一种基于LAMP扩增的荧光检测与CRISPR/Cas检测的双重检测体系及应用
文献发布时间:2023-06-19 19:04:00
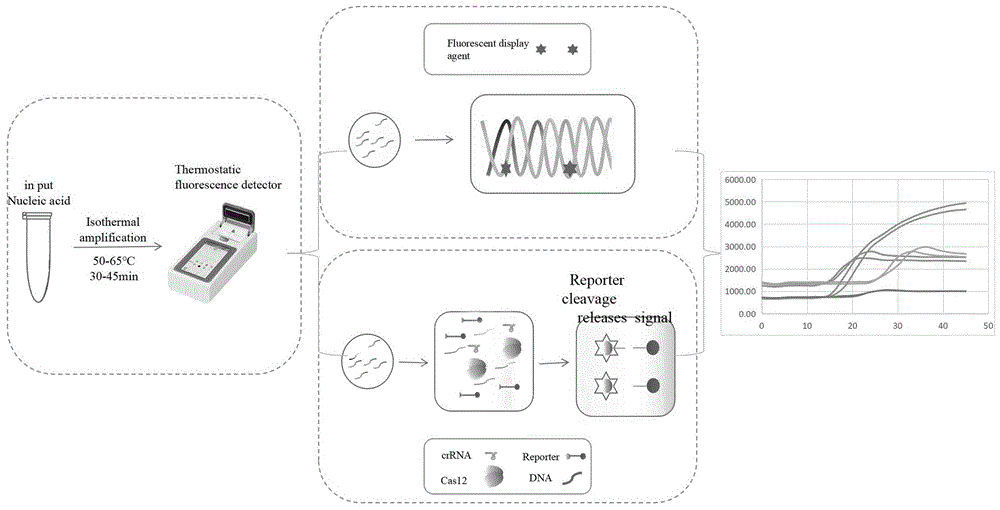
技术领域
本发明涉及基于LAMP扩增的荧光检测与CRISPR/Cas检测的双重检测体系及应用;属于核酸检测技术领域。
背景技术
等温扩增技术(isothermal amplification technology,ITA)是近年来发展起来的基于恒温扩增的新型核酸扩增技术,被许多学者认为是一种有可能与PCR媲美的检测方法。PCR技术原理是利用94℃高温使DNA双链解开,并在耐高温的DNA聚合酶的作用下扩增DNA片段。至少需要2个不同的温度,经过1小时以上特异性的扩增得到目的DNA。而恒温核酸扩增是利用各种酶与DNA之间的反应使核酸在同一温度下得到扩增。目前常见的等温核酸扩增技术包括LAMP、NERA、NASBA、RCA、HDA、RPA和ERA。在这些扩增技术中LAMP扩增DNA的效率高,能够在1h内有效的扩增1-10拷贝的目的基因,扩增效率是普通PCR的10倍-100倍,反应时间短,特异性强,不需要特殊的设备。若在体系中加入逆转录酶,能够实现RNA的扩增,对样本的包容性强。
LAMP的扩增产物检测手段主要包括:
1)、浊度法:通过肉眼观察或者浊度仪检测焦磷酸沉淀,此检测方法无特异性;
2)、普通电泳法:将LAMP扩增产物进行普通凝胶电泳,此检测方法也无特异性,且操作复杂耗时,容易形成气溶胶,造成假阳性影响检测结果;
3)、荧光染料染色法:反应中加入荧光染料通过荧光检测仪收集荧光进行结果的判定。染料主要包括:SYBR Green I、羟基萘酚蓝(Hydroxynaphthol blue,HNB)、钙黄绿素。但是SYBR Green I染料只要扩增出核酸便显示阳性,特异性弱;羟基萘酚蓝与钙黄绿素均是指示LAMP的副产物镁离子的变化,也无特异性,且多为单个靶点的检测;
4)、TaqMan荧光探针检测法:基于PCR扩增时Taq酶的5′→3′外切酶活性将探针酶切降解。但LAMP扩增用的DNA聚合酶大多自然缺失5′→3′外切酶活性,很难或者无法进行探针的酶切和降解;
5)、双重信标检测法:分子信标在复性温度下,因为模板不存在时形成茎环结构,当加热变性会互补配对的茎环双链解开,如果有模板存在环序列将与模板配对。与模板配对后,分子信标将成链状而非发夹状,使得荧光基团与淬灭剂分开。等温扩增的温度可能达不到变性温度,而且分子信标探针杂交时,探针不能完全肯定与模板结合,稳定性差;
6)、校对酶介导的探针裂解检测技术:在TaqMan荧光探针基础上,引入一个校对酶,补足BST酶缺失5′→3′外切酶活性。校对聚合酶是一种以比标准Taq更高的保真度复制DNA的酶。但是在某些时候校对聚合酶的校对功能会将不正确的核苷酸添加到正在生长的DNA链上,导致模板与新合成的链之间不匹配,会对LAMP扩增产生影响。
以上检测手段在一定程度上优化了LAMP检测方法,但这些方法依旧存在缺点和局限性。而随着CRISPR/Cas检测技术的发展,尝试通过CRISPR/Cas检测技术,提高LAMP检测的特异性性,打破检测的局限性成为一个新的突破点。
