一种马克斯克鲁维单倍体酵母及其构建方法与应用
文献发布时间:2023-06-19 19:27:02
技术领域
本发明属于本发明属于生物工程技术领域,具体涉及一种马克斯克鲁维单倍体酵母及其构建方法与应用。
背景技术
近年来随着合成生物学的蓬勃发展,正极大的推动和完善各种微生物细胞工厂的开发和高效生物制造能力。而工业微生物底盘细胞作为生物制造最核心的环节,对其理性设计与改造正发挥着越来越重要的作用。借助合成生物学技术,非常规酵母因具其独特的性状,如高密度发酵、耐受温度和pH范围广,底物谱广泛等,逐渐成为极具开发潜力的微生物细胞而用于多种天然产物、生物燃料和大量大宗化学品的绿色生物制造中。
马克斯克鲁维酵母(Kluyveromyces marxianus),广泛存在于酸奶、水果、酸乳酒等环境中,是自然界中存在的安全酵母。作为新型工业微生物底盘细胞,马克思克鲁维酵母是一种GRAS级的酵母,已得到欧盟食品安全监督局、美国FDA的安全认证,也被中国卫生委批准为新食品原料(2013年),因此可将其用于营养化学品、蛋白、药品等的生产。相比于酿酒酵母,马克思克鲁维酵母具有更高的生长温度、更快的生长速率和更广的底物利用谱等优势,在食品饲料工业、环境领域、生物医药等有着广泛应用,是一类极具潜力的酵母细胞工厂宿主。
K.marxianus在自然环境中通常能以单倍体细胞存在又能以二倍体形式存在。且单倍体细胞存在两种不同性别,主要包括交配型a(mating-type a,MATa)型与交配型α(mating-type,MATα)型。目前一些性能优良的工业化马克斯克鲁维酵母,如菌株K.marxianus ATCC36534、K.marxianus NRRL Y-6860均以二倍体的形式存在,这大大阻碍了该酵母在合成生物学领域进一步工程化改造和应用。如何开发野生型的二倍体酵母菌株而获得稳定、性能优良的单倍体菌株已成为了打造马克斯克鲁维酵母细胞工厂的核心问题之一。而当前针对马克思克鲁维酵母细胞工厂的开发仍显不足,现有的报道仅停留在野生发酵菌株的选育、常规的敲除和表达工具方法的建立(CN201610944729.3;CN201610944729.3)等。与此同时,马克斯克鲁维酵母主要应用停留在传统食品工业和一些大宗化学品包括乙醇、酶制剂的合成。因此,构建更方便、高效的马克斯克鲁维酵母底盘菌株及开发更多高附加的产品将进一步推动该酵母的工业化应用。
发明内容
本发明所要解决的技术问题是针对现有技术的不足,提供一种马克斯克鲁维单倍体酵母。
本发明还要解决的技术问题是提供上述马克斯克鲁维单倍体酵母的构建方法。
本发明最后要解决的技术问题是提供上述马克斯克鲁维单倍体酵母在实现麦角硫因生物合成领域和实现透明质酸降解酶的胞外分泌表达领域中的应用。
为了解决上述技术问题,本发明公开了采用的技术方案如下:
一种马克斯克鲁维单倍体酵母,所述的马克斯克鲁维单倍体酵母是一种底盘菌株,是以马克斯克鲁维酵母为宿主,在构建MATα3基因缺陷菌株的基础上,通过诱导产孢分离法获得的。
其中,所述的马克斯克鲁维酵母为马克斯克鲁维酵母Kluyvomyces marxianusATCC36534,于2019年9月从北纳生物购买得到。
其中,所述的马克斯克鲁维单倍体酵母的构建方法,包括如下步骤:
(1)MATα3基因缺陷菌株的构建:通过敲除马克斯克鲁维酵母Kluyvomycesmarxianus ATCC 36534的MATα3基因,获得MATα3基因缺陷菌株;
