本进行扩增。通过大量样本的结果以证明本发明方法的普适应和灵敏性。
设计针对HLA不同基因的特异性扩增引物,引物合成选择5’P,以下表1为引物序列:
表1
引物使用EB buffer配置与稀释,先分别稀释F和R引物,将F和R混合之后,按照下表2引物配比配置引物MIX。
表2
配置完引物MIX后,根据下表3PCR扩增体系,按照顺序加好后,扣上管盖混匀离心。
表3
根据下表4参数设置好PCR仪的程序进行扩增,热盖:105℃,升降温速率:2.0℃/S。
表4
扩增后的结果使用1%琼脂糖凝胶进行电泳实验,以确定目的基因是否扩增。电压:150V时间:50min,下图为Coriell gDNA扩增结果。(5ul扩增产物+5ul 6×loadingbuffer进行跑胶)
图1~9为跑胶结果,使用的Marker为Vazyme DL5000(扩增不佳的样本有做再次扩增)。
图中各泳道分别为:
图1.(1)NA16689(2)NA17212(3)NA17272(4)NA17222(5)NA17283(6)NA17218(7)NA17215(8)NA17119(9)NA17084(10)NA17466(11)NA17240(12)NA17292(13)NA17231(14)NA17263(15)NA17201。
图2.(1)NA17249(2)NA17618(3)NA17230(4)NA17269(5)NA17129(6)NA17130(7)NA17264(8NA)17203(9)NA17247(10)NA17242(11)NA17438(12)NA17262(13)NA02016(14)NA17236(15)NA17217。
图3.(1)NA17286(2)NA17248(3)NA17282(4)NA17052(5)NA17019(6)NA17291(7)NA17235(8)NA16654(9)NA17287(10)NA17275(11)NA17261(12)NA17216(13)NA17256(14)NA17286(15)NA17221。
图4.(1)NA17075(2)NA17224(3)NA13000(4)NA17039(5)NA17281(6)NA12244(7)NA17206(8)NA17245(9)NA17208(10)NA17293(11)NA17274(12)NA17234(13)NA17265(14)NA17276(15)NA17289(16)NA17114(17)NA17237。
图5.(1)NA17279(2)NA23090(3)NA17228(4)NA07439(5)NA17298(6)NA17229(7)NA17248(8)NA17214(9)NA16688(10)NA17254(11)NA23093(12)NA17207(13)NA17246(14)NA17209(15)NA17073(16)NA17285(17)NA12273(18)NA17260。
图6.(1)NA17115(2)NA17204(3)NA17243(4)NA17267(5)NA17058(6)NA17232(7)NA17075(8)NA17295(9)NA17268(10)NA17440(11)NA17290(12)NA17296(13)NA17226(14)NA17257(15)NA17280(16)NA10005(17)NA17213。
图7.(1)NA17261(2)NA09301(3)NA17219(4)NA17227(5)NA17287(6)NA17244(7)NA17288(8)NA17234(9)NA17211(10)NA17256。
图8.(1)NA17298(2)NA17291(3)NA09301(4)NA17262(5)NA17242(6)NA17214(7)NA17206(8)NA17269(9)NA17230(10)NA17438(11)NA17114(12)NA17073(13)NA17247(14)NA17229。
图9.(1)NA17264(2)NA17236(3)NA17265(4)NA17237(5)NA17233(6)NA17277(7)NA17057(8)NA17020(9)NA17205(10)NA17220(11)NA17209(12)NA17087(13)NA17212。
图10为使用9组引物组实现的扩增区域。
二.建库
扩增结束后,对扩增产物震荡瞬离,离心结束后水平静置放置,准备进行接头连接。
根据传统PacBio接头退火程序,对Barcode接头退火后备用。
准备配置连接酶Mix,根据下表5进行配置。
表5
根据下表6配制连接体系。
表6
配制完后扣上管盖,瞬离,振荡混匀,再次瞬离,然后放入表7预先设置好程序的PCR仪上开始连接反应;
反应程序:热盖75℃,升降温速率:2.5℃/S。
表7
连接反应后,配制根据下表8配制外切酶Mix。
表8
将装有连接产物的管放置于冰上,每管加入1μL外切酶mix。
扣上管盖,瞬离,振荡混匀,再次瞬离,然后放入表9预先设置好程序的PCR仪上开始消化反应;
反应程序:热盖45℃,升降温速率:2.5℃/S。
表9
三.纯化
将PB beads提前半小时从冰箱中取出,振荡混匀,然后放置于垂直混匀仪上室温平和30min;
1.在消化后的产物中加入6.6μL PB beads(0.6x),轻弹混匀或低速振荡混匀,瞬离,室温放置10min,期间轻弹混匀2-3次;
2.将PCR管瞬离,然后放置于磁力架上,吸附磁珠10min,期间配制70%酒精,酒精需要现用现配;
3.弃上清,然后沿吸附磁珠的对侧管壁加入200μL 70%酒精,切勿冲散磁珠;
4.重复上一步操作,进行第二次酒精清洗;
5.弃去酒精,将PCR管离心,然后重新放回磁力架,弃去残液;
6.PCR管开盖干燥不超过30s,然后加入20μL EB;
7.振荡悬浮磁珠,保持体系磁珠呈混匀状态,室温放置10min,洗脱DNA,期间轻弹2-3次;
8.将PCR管瞬离,然后放置于磁力架上,吸附磁珠10min;
9.吸取上清20μL转移至新的PCR管内;
10.利用Qubit对单样本文库进行定量,针对88个样本,每个单样本文库取相同质量3ng文库DNA形成混合文库。