CRISPR/Cas系统的检测技术主要是利用CRISPR/Cas系统原有的适应、表达和干扰3个阶段,进行针对性的改造以实现特异性靶标的检测。通过人工设计向导RNA(Single-guide RNA,sgRNA),使其特异性识别靶序列后,Cas蛋白裂解靶序列从而形成特异性的双链断裂(Double strand breaks,DSB),而sgRNA的结构取决于所用的Cas蛋白。基于Cas12、Cas13及Cas14的核酸检测方法依赖于Cas蛋白的附属切割活性,其中Cas13靶向ssRNA,Cas12和14靶向ssDNA。当Cas蛋白与sgRNA、靶序列形成效应复合物时,其附属切割活性被激活,对已被标记的ssRNA或ssDNA报告基因进行切割,从而释放出信号达到检测效果。
利用CRISPR/Cas系统特异的序列识别及切割活性,将其应用于与LAMP扩增相结合,为提高检测灵敏度、特异性等性能指标以及实现双重检测提供了一种新思路。据目前所了解,LAMP扩增敏感性强,特别容易形成气溶胶,造成假阳性影响检测结果。其检测方式多,但存在特异性差以及不稳定的问题。此外,目前的LAMP扩增检测多数为单靶点检测,缺少对扩增体系监测、区分种属的多重检测方法。而待检样本通常是从比较复杂的物质中提取出来的核酸,易对反应体系产生影响,如抑制等温扩增反应。当检测结果为阴性时,我们无法判断是待测样本中无靶标核酸检不出,还是由于扩增受抑制导致检不出。故目前在分子诊断的应用中,往往都需要对扩增反应本身进行质控,比如对人源内标或自行投入内质控品等同时进行扩增检测,以判断反应本身的有效性,而现有的单靶标检测方法都不能很好的满足这个需求。
发明内容
要解决上述技术问题,本发明第一方面提供了一种基于LAMP扩增的荧光检测和CRISPR/Cas检测的双重检测体系。本发明的双重检测体系可以同时对同一样本以两个靶标的方式进行检测,其中一个靶标对样本进行是否扩增出靶序列核酸的荧光检测;另一靶标利用CRISPR/Cas对样本进行是否包含靶序列的包含内参检验的特异性检测。最后由荧光定量仪实时读取荧光数值,根据荧光起始值及信号强弱对检验结果进行分析判定。从而在单管中同时实现:a)、检测内标和靶标、b)区分样本病原体的属和种。
本发明解决上述问题的技术方案如下:
一种基于LAMP扩增的CRISPR/Cas和荧光检测的双重检测体系,包括靶序列扩增引物、能够识别靶序列的荧光报告探针、指示等温扩增反应是否有目标产物产生的荧光显示剂、反应缓冲液、增强缓冲液、包含DNA聚合酶和CRISPR/Cas蛋白酶的酶混合液、能够引导所述的CRISPR/Cas蛋白酶识别靶序列的sgRNA。
本发明提出的检测体系,操作简单,能够在单个反应管内同时进行双靶标的扩增和检测,减少开管反应的污染风险,对温控要求低,可降低实验室设备要求,缩短检测时间,自动化程度高。
本发明提出的检测体系是对待测样本中脱氧核糖核苷酸或者核糖核苷酸(还需加逆转录酶)进行扩增检测,具有灵敏度高,特异性强,适用于简便快速的核酸检测。
所述的DNA聚合酶,是一种具有链置换作用的DNA聚合酶,可置换下游的DNA链而不需要模板变性。可以是BST 2.0DNA聚合酶,BST 3.0DNA聚合酶、Warmstar BST 2.0DNA聚合酶、BST LF DNA聚合酶、OmniAmp DNA聚合酶。
所述的CRISPR/Cas蛋白酶,是一种可耐受55℃以上反应温度,在sgRNA的引导下能够特异性识别目标DNA,同时具有顺式和反式切割活性的CRISPR/Cas蛋白,包括但不限于AapCas12b、BrCas12b、AacCas12b蛋白。
优选的,CRISPR/Cas蛋白酶为AapCas12b、BrCas12b蛋白。
作为上述技术方案的优选,所述CRISPR/Cas蛋白与sgRNA的用量比为1:2。
本发明上述技术方案中,待检测核酸样本可以是从血液、唾液、组织、脑脊液、痰液等临床样本中,通过磁珠法或柱式法提取得到核酸。