(2)马克斯克鲁维单倍体酵母的构建:将步骤(1)获得的MATα3基因缺陷菌株涂布于诱导产孢培养基中培养,并收集细胞,使用溶壁酶处理收集的细胞获得孢子悬浮液,将孢子悬浮液涂布于YPD平板并验证,得到马克斯克鲁维单倍体酵母细胞。
具体的,步骤(1)是将SEQ ID NO.1-3所示的核苷酸序列克隆到表达质粒pMLG中得到pMLG-sgRNA-UP-DOWN,再将重组质粒pMLG-sgRNA-UP-DOWN通过化学转化法转化至马克斯克鲁维酵母中,得到敲除α3基因的工程菌株。
其中,步骤(2)中,所述的诱导产孢培养基配方为葡萄糖1g/L,KCl 1.8g/L,NaAC8.2g/L,酵母膏2.5g/L,琼脂20g/L。
其中,步骤(2)中,所述的培养,其培养条件为:30℃、培养3-5天。优选的,30℃恒温培养3天。
上述马克斯克鲁维单倍体酵母作为底盘菌株在微生物菌株基因工程菌构建中的应用也在本发明所保护的范围之内。
其中,所述的马克斯克鲁维单倍体酵母作为底盘菌株在构建生物合成麦角硫因的基因工程菌中的应用也在本发明所保护的范围之内。
具体的,通过醋酸锂转化法将麦角硫因合成基因表达质粒转化至马克斯克鲁维单倍体酵母构建生产麦角硫因的基因工程菌,并使用该基因工程菌实现了麦角硫因的发酵制备。
具体的,以粟酒裂殖酵母(Schizosaccharomyces pombe)基因组为模版,利用引物SpEgt1-F和引物SpEgt1-R扩增SEQ ID NO.4所示的SpEgt1的核苷酸序列。将PCR产物SpEgt1与经过限制性内切酶Nde I和Xhol I双酶切后的质粒pRSG,使用一步克隆法在ExnaseⅡ作用下进行连接,得到重组质粒pRSG-SpEgt1并验证。以粟酒裂殖酵母(Schizosaccharomycespombe)基因组为模版,利用引物SpEgt2-F和引物SpEgt2-R扩增SEQ ID NO.5所示的SpEgt2的核苷酸序列。将PCR产物SpEgt2与经过限制性内切酶Sal I和Sma I双酶切后的质粒pRSG-SpEgt1,使用一步克隆法在ExnaseⅡ作用下进行连接,得到重组质粒pRSG-SpEgt1-SpEgt2并验证(图4,图5)。将验证成功后的重组质粒pRSG-SpEgt1-SpEgt2进行质粒抽提,采用醋酸锂转化法转化到筛选得到的马克斯克鲁维酵母单倍体HP11中,将复苏转化液涂布到含有G418(100μg/mL)的YPD琼脂平板,37℃,静置培养12h。从平板上挑取长出来的单菌落观察重组菌菌株菌落形态与菌落PCR验证阳性菌株,即为具有麦角硫因表达质粒的马克斯克鲁维酵母工程菌。
具体的,在马克斯克鲁维酵母工程菌摇瓶发酵考察中,发现在发酵到48h时,OD
具体的,马克斯克鲁维酵母工程菌发酵罐发酵考察中,在10L发酵罐中,经过90h发酵,发酵罐中菌体细胞干重DCW最高可至47.65g/L,发酵液残糖量由200g变为7.8g,麦角硫因的产量为835.6±31.6mg/L。通过基因工程改造马克斯克鲁维酵母单倍体,初步实现了麦角硫因在马克斯克鲁维酵母单倍体HP11中的表达。
其中,所述的马克斯克鲁维单倍体酵母作为底盘菌株在构建胞外分泌表达透明质酸降解酶的基因工程菌中的应用也在本发明所保护的范围之内。。
具体的,通过醋酸锂转化法将透明质酸降解酶基因表达质粒转化至马克斯克鲁维单倍体酵母,并使用该基因工程菌实现了透明质酸降解酶的胞外分泌表达。
