一种检测非整倍体缺失染色体信息的方法
文献发布时间:2023-06-19 19:27:02
技术领域
本发明属于基因组测序及生物信息学领域,特别是涉及一种检测非整倍体缺失染色体信息的方法。
背景技术
在大多情况下,非整倍体对于动物及人类是致命的,但植物对于非整倍体却通常表现出较强的耐受力,特别是在异源多倍体植物中。非整倍体在基因及分子标记的物理位置确定、基因转移、连锁群与染色体的对应关系的确立上具有无可比拟的优势,对于植物的遗传、育种的研究有着重要的意义,同时在实际育种中的应用中非整倍体也取得了不少成果。
通过非整倍体研究,可以更快地、有系统地梳理清植物的各种性状之间的遗传规律,并确定其染色体与其近缘植物间的关系,从而更有计划地选育出各种特殊优良的新品种。但由于这种研究涉及大量的杂交实验,工作量大且时间长,在林木领域研究略显匮乏。由于林木生长时期较长,难以在育种过程中调整方向,所以在开展正式实验前必须得到足够的染色体信息。
如今基于个体染色体组的核型分析,如C带法、G带法、流式细胞术以及基于染色体特异探针的荧光原位杂交技术(FISH)都是较为常见的非整倍体鉴定方法。但上述方法大多对实验材料类型有较强的偏好性并需要长时间的实验制备,在面对大批量实验材料的筛选工作时将略显吃力。倍性分析仪可快速确定所创制群体是否为非整倍体,但难以确定每个个体的具体染色体组成情况。
此外,传统高通量测序后利用T检验检测样品与标准参照样覆盖深度是否有显著性差异的方法现已在唐氏综合征、18-三体综合征等人类疾病的临床中应用。但在植物非整倍体育种中,由于杂交品种繁多、基因组差异大、染色体增减数目多、标准参照获取困难等因素,同样难以展开。因此,亟需提供一种检测非整倍体缺失染色体信息的方法。
发明内容
本发明的目的是提供一种检测非整倍体缺失染色体信息的方法,以解决上述现有技术存在的问题。
为实现上述目的,本发明提供了一种检测非整倍体缺失染色体信息的方法,包括以下步骤:
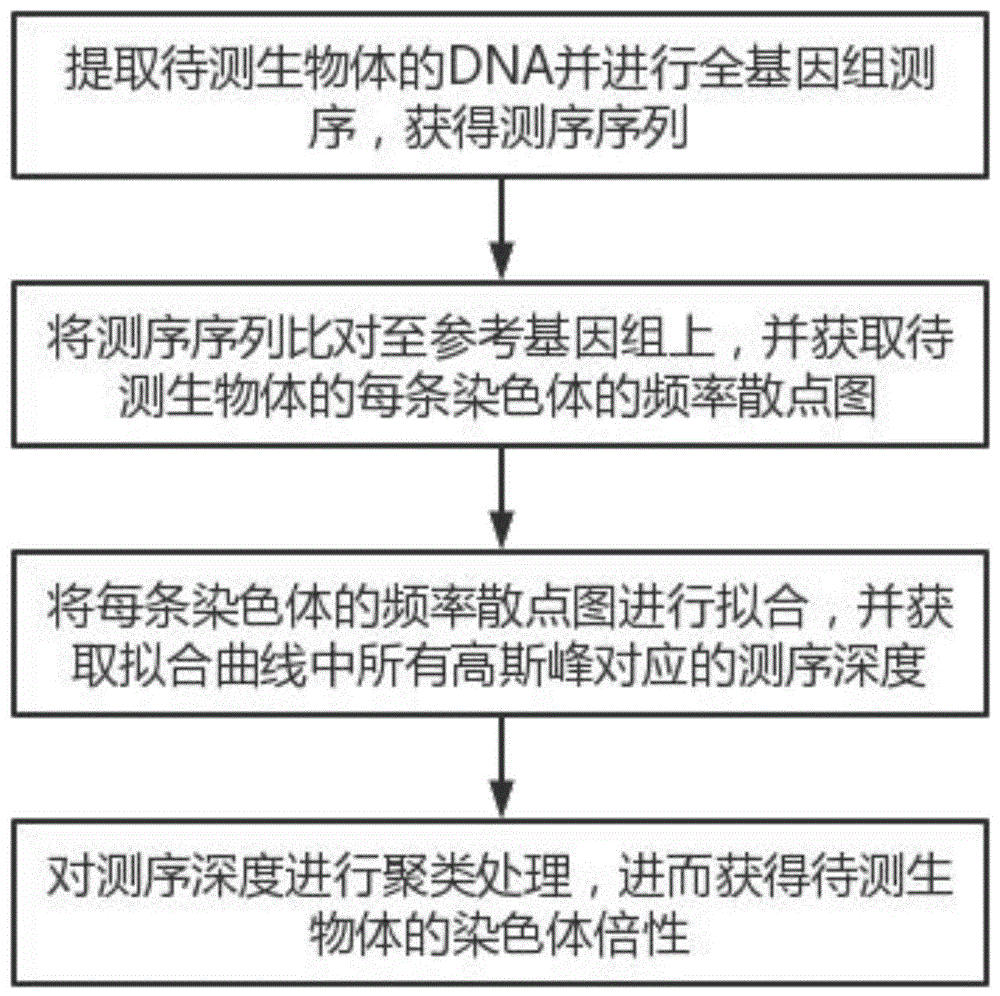
提取待测生物体的DNA并进行全基因组测序,获得测序序列;
将所述测序序列比对至参考基因组上,并获取待测生物体的每条染色体的频率散点图;
将所述每条染色体的频率散点图进行拟合,并获取拟合曲线中所有高斯峰对应的测序深度;
对所述测序深度进行聚类处理,进而获得待测生物体的染色体倍性。
可选地,对提取的DNA进行全基因组测序之前还包括:基于琼脂糖凝胶电泳检测DNA的完整性,并采用酶标仪对DNA含量进行浓度检测。
可选地,所述参考基因组选用待测生物体的物种本身或近源物种的基因组,且已挂载至染色体级别。
可选地,获取每条染色体的频率散点图的过程包括:获取每条染色体上每个碱基的测序深度,并统计每种测序深度的出现频率,进而获得每条染色体的频率散点图。
可选地,将所述每条染色体的频率散点图进行拟合的过程包括:将所述每条染色体的频率散点图拟合成单条高斯曲线或x条高斯曲线的叠加所形成的混合高斯模型,其中,x为每条染色体的频率散点图中峰的数量。