所述的荧光报告探针为一段单链DNA,其5’端标记有包括但不限于FAM、HEX、VIC、CY5、CY3、ROX、TAMRA的荧光基团,3’端标记有包括但不限于BHQ1、BHQ2的可以与5’端任一荧光基团进行组合的淬灭基团。
所述的单链DNA序列,可以是TTATTATT或TTTTT或TTTTTTTT或其他富含TA的单链DNA序列,优选的序列为TTATTATT;该序列可以被CRISPR/Cas蛋白识别并切割。
作为上述技术方案的优选,所述荧光染料和荧光报告探针5’端荧光基团的激发光不同。
作为上述技术方案的优选,所述的荧光显示剂包括但不限于SYBRGreen、EvaGreen。
所述的荧光显示剂,即荧光染料,染料分子可以嵌入双链DNA分子中且不影响扩增效率以及CRISPR/Cas蛋白的活性。
作为上述技术方案的优选,荧光染料为EvaGreen,其终浓度用量为0.1x~1x;更优选的用量为0.1x~0.5x。
作为上述技术方案的优选,所述的反应增强剂包括但不限于牛磺酸、甜菜碱、海藻糖或BSA。
作为上述技术方案的优选,所述的反应增强剂选自以下选项中的至少一种:i)300~1500mM的甜菜碱或100~500mM海藻糖;ii)0.5~2mg/mL BSA;iii)200~1000mMTaurine。
作为上述技术方案的优选,所述的反应缓冲液包括:Tris-HCl,dNTPs,Tween-20或
作为上述技术方案的优选,所述的反应缓冲液包括:0.01~0.04M Tris-HCl,1~2.8mM dNTPs,0.01~0.2% Tween-20或
2x反应缓冲液包括:20~80mM Tris-HCl,2~4mM dNTPs,0.1%-0.4%Tween-20或
作为上述技术方案的优选,所述的DNA聚合酶为10~100ng/μL,CRISPR/Cas蛋白酶1~20ng/μL。
作为上述技术方案的优选,所述的靶序列扩增引物为1x;内参扩增引物为1x;sgRNA为10~100nM,进一步优选为20~60nM;
作为上述技术方案的优选,所述的酶混液包含10~40ng/μL DNA聚合酶和1~10ng/μL CRISPR/Cas蛋白酶。
10x酶混合液包括:100~400ng/μL DNA聚合酶和10~100ng/μL CRISPR/Cas蛋白酶。
作为上述技术方案的优选,所述的酶混液中,DNA聚合酶与CRISPR/Cas蛋白酶的用量比为DNA聚合酶:CRISPR/Cas蛋白酶=20:1。
作为上述技术方案的另一种优选,所述的酶混合液还包括逆转录酶。
所述的逆转录酶,当检测样本为RNA核酸时使用;可耐受55℃以上反应温度,包括但不限MMLV,AMV等。
作为上述另一种优选技术方案的进一步优选,所述的酶混液中,DNA聚合酶、CRISPR/Cas蛋白酶的用量比为DNA聚合酶:CRISPR/Cas蛋白酶=20:1,逆转录酶投入终浓度为1~30U。
用于RNA样本检测时,10x酶混合液还包括10~300U/μL逆转录酶。
更进一步的,逆转录酶投入终浓度为1-15U。
本发明的第二方面是提供上述双重检测体系的一种应用。
上述双重检测体系在同时a)、检测内标和靶标,和b)、区分样本病原体的属和种方面的应用。
所述双重检测体系的使用方法,具体检测某样本时,将内参引物和指示等温扩增反应是否有目标产物产生的荧光显示剂混成检测引物1;将靶序列扩增引物和能够引导所述的CRISPR/Cas蛋白酶识别靶序列的sgRNA混成检测引物2;然后,将所有的组分加入反应管中,置于荧光定量PCR仪或恒温扩增荧光检测仪上,于55~65℃进行等温扩增检测;根据实时或终点荧光信号强弱进行结果分析,进行判断,同时实现:a)、检测内标和靶标,b)、区分样本病原体的属和种。