具体的,以Kluyveromyces marxianus ATCC 36534基因组为模板,利用引物Afactor-F和引物Afactor-R扩增菊粉酶信号肽的核苷酸序列;将来源于数据库日本医蛭基因组模版DNA的Leecho透明质酸降解酶(NCBI登录号:X4Y2L4.1)的密码子优化后的核苷酸序列委托通用生物(安徽)股份有限公司进行全基因合成,并通过引物Leecho-F和引物Leecho-R进行PCR扩增。将PCR产物α-Leecho与经过限制性内切酶Nde I和Xhol I双酶切后的质粒pRSG,使用一步克隆法在ExnaseⅡ作用下进行连接,得到重组质粒pRSG-α-Leecho并验证。将验证成功后的重组质粒pRSG-α-Leecho进行质粒抽提,采用化学转化法转化到筛选得到的马克斯克鲁维酵母单倍体HP11中,将复苏转化液涂布到含有G418(100μg/mL)的YPD琼脂平板,37℃,静置培养12h。从平板上挑取长出来的单菌落观察重组菌菌株菌落形态与菌落PCR验证阳性菌株,即为具有透明质酸降解酶表达质粒的马克斯克鲁维酵母单倍体工程菌。
具体的,在具有透明质酸降解酶表达质粒的马克斯克鲁维酵母单倍体工程菌产酶发酵考察中,对发酵液中的蛋白含量及酶活进行检测。根据蛋白浓度标准曲线计算发酵液中蛋白含量,发酵液中的胞外蛋白含量为0.297mg/mL,根据酶活计算公式,可计算出酶活2643±21U/mL(图8)。酶活定义如下:给定条件下,每分钟降解透明质酸形成1μmol不饱和双键所需要的酶量。通过基因工程改造,初步实现了日本医蛭透明质酸降解酶在马克斯克鲁维酵母单倍体HP11中的胞外表达。
有益效果:
1、本发明构建了一种获得稳定、性能优良、方便高效的马克思克鲁维单倍体酵母HP11,其作为底盘菌株,可开发更多高附加的产品,将进一步推动该酵母的工业化应用。
2、本发明利用构建的马克思克鲁维单倍体酵母HP11,通过麦角硫因合成途径重构,提供了一种食品级微生物发酵生产麦角硫因的新方法。本发明所述一株稳定高效生产麦角硫因的基因工程菌,经90h发酵后,麦角硫因的产量可达到835.6±31.6mg/L。该菌株安全性高、易于遗传操作、发酵周期短,具有巨大的应用价值和工业化潜力。
2、本发明利用构建的马克思克鲁维单倍体酵母HP11,通过透明质酸降解酶途径重构,提供了一种食品级微生物发酵生产透明质酸降解酶的新方法。本发明所述一株稳定高效生产透明质酸降解酶的基因工程菌,经72h发酵后,发酵液中透明质酸降解酶酶活2643±21U/mL,该菌株安全性高、易于遗传操作、发酵周期短,具有巨大的应用价值和工业化潜力。
附图说明
下面结合附图和具体实施方式对本发明做更进一步的具体说明,本发明的上述和/或其他方面的优点将会变得更加清楚。
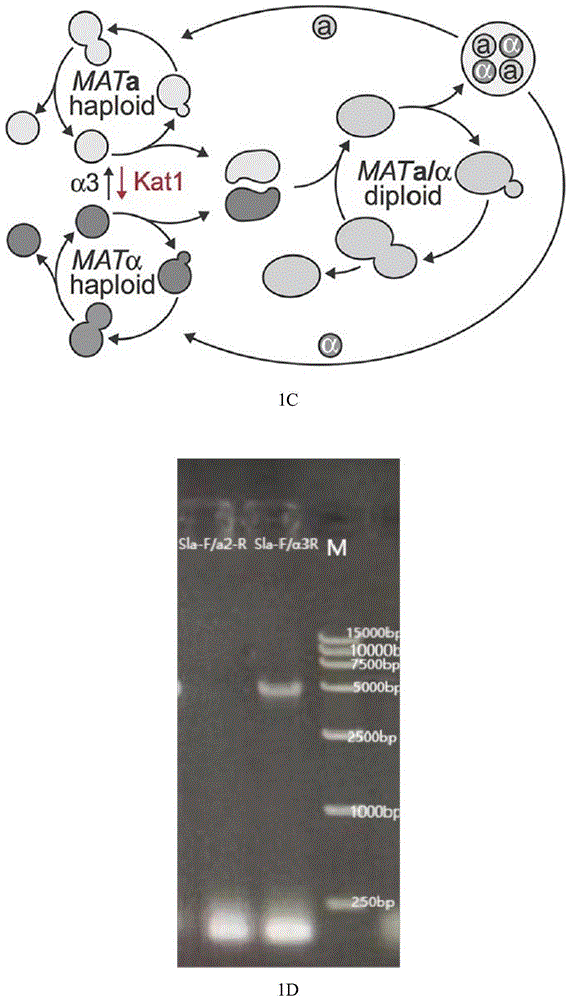
图1 1A为质粒pMLG-sgRNA-UP-DOWN构建过程图;1B为野生菌与敲除株经过PCR验证的电泳检测示意图,注:泳道1:DL 5000Marker;泳道2:野生菌;泳道3:敲除株;1C为马克斯克鲁维酵母配型交替世代图;1D为经过Sla-F/a2-R和Sla-F/α3-RPCR验证的电泳检测图谱,注:泳道1:Sla-F/a2-R;泳道2:Sla-F/α3-R,泳道3:DL 15000Marker。
图2为麦角硫因结构示意图;
图3为麦角硫因在真核生物中的合成途径示意图;
图4 4A为质粒pRSG-SpEgt1-SpEgt2构建过程图(图中Sp1即为SpEgt1,Sp2即为SpEgt2);4B为重组质粒pRSG-SpEgt1-SpEgt2的电泳检测示意图,注:泳道1:DL5000Marker;泳道2:pRSG-SpEgt1-SpEgt2经Nde 1和Xho l酶切验证(目的条带2322bp);泳道3:pRSG-SpEgt1-SpEgt2经Sal l和Sma l酶切验证(目的条带1179bp);