四.混库
1.对混合文库用枪量取准确体积未204μL(取3ng),向其中加入122.4μL PB beads(0.6x),轻弹混匀或低速振荡混匀,瞬离,室温放置10min,期间轻弹混匀2-3次;
2.将EP管瞬离,然后放置于磁力架上,吸附磁珠10min;
3.弃上清,然后沿吸附磁珠的对侧管壁加入200μL 70%酒精,切勿冲散磁珠;
4.重复上一步操作,进行第二次酒精清洗;
5.弃去酒精,将PCR管离心,然后重新放回磁力架,弃去残液;
6.EP管开盖干燥不超过30s,然后加入15μL EB(根据样本数量和beads体积适当调整EB体积);
7.振荡悬浮磁珠,室温放置10min,洗脱DNA,期间轻弹2-3次;
8.将EP管瞬离,然后放置于磁力架上,吸附磁珠5min;
9.吸取所有上清转移至新的PCR管内;
10.利用Qubit对终文库进行定量,浓度为14ng/ul。
11.使用5200Fragment Analyzer System检测文库,并且计算平均片段大小,图11为分析结果。结果显示的片段大小与数量的关系与实际样本的情况一致。
五.上机测序
上机测序根据PacBio Sequel ll推荐的扩增子上机流程进行操作。结果如下表10-12所示。
表10
表11
表12
备注:加粗部分数据为本产品分型精度相比参考分型精度有提升的HLA基因。结果汇总如表13所示。
表13
以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
以上所述实施例仅表达了本发明的几种实施方式,便于具体和详细地理解本发明的技术方案,但并不能因此而理解为对发明专利保护专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准,说明书可以用于解释权利要求的内容。
序列表
<110> 西安浩瑞基因技术有限公司
<120> HLA基因扩增引物、试剂盒、测序文库构建方法及测序方法
<160> 18
<170> SIPOSequenceListing 1.0
<210> 1
<211> 23
<212> DNA
<213> Artificial Sequence
<400> 1
gggttcagac atgatgactg agc 23
<210> 2
<211> 25
<212> DNA
<213> Artificial Sequence
<400> 2
ggatttaggg ttctttgcac caaac 25
<210> 3
<211> 26
<212> DNA
<213> Artificial Sequence
<400> 3
tcatgtgctt ctcttgagca gtctga 26
<210> 4
<211> 24
<212> DNA
<213> Artificial Sequence
<400> 4
trtgacagca attttctctc ccct 24
<210> 5
<211> 27
<212> DNA
<213> Artificial Sequence
<400> 5
cagactctgt ccaatcccag ggtcaca 27
<210> 6
<211> 28
<212> DNA
<213> Artificial Sequence
<400> 6
gagcacagta gctttcggga attgacca 28
<210> 7
<211> 27
<212> DNA
<213> Artificial Sequence
<400> 7
gaaaacattt gccgtcagaa gaaggtg 27
<210> 8
<211> 27
<212> DNA
<213> Artificial Sequence
<400> 8
ccttttgtac ctgaggtcca gcatgaa 27
<210> 9
<211> 24
<212> DNA
<213> Artificial Sequence
<400> 9
atcctggata ctcacgacgc ggac 24
<210> 10
<211> 25
<212> DNA
<213> Artificial Sequence
<400> 10
catcaacctc tcatggcaag aattt 25
<210> 11
<211> 28
<212> DNA
<213> Artificial Sequence
<400> 11
tgaatggctc tgaaaatttg tctcagaa 28
<210> 12
<211> 25
<212> DNA
<213> Artificial Sequence
<400> 12
ggggtctctc cctggtttcc acaga 25
<210> 13
<211> 28
<212> DNA
<213> Artificial Sequence
<400> 13
tgamtgggtc tgaaaatttg tctcagaa 28
<210> 14
<211> 25
<212> DNA
<213> Artificial Sequence
<400> 14
ctctccattg cayataggtg gcccc 25
<210> 15
<211> 29
<212> DNA
<213> Artificial Sequence
<400> 15
atgcttwttg ggttagtgtc tgtctcctc 29
<210> 16
<211> 26
<212> DNA
<213> Artificial Sequence
<400> 16
caghrgastg gagaggtctg ttttcc 26
<210> 17
<211> 27
<212> DNA
<213> Artificial Sequence
<400> 17
tctkcayttc agctcaggaa tcctgca 27
<210> 18
<211> 27
<212> DNA
<213> Artificial Sequence
<400> 18
tctgcacttc agctcamgag tcctgca 27