人源内标扩增引物序列可以基于ACTB或者其他管家基因如GAPDH进行设计。
所述的ACTB引物组包括如下表格中的3组,优选的引物组为引物组1。
综上所述,本发明具有以下有益效果:
1)非必要使用昂贵且精密的荧光定量PCR仪器;
2)使用普通的PCR管,一次性完成加样,全过程闭管反应,不易产生污染;
3)灵敏度高,筛选合适的引物和sgRNA可实现高灵敏度的检测;
4)特异性高,通过sgRNA靶向指导CRIPSR/Cas蛋白精准切割待测靶点;
5)实现了等温LAMP扩增的双重检测,可以同时实现:a)、检测内标,和b)检测靶标或区分样本病原体的属和种。
附图说明
图1是双重LAMP扩增检测原理流程图;
图2是核酸扩增显示剂对双重LAMP内标扩增的影响图;
图3是核酸扩增显示剂对双重LAMP扩增靶点检测的影响图;
图4是增强缓冲液对双重LAMP扩增靶点检测的影响图;
图5是结核分枝杆菌阳性样本和人源细胞核酸内标扩增曲线图;
图6是结核分枝杆菌阳性样本检测曲线图;
图7是结核分枝杆菌灵敏度检测结果;
图8是结核分枝杆菌特异性检测结果;
图9是甲型流感病毒阳性样本和人源细胞核酸内标扩增曲线图;
图10是甲型流感病毒阳性样本检测曲线图;
图11是甲型流感病毒灵敏度检测结果。
具体实施方式
下面结合具体实施例及附图,对本发明提供的技术方法进行详细的阐述与说明,所述实施例仅仅是本发明的一部分实施例,并不局限于本发明全部的实施例。以下实施例中所用到的试剂组分除特别说明外,均为本发明试剂盒中的组分。本领域的专业人员在本发明基础上的修改或者改动,这些等价修改的方式同样落在本发明权利要求书中所述的权利要求范围。
实施例一结核分枝杆菌的检测
1.1、实验材料
1.1.1、2x反应缓冲液
40mM Tris-HCl,2.8mM dNTPs,0.2%Tween-20或
1.1.2、10x酶混合液
270ng/μL DNA聚合酶和40ng/μL CRISPR/Cas蛋白酶。可选的用于RNA样本检测时,10x酶混合液包括270ng/μL DNA聚合酶、40ng/μL CRISPR/Cas蛋白酶、80U/μL逆转录酶。
1.1.3、增强缓冲液
增强缓冲液1是500mM Taurine;
增强缓冲液2是1000mM甜菜碱;
增强缓冲液3是1mg/mL BSA;
增强缓冲液4是160mM海藻糖。
1.1.4、核酸扩增显示剂
核酸扩增显示剂-1是荧光染料EvaGreen;
核酸扩增显示剂-2是荧光染料SYBRGreen。
1.1.5、扩增引物和sgRNA
检测引物1是ACTB引物(见上述表格)与核酸扩增显示剂的预混物;其中,检测引物1-1是ACTB引物与核酸扩增显示剂1(EvaGreen)的预混物;检测引物1-2是ACTB引物与核酸扩增显示剂1(SYBRGreen)的预混物;
检测引物2是筛选得到的结合分支杆菌IS6110靶点的最优引物组(ID NO.15~20)与sgRNA的预混物;
查找结核分枝杆菌IS6110靶点引物扩增区域中的PAM识别点(TTN),取PAM识别点后20-23bp的序列设计sgRNA扩增下游引物(ID NO.21ACCGTCGAACGGCTGATGACGTGCCACTTCTCAGATTT);
所述sgRNA的制备方法如下:
以人工合成的含Cas蛋白序列(ID NO.22GTCTAGAGGACAGAATTTTTCAACGGGTGTGCCAATGGCCACTTTCCAGGTGGCAAAGCCCGTTGAGCTTCTCAAATCTGAGAAGTGGCAC)的质粒作为模板,上游引物(ID NO.23GTAAAACGACGGCCAGT),下游引物(ID NO.21同上);通过PCR反应,得到crDNA;经体外转录试剂盒得到sgRNA产物,通过RNA纯化试剂盒进行纯化,Qubit检测浓度;
体外转录试剂盒使用的是HiScribeT7 Quick High Yield RNA Synthesis kit(New England Biolabs);RNA纯化试剂盒使用的是RNA Clean&Concentrator-5kit(ZymoResearch)。
1.1.6、阴性样本和阳性样本
阴性样本:提取的人源细胞核酸,浓度0.1~10ng/μL;
阳性样本:人工合成的含待检测结核分枝杆菌IS6110靶点基因片段(ID NO.14)的质粒;并用0.1~10ng/μL的人源细胞核酸稀释至要求浓度。(ID NO.14AAAGACCGCGTCGGCTTTCTTCGCGGCCGAGCTCGACCGGCCAGCACGCTAATTACCCGGTTCATCGCCGATCATCAGGGCCACCGCGAGGGCCCCGATGGTTTGCGGTGGGGTGTCGAGTCGATCTGCACACAGCTGACCGAGCTGGGTGTGCCGATCGCCCCATCGACCTACTACGACCACATCAACCGGGAGCCCAGCCGCCGCGAGCTGCGCGATGGCGAACTCAAGGAGCACATCAGCCGCGTCCACGCCGCCAACTACGGTGTTTACGGTGCCCGCAAAGTGTGGCTAACCCTGAACCGTGAGGGCATCGAGGTGGCCAGATGCACCGTCGAACGGCTGATGACCAAACTCGGCCTGTCCGGGACCACCCGCGGCAAAGCCCGCAGGACCACGATCGCTGATCCGGCCACAGCCCGTCCCGCCGATCTCGTCCAGCGCCGCTTCGGACCACCAGCACCTAACCGGCTGTGGGTAGCAGACCTCACCTATGTGTCGACCTGGGCAGGGTTCGCCTACGTGGCCTTTGTCACCGACGCCTACGCTCGCAGGATCCTGGGCTGGCGGGTCGCTTCCACGATGGCCACCTCCATGGTCCTCGACGCGATCGAGCAAGCCATCTGGACCCGCCAACAAGAAGGCGTACTCGACCTGAAAGACGTTATCCACCATACGGATAGGGGATCTCAGTACACATCGATCCGGTTCAGCGAGCGGCTCGCCGAGGCAGGCATCCAACCGTCGGTCGGAGCGGTCGGAAGCTCCTATGACAATGCACTAGCCGAGACGATCAACGGCCTATACAAGACCGAGCTGATCAAACCCGGCAAGCCCTGGCGGTCCATCGAGGATGTCGAGTTGGCCACCGCGCGCTGGGTCGACTGGTTCAACCATCGCCGCCTCTACCAGTACTGCGGCGACGTCCCGCCGGTCGAACTCGAGGCTGCCTACTACGCTCAACGCCAGAGACCAGCCGCCGGCTGA)
1.2、核酸扩增显示剂对双重LAMP扩增靶点检测和内标扩增的影响
1.2.1、预处理一