图5为添加甲硫氨酸,组氨酸甜菜碱等前体对工程菌产麦角硫因影响的示意图;
图6为工程菌在10L发酵罐中的麦角硫因产量示意图;
图7为重组质粒pRSG-Leecho的电泳示意图。注:泳道1:DL 5000Marker;泳道2:pRSG-Leecho经过Nde 1和Xho l酶切验证(目的条带1535bp);
图8为工程菌生长量和发酵液中酶活示意图。
具体实施方式
下述实施例中所述实验方法,如无特殊说明,均为常规方法;所述试剂和材料,如无特殊说明,均可从商业途径获得。
主要实验材料和实验试剂盒:E.coli DH5α感受态细胞(南京诺唯赞生物科技有限公司),引物合成(南京金斯瑞生物科技有限公司),DNA测序(生工生物工程(上海)股份有限公司),2×Phanta Max Master Mix(Dye Plus)高保真酶(南京诺唯赞生物科技有限公司),限制性内切酶Sph I,Bcl I,Nde I,Xho I,Sal I,Sma I(Takara),DNA marker(南京诺唯赞生物科技有限公司),质粒提取试剂盒(南京诺唯赞生物科技有限公司),胶回收试剂盒(南京诺唯赞生物科技有限公司),一步克隆连接ExnaseⅡ试剂盒(南京诺唯赞生物科技有限公司),pMLG质粒(由实验室自主构建,在pML质粒的基础上将筛选标记由URA3替换为G418,质粒上原有2μori复制元件替换为自主复制序列ARS1,pMLG质粒的核苷酸序列如SEQ ID NO.8所示),pRSG质粒(由实验室自主构建,在pRS426质粒的基础上,将筛选标记URA3替换为G418,质粒上原有2μori复制元件替换为自主复制序列ARS1,并通过添加酶切位点和T
发酵液中麦角硫因检测方法:产物使用HPLC系统测定,Hypersil ODS C18柱(250mm×4.6mm×5μm);柱温:30℃;流动相:甲醇:水(1:99v/v);流速:0.7mL/min;检测波长:257nm;进样量:5μL;检测时间:20min。
发酵液中透明质酸降解酶酶活测定方法:HAase专一作用于HA的葡萄糖苷酸键,产生具有还原端为葡萄糖醛酸小分子多糖。采用3,5-二硝基水杨酸(DNS)比色法测定水解HA产生的还原糖当量,用分析纯葡萄糖作标准曲线,用考马斯亮蓝法测定蛋白质含量,计算酶比活力。用pH 5.5、50mM的柠檬酸-磷酸氢二钠缓冲液配制0.2%的HA底物,1mL反应体系含有800μL的HA溶液、100μL的酶液,缓冲液补足至1mL。在37℃反应20min,立即沸水终止反应,采用DNS法测定产生的还原糖当量(相当于等量葡萄糖的还原力),计算酶活比。
下述实施例中,所用菌株为马克斯克鲁维酵母(Kluyvomyces marxianus),菌株号:ATCC 36534,于2019年9月从北纳生物购买得到。
文中所述的MATα3基因缺陷菌株与所述的敲除α3基因的工程菌株,二者所表示的意思相同。
实施例1:α3基因敲除菌和单倍体菌株的获得
用于敲除的重组片段由需敲除基因的上下游同源臂组成(上游同源臂-下游同源臂)。将SEQ ID NO.1-3所示的核苷酸序列克隆到表达质粒pMLG中得到pMLG-sgRNA-UP-DOWN,再将重组质粒pMLG-sgRNA-UP-DOWN通过化学转化法转化至马克斯克鲁维酵母中,得到敲除α3基因的工程菌株,具体过程如下:
(1)利用引物UP-F和引物UP-R扩增SEQ ID NO.1所示的UP的核苷酸序列。利用引物DOWN-F和引物DOWN-R扩增SEQ ID NO.2所示的DOWN的核苷酸序列。利用引物sgRNA-F和sgRNA-R扩增SEQ ID NO.3所示的sgRNA的核苷酸序列。
表1实施例1所用引物序列-1
PCR扩增体系为:基因组模版DNA:2μL,引物F和引物R:各2μL,2×Phanta MaxMaster Mix(Dye Plus)高保真酶:12.5μL,ddH
PCR反应程序为:95℃预变性5min,95℃变性30s;然后55℃退火30s,72℃延伸1min,循环35次;回收PCR扩增产物,将三个片段重叠连接;
重叠PCR扩增体系:三种片段各2μL,引物sgRNA-F和DN-R:各2μL,2×Phanta MaxMaster Mix(Dye Plus)高保真酶:12.5μL,ddH
重叠PCR反应程序为:95℃预变性10min,95℃变性30s;然后55℃退火1min,72℃延伸3min,循环35次;回收PCR扩增产物;
将重叠PCR产物与经过限制性内切酶Sph I和Bcl I双酶切后的质粒pMLG,使用一步克隆法在ExnaseⅡ作用下进行连接,得到重组质粒pMLG-sgRNA-UP-DOWN;
(2)将重组质粒pMLG-sgRNA-UP-DOWN转化至感受态大肠杆菌DH5α中,涂布在含有100μg/mL氨苄青霉素的LB固体培养基上,37℃恒温培养12~16h得到初步阳性克隆;分别挑取初步阳性克隆于5mL含有100μg/mL氨苄青霉素的LB液体培养基中,37℃、200rpm培养过夜,提取质粒,经限制性内切酶Sph I和Bcl I双酶切质粒,根据电泳结果判断具有序列表SEQ ID NO:1-3的DNA重叠片段的质粒为重组质粒pMLG-sgRNA-UP-DOWN(图1A)。
(3)将验证成功后的重组质粒pMLG-sgRNA-UP-DOWN进行质粒抽提,采用化学转化法转化到马克斯克鲁维酵母(Kluyveromyces marxianus)菌株中,将复苏转化液涂布到含有G418(100μg/mL)的YPD琼脂平板,37℃,静置培养12h。从平板上挑取长出来的单菌落观察重组菌菌株菌落形态与菌落PCR验证阳性菌株,即为α3基因敲除菌株(图1B),所使用的验证引物为α3Out-F和α3Out-R。
(4)将α3基因敲除株稀释涂布到诱导产孢培养基上,所述的产孢培养基为葡萄糖1g/L,KCl 1.8g/L,NaAC 8.2g/L,酵母膏2.5g/L,琼脂20g/L。30℃恒温培养3天,刮取菌落外围菌泥,无菌水清洗两至三次,用1.2M山梨醇配制100mg/mL蜗牛酶溶液,加入200μL蜗牛酶溶液,37℃水解1h,55℃灭酶10min,用涡旋仪充分震荡5-10min,4000rpm离心5min,用200μL无菌水重悬,稀释涂布到YPD平板,用SLA-F和α3-R鉴定酵母MATα配型,用SLA-F和a2-R鉴定MATa配型。根据图1C的酵母配型世代图可知,在敲除α3基因后,配型为MATα的单倍体会保持MATα。通过PCR鉴定出一株MATα配型的马克斯克鲁维酵母单倍体,如图1D。
表2实施例1所用引物序列-2
实施例2:构建马克斯克鲁维酵母单倍体中麦角硫因的表达途径
麦角硫因(2-巯基-L-组氨酸三甲基内盐,Ergothioneine,EGT)是1909年发现的一种由组氨酸衍生而来的特殊氨基酸,在咪唑环上第二个C原子上连着一个巯基,结构如图2所示。图3展示了麦角硫因在真核生物中的合成途径。在真核生物中,Egt1将三个甲基从S-腺苷甲硫氨酸(SAM)转移到组氨酸中形成组氨酸三甲内盐,并利用氧和半胱氨酸进一步催化组氨酸三甲内盐生成海西烯半胱氨酸亚砜,最后通过Egt2催化生成L-EGT。