可选地,对所述测序深度进行聚类处理之前还包括:将所述测序深度进行排序,获得每条染色体中测序深度最大的高斯峰。
可选地,获得待测生物体的染色体倍性的过程包括:对所述测序深度进行一维数组聚类,获得不同聚类组;基于所述不同聚类组的中位数之间的倍性关系,获得不同聚类组的测序深度的中位数;基于所述不同聚类组的测序深度的中位数及每条染色体中测序深度最大的高斯峰,对不同的染色体进行聚类,进而获得待测生物体的染色体倍性。
本发明的技术效果为:
本发明利用统计基因组上每种测序深度出现频率的方法识别出染色体的真实测序深度,使用混合高斯拟合模型对测序深度频率曲线进行拆分,以梳理多种造成染色体测序深度不稳定的因素,并将聚类算法应用到对所有测序深度峰值进行分组,以确定单体的测序深度,从而提高染色体倍性的检测精度。
本发明以二代高通量测序技术为基础,在面对大批量的样品时,相较于传统检测方法可大大缩短时间,同时可实现自动化操作,具有标准性和可重复性等优势。
附图说明
构成本申请的一部分的附图用来提供对本申请的进一步理解,本申请的示意性实施例及其说明用于解释本申请,并不构成对本申请的不当限定。在附图中:
图1为本发明实施例中的检测非整倍体缺失染色体信息的方法流程图;
图2为本发明实施例中的19条染色体的频率散点图;
图3为本发明实施例中的1号样品进行高斯拟合后的拟合曲线图。
具体实施方式
需要说明的是,在不冲突的情况下,本申请中的实施例及实施例中的特征可以相互组合。下面将参考附图并结合实施例来详细说明本申请。
需要说明的是,在附图的流程图示出的步骤可以在诸如一组计算机可执行指令的计算机系统中执行,并且,虽然在流程图中示出了逻辑顺序,但是在某些情况下,可以以不同于此处的顺序执行所示出或描述的步骤。
实施例一
如图1所示,本实施例中提供一种检测非整倍体缺失染色体信息的方法,包括以下步骤:
DNA提取:
根据待测动植物样本特性,选取适当的DNA提取方案,并进行DNA含量及纯度的检测,样品质量以符合测序仪官方上机标准为准。
实施例选用通过杂交获得的6株非整倍体植株作为实验样品以及两株整倍体植株作为对照,使用MGIEasy通用DNA文库制备试剂盒进行标准DNA的提取。提取完成后使用琼脂糖凝胶电泳检测样品完整性,并使用酶标仪进行浓度检测,检测试剂盒选用DNABR。结果显示提取样本质量均符合测序平台上机标准。
全基因组测序:
基于二代高通量测序技术,根据官方指导手册在Illumina或BGI测序平台进行测序文库制备以及上机检测,仪器参数及操作方法均严格参照对应测序平台的指导手册进行。
基于二代高通量测序技术,根据官方指导手册在MGISEQ-2000测序平台进行测序文库制备以及上机检测。建库类型为DNBSEQ WGS,测序模式选择为PE150全基因组测序,仪器参数及操作方法均严格参照对应测序平台的指导手册进行。
测序序列与参考基因组比对及测序深度统计:
得到下机数据后,将双端测序得到的序列比对至参考基因组上,参考基因组选用物种本身或近源物种的基因组均可,但必须已挂载至染色体级别。鉴于种内不同单株间等位基因的差异,比对方案需尽可能选择误差容忍度较高的方法。完成比对后,分别计算参考基因组上每个核苷酸的测序深度,并以染色体为单位统计每种测序深度的出现频率。结果以散点图呈现,横坐标为测序深度,纵坐标为该种测序深度所对应的出现频率。
以1号样品为例,下机共得到266.24M双端序列。在过滤掉低质量序列后,使用BWA-MEM将测序得到的序列比对至杨树参考基因组上,比对率为92.06%。随后计算染色体上每个碱基的测序深度,并以染色体为单位统计频率。如图2所示为19条染色体的线条连接散点图,横坐标为测序深度,纵坐标为该种测序深度的出现频率。
以染色体为单位绘制拟合曲线并进行峰值的计算:
利用高斯拟合原理将每条染色体的频率散点图拟合成单条高斯曲线或x条高斯曲线的叠加所形成的混合高斯模型,x为频率曲线中出现峰的数量。在完成拟合后,记录所有高斯峰所对应的测序深度,并按从小到大排列。
对每条染色体进行曲线拟合,如图3所示,以1号样品的1号染色体为例,根据峰的数量进行高斯拟合可得到2条正态分布曲线,R-Square(R
判断染色体在生物体内的具体倍性:
对上一步所记录的所有数值,使用DBSCAN或其他无需提前设置组数的聚类算法进行一维数组聚类。不同聚类组的中位数之间应存在倍性关系,假设第一组峰(即单体)对应测序深度为y,则第二组峰对应测序深度应为2y,第三组峰对应测序深度应为3y,第n组峰对应测序深度应为n×y。随后确定每条染色体中测序深度最大的高斯峰,若其对应测序深度被聚类至第n组,则该条染色体在生物体内的倍性即为n。
利用DBSCAN算法对上述记录结果进行一维数组聚类,可将这些曲线分成三组,组内测序深度的中位数分别为34X、68X和101X。其中1号染色体的最后一个峰(测序深度:68X)最终被聚类至第二组,代表该条染色体在生物体内的倍性为2,即二体。而5号、8号、13号和19号染色体的最后一个峰都被聚类至第三组,代表以上染色体在生物体内的倍性为3,即三体。通过这种方式,最终判断1号样品为5号、8号、13号、19号染色体三体植株,细胞内共计有42条染色体。对于本实施例样品的检测结果如表1所示:
表1
/>
通过表1可以看出,本实施例的检测结果与倍性分析仪的结果具有很好的一致性。由于本实施例以二代高通量测序技术为基础,在面对大批量的样品时,相较于传统检测方法可大大缩短时间,同时可实现自动化操作,具有标准性和可重复性等优势。
以上所述,仅为本申请较佳的具体实施方式,但本申请的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本申请揭露的技术范围内,可轻易想到的变化或替换,都应涵盖在本申请的保护范围之内。因此,本申请的保护范围应该以权利要求的保护范围为准。
- 一种用于无创检测待测胎儿染色体是否为非整倍体的试剂盒及其专用探针组
- 一种基于扩增子测序检测染色体杂合性缺失的试剂盒及方法
- 一种利用染色体特异位点在单细胞扩增基础上进行染色体非整倍体检测的方法
- 一种染色体非整倍体异常的检测方法及检测试剂盒