将2x反应缓冲液、10x酶混合液、增强缓冲液(增强缓冲液1)、检测引物2按照下表中各组分用量,在1~8号离心管中均配制1份40μL反应预混液,共8份;
1.2.2、预处理二
向1号管和2号管均加入5μL阴性样本和5μL检测引物1-1;
向3号管和4号管均加入5μL阳性样本和5μL检测引物1-1;
向5号管和6号管均加入5μL阴性样本和5μL检测引物1-2;
向7号管和8号管均加入5μL阳性样本和5μL检测引物1-2;
本例中,阳性样本浓度均为1fg/μL(2*10
1.2.3、反应
瞬时离心后将反应管置于扩增设备(如QPCR、恒温荧光检测仪,本实施例采用恒温荧光检测仪)。
1.2.4、结果判断
靶标扩增检测荧光呈显著上升趋势判定为阳性;荧光上升趋于平缓或不显著状态判定为阴性。内标扩增检测曲线呈显著上升趋势判定内标和靶标扩增成功,扩增有效。
1.2.5、结果分析
如图2和3所示,核酸扩增显示剂-1对双重LAMP内标扩增以及靶点检测的影响最小。
1.3、增强缓冲液对双重LAMP扩增靶点检测的影响
1.3.1、预处理一
将2x反应缓冲液、10x酶混合液、检测引物1、检测引物2按照下表中各组分用量,在1~20号离心管中均配制1份40μL反应预混液,共20份;
1.3.2、预处理二
向1号管和2号管均加入5μL阴性样本和增强缓冲液1;
向3号管和4号管均加入5μL阳性样本和增强缓冲液1;
向5号管和6号管均加入5μL阴性样本和增强缓冲液2;
向7号管和8号管均加入5μL阳性样本和增强缓冲液2;
向9号管和10号管均加入5μL阴性样本和增强缓冲液3;
向11号管和12号管均加入5μL阳性样本和增强缓冲液3;
向13号管和14号管均加入5μL阴性样本和增强缓冲液4;
向15号管和16号管均加入5μL阳性样本和增强缓冲液4;
向17号管和18号管仅加入5μL阴性样本;
向19号管和20号管仅加入5μL阳性样本;
本例中,阳性样本浓度均为1fg/μL(2*10
1.3.3、反应
瞬时离心后将反应管置于扩增设备(如QPCR、恒温荧光检测仪,本实施例采用恒温荧光检测仪)。
1.3.4、结果分析
如图4所示,增强缓冲液1、2、3能够提高靶点的检出时间,增强缓冲液4对整个反应无明显变化。
1.4、结核分支杆菌阳性样本和阴性样本检测
1.4.1、预处理一
将2x反应缓冲液、10x酶混合液、增强缓冲液(增强缓冲液1)、检测引物1、检测引物2、按照下表中各组分用量,在1~4号离心管中均配制1份45μL反应预混液,共4份;
1.4.2、预处理二
向1号管和2号管均加入5μL阴性样本;
向3号管和4号管均加入5μL阳性样本;与前述不同,本例中阳性样本的浓度为1pg/μL(2*10
1.4.3、反应
瞬时离心后将反应管置于扩增设备(如QPCR、恒温荧光检测仪,本实施例采用恒温荧光检测仪)。
1.4.4、结果判断
靶标荧光检测曲线呈上升趋势判定为阳性;荧光检测曲线呈水平状态判定为阴性。内标扩增曲线呈上升趋势判定内标和靶标扩增成功,扩增有效。
1.4.5、结果分析
从图5和6可以看出,靶标荧光检测曲线,阳性样本荧光检测曲线呈明显上升趋势,而内标样本荧光检测曲线平缓基本呈水平状态。内标扩增曲线呈上升趋势判定内标扩增成功,扩增有效。此检测与判断方法可正确快速的区分待检测靶点和非检测靶点以及扩增体系的有效性。
1.5、结核分枝杆菌灵敏度检测
1.5.1、预处理一
将2x反应缓冲液、10x酶混合液、增强缓冲液(增强缓冲液1)、检测引物1、检测引物2、按照下表中各组分用量,在1~12号离心管中均配制1份45μL反应预混液,共12份;
1.5.2、预处理二