(1)构建SpEgt1表达质粒
以粟酒裂殖酵母(Schizosaccharomyces pombe)基因组为模版,利用引物SpEgt1-F和引物SpEgt1-R扩增SEQ ID NO.4所示的SpEgt1的核苷酸序列。
PCR扩增体系为:粟酒裂殖酵母(Schizosaccharomyces pombe)基因组模版DNA:2μL,引物F和引物R:各2μL,2×Phanta Max Master Mix(Dye Plus)高保真酶:12.5μL,ddH2O:6.5μL。
PCR反应程序为:95℃预变性5min,95℃变性30s;然后55℃退火30s,72℃延伸2min30s,循环35次;回收PCR扩增产物。
将PCR产物SpEgt1与经过限制性内切酶Nde I和Xhol I双酶切后的质粒pRSG,使用一步克隆法在ExnaseⅡ作用下进行连接,得到重组质粒pRSG-SpEgt1。
将重组质粒pRSG-SpEgt1转化至感受态大肠杆菌DH5α中,涂布含有100μg/mL氨苄青霉素的LB固体培养基上,37℃恒温培养12~16h得到初步阳性克隆;分别挑取初步阳性克隆于5mL含有100μg/mL氨苄青霉素的LB液体培养基中,37℃、200rpm培养过夜,提取质粒,经限制性内切酶Nde I和Xhol I双酶切质粒,根据电泳结果判断具有序列表SEQ ID NO:4的SpEgt1质粒为重组质粒pRSG-SpEgt1,具有该质粒的菌落为阳性克隆菌株。
(2)构建SpEgt2表达质粒
以粟酒裂殖酵母(Schizosaccharomyces pombe)基因组为模版,利用引物SpEgt2-F和引物SpEgt2-R扩增SEQ ID NO.5所示的SpEgt2的核苷酸序列。
PCR扩增体系为:粟酒裂殖酵母(Schizosaccharomyces pombe)基因组模版DNA:2μL,引物F和引物R:各2μL,2×Phanta Max Master Mix(Dye Plus)高保真酶:12.5μL,ddH
PCR反应程序为:95℃预变性5min,95℃变性30s;然后55℃退火30s,72℃延伸2min30s,循环35次;回收PCR扩增产物。
将PCR产物SpEgt2与经过限制性内切酶Sal I和Sma I双酶切后的质粒pRSG-SpEgt1,使用一步克隆法在ExnaseⅡ作用下进行连接,得到重组质粒pRSG-SpEgt1-SpEgt2(图4,图5)。
表3实施例2所用引物序列
将重组质粒pRSG-SpEgt1-SpEgt2转化至感受态大肠杆菌DH5α中,涂布含有100μg/mL氨苄青霉素的LB固体培养基上,37℃恒温培养12~16h得到初步阳性克隆;分别挑取初步阳性克隆于5mL含有100μg/mL氨苄青霉素的LB液体培养基中,37℃、200rpm培养过夜,提取质粒,经限制性内切酶Sal I和Sma I双酶切质粒,根据电泳结果判断具有序列表SEQ IDNO:5的SpEgt2质粒为重组质粒pRSG-SpEgt1-SpEgt2,具有该质粒的菌落为阳性克隆菌株。
(3)将验证成功后的重组质粒pRSG-SpEgt1-SpEgt2进行质粒抽提,采用醋酸锂转化法转化到筛选得到的马克斯克鲁维酵母单倍体HP11中,将复苏转化液涂布到含有G418(100μg/mL)的YPD琼脂平板,37℃,静置培养12h。从平板上挑取长出来的单菌落观察重组菌菌株菌落形态与菌落PCR验证阳性菌株,即为具有麦角硫因表达质粒的马克斯克鲁维酵母工程菌。