向1号管和2号管均加入5μL阴性样本;
向3号管和4号管均加入5μL阳性样本1;
向5号管和6号管均加入5μL阳性样本2;
向7号管和8号管均加入5μL阳性样本3;
向9号管和10号管均加入5μL阳性样本4;
向11号管和12号管均加入5μL阳性样本5;
与前述不同,本例中阳性样本的浓度分为5个梯度,依次是:
阳性样本1:1pg/μL(约2*10
阳性样本2:100fg/μL(约2*10
阳性样本3:10fg/μL(2*10
阳性样本4:1fg/μL(2*10
阳性样本5:100ag/μL(2*10
1.5.3、反应
瞬时离心后将反应管置于扩增设备(如QPCR、恒温荧光检测仪,本实施例采用恒温荧光检测仪)。
1.5.4、结果与分析
图7为结合分枝杆菌的灵敏度检测结果;从图7可以看出,最低检测限为1fg/μL(2*10
需要说明的是,试剂盒的检测限有部分依赖于靶点引物的设计以及sgRNA的效率,所以该靶点的灵敏度并不能完全代表所有靶点检测的灵敏度,但具有较高的参考价值。
1.6、结核分枝杆菌特异性检测
1.6.1、预处理一
将2x反应缓冲液、10x酶混合液、增强缓冲液(增强缓冲液1)、检测引物1、检测引物2、按照下表中各组分用量,在1~12号离心管中均配制1份45μL反应预混液,共12份;
1.6.2、预处理二
向1号管和2号管均加入5μL阴性样本;
向3号管和4号管均加入5μL阳性样本(1pg/μL);
向5号管和6号管均加入5μL干扰样本1(1pg/μL);
向7号管和8号管均加入5μL干扰样本2(1pg/μL);
向9号管和10号管均加入5μL干扰样本3(1pg/μL);
向11号管和12号管均加入5μL干扰样本4(1pg/μL);
向13号管和14号管均加入5μL干扰样本5(1pg/μL);
向15号管和16号管均加入5μL干扰样本6(1pg/μL);
干扰样本1:蜡样芽孢杆菌(Bacillus cereus);
干扰样本2:溶血葡萄球菌(Staphylococcus haemolyticus);
干扰样本3:铜绿假单胞菌(Pseudomonas aeruginosa);
干扰样本4:鲍曼不动杆菌(Acinetobacter baumannii);
干扰样本5:屎肠球菌(Enterococcus Faecium);
干扰样本6:表皮生长因子EGFR靶点;
1.6.3、反应
瞬时离心后将反应管置于扩增设备(如QPCR、恒温荧光检测仪,本实施例采用恒温荧光检测仪)。
1.6.4、结果分析
图8是结核分枝杆菌特异性检测结果;从图8可以看出,除阳性样本的检测荧光信号上升外,其余样本检测荧光均为无明显变化;由此可见,本检测体系的特异性高,能够准确的区分待检基因和非检测基因,在常见的其他呼吸道或肺部感染的病原体中均不会产生交叉信号。
实施例二
2.1实验材料
2.1.1、2x反应缓冲液
40mM Tris-HCl,2.8mM dNTPs,0.2%Tween-20或
2.1.2、10x酶混合液
270ng/μL DNA聚合酶和40ng/μL CRISPR/Cas蛋白酶。可选的用于RNA样本检测时,10x酶混合液包括270ng/μL DNA聚合酶、40ng/μL CRISPR/Cas蛋白酶、80U/μL逆转录酶;
2.1.3、增强缓冲液
增强缓冲液1是500mM Taurine;
增强缓冲液2是1000mM甜菜碱;
增强缓冲液3是1mg/mL BSA;
增强缓冲液4是160mM海藻糖;
2.1.4、核酸扩增显示剂
核酸扩增显示剂-1是荧光染料EvaGreen;
核酸扩增显示剂-2是荧光染料SYBRGreen;
2.1.5、扩增引物和sgRNA
检测引物1是ACTB引物与核酸扩增显示剂(EvaGreen)的预混物;
检测引物3是M1靶点的引物组与sgRNA的预混物;
M1靶点的引物组序列(ID NO.24~29);所述M1靶点是购买的甲型流感病毒假病毒的M1靶点。
查找M1靶点引物扩增区域中的PAM识别点,取PAM识别点后20-23bp的序列设计sgRNA扩增下游引物(ID NO.30TCACTGGGCACGGTGAGCGT-GTGCCACTTCTCAGATTT);
所述sgRNA的制备方法如下:
以人工合成的含Cas蛋白序列(ID NO.22同上)的质粒作为模板,上游引物(IDNO.23同上),下游引物(ID NO.30同上);通过PCR反应,得到crDNA;经体外转录试剂盒得到sgRNA产物,通过RNA纯化试剂盒进行纯化,Qubit检测浓度;
体外转录试剂盒使用的是HiScribeT7 Quick High Yield RNA Synthesis kit(New England Biolabs);RNA纯化试剂盒使用的是RNA Clean&Concentrator-5kit(ZymoResearch)。
2.1.6、阴性样本和阳性样本
阴性样本:提取的人源细胞核酸,浓度0.1~10ng/μL;
阳性样本:含待检测M1靶点基因片段的甲型流感病毒;并用0.1~10ng/μL的人源细胞核酸稀释至约10
2.2甲型流感病毒阳性样本和阴性样本检测
2.2.1、预处理一
将2x反应缓冲液、10x酶混合液、增强缓冲液(增强缓冲液1)、检测引物1、检测引物3、按照下表中各组分用量,在1~4号离心管中均配制1份45μL反应预混液,共4份;
2.2.2、预处理二
向1号管和2号管均加入5μL阴性样本;
向3号管和4号管均加入5μL阳性样本;本例中阳性样本的浓度为10
2.2.3、反应
瞬时离心后将反应管置于扩增设备(如QPCR、恒温荧光检测仪,本实施例采用恒温荧光检测仪)。
2.2.4、结果分析
图10为甲型流感病毒阳性样本检测曲线图;从图10可以看出,内标扩增曲线呈上升趋势判定内标和靶标扩增成功,扩增有效。荧光检测曲线中,阳性样本荧光检测曲线呈明显上升趋势,内标样本荧光检测曲线平缓基本呈水平状态。
2.3甲型流感病毒灵敏度检测
2.3.1、预处理一
将2x反应缓冲液、10x酶混合液、增强缓冲液(增强缓冲液1)、检测引物1、检测引物3、按照下表中各组分用量,在1~14号离心管中均配制1份45μL反应预混液,共14份;
2.3.2、预处理二
向1号管和2号管均加入5μL阴性样本;
向3号管和4号管均加入5μL阳性样本1;
向5号管和6号管均加入5μL阳性样本2;
向7号管和8号管均加入5μL阳性样本3;
向9号管和10号管均加入5μL阳性样本4;
向11号管和12号管均加入5μL阳性样本5;
向13号管和14号管均加入5μL阳性样本6;
与前述不同,本例中阳性样本的浓度分为6个梯度,依次是:
阳性样本1:10
阳性样本2:10
阳性样本3:10
阳性样本4:10
阳性样本5:10
阳性样本6:10
2.3.3、反应
瞬时离心后将反应管置于扩增设备(如QPCR、恒温荧光检测仪,本实施例采用恒温荧光检测仪)。
2.3.4、结果与分析
图11为甲型流感病毒的灵敏度检测结果;从图11可以看出,最低检测限为10
- 一种基于RT-RPA体系和CRISPR/Cas体系的基因检测方法及系统及其应用
- 一种基于PCR扩增和CRISPR-Cas12a的核酸检测方法及应用