实施例3:马克斯克鲁维酵母工程菌摇瓶发酵考察
将麦角硫因表达质粒的马克斯克鲁维酵母工程菌的甘油管种子接种于含有G418抗生素的种子培养基中(葡萄糖20g/L,酵母粉10g/L,蛋白胨20g/L),于30℃,220rpm过夜培养12-16h,转接到装有50mL发酵培养基的250mL三角瓶中(葡萄糖20g/L,酵母粉10g/L,蛋白胨20g/L),接种量2%v/v,于30℃,220rpm,培养48h。在培养到12h时,向发酵培养基中添加浓度为2-10g/L甲硫氨酸,浓度为10-15g/L组氨酸甜菜碱﹑组氨酸、半胱氨酸。摇瓶发酵结束后,对发酵液的麦角硫因产量进行检测。结果如图5,可知在发酵到48h时,OD
实施例4:工程菌发酵罐发酵考察
从工程菌划线平板上,使用接种环取单菌落接于含有G418抗生素的种子培养基(葡萄糖20g/L,酵母粉10g/L,蛋白胨20g/L),于30℃,220rpm过夜培养12-16h;按5%v/v的接种量接种于种子培养基摇瓶,250ml三角瓶装液50mL,30℃,220rpm摇床过夜,准备种子。将种子液按10%v/v的接种比例,转接至装有发酵培养基(葡萄糖20g/L,酵母粉10g/L,蛋白胨20g/L)的10L发酵罐中。
发酵罐的搅拌转速为450-800rpm,溶氧偶联,溶氧控制在40%,温度为30℃,空气流量3L/min。在发酵至24h,在发酵培养基中添加浓度为5g/L甲硫氨酸、浓度为10g/L组氨酸甜菜碱﹑组氨酸、半胱氨酸。发酵罐发酵结束后,对发酵液的麦角硫因产量进行检测。结果如图6,在10L发酵罐中,经过90h发酵,发酵罐中菌体细胞干重DCW最高可至47.65g/L,麦角硫因的产量为835.6±31.6mg/L。通过基因工程改造马克斯克鲁维酵母单倍体,初步实现了麦角硫因在马克斯克鲁维酵母单倍体HP11中的表达,后续可通过培养基优化,启动子优化等发酵优化技术进一步提高麦角硫因产量。
实施例5:构建马克斯克鲁维酵母单倍体中透明质酸降解酶的表达途径
(1)以Kluyveromyces marxianus ATCC 36534基因组为模板,利用引物Afactor-F和引物Afactor-R扩增菊粉酶信号肽的核苷酸序列;将来源于数据库日本医蛭基因组模版DNA的Leecho透明质酸降解酶(NCBI登录号:X4Y2L4.1)的密码子优化后的核苷酸序列委托通用生物(安徽)股份有限公司进行全基因合成,并通过引物Leecho-F和引物Leecho-R进行PCR扩增。
表4实施例5所用引物序列
PCR扩增体系为:数据库日本医蛭基因组模版DNA:2μL,引物F和引物R:各2μL,2×Phanta Max Master Mix(Dye Plus)高保真酶:12.5μL,ddH
PCR反应程序为:95℃预变性5min,95℃变性30s;然后55℃退火30s,72℃延伸2min30s,循环35次;回收PCR扩增产物。
将回收到的菊粉酶信号肽基因(SEQ ID NO:6)与Leecho降解酶基因(SEQ ID NO:7)重叠,重叠PCR扩增体系为:菊粉酶信号肽基因:2μL;Leecho降解酶基因:2μL,引物F和引物R:各2μL;2×Phanta Max Master Mix(Dye Plus)高保真酶:12.5μL,ddH
PCR反应程序为:95℃预变性10min,95℃变性30s;然后55℃退火1min,72℃延伸2min,循环35次;回收PCR扩增产物。
将PCR产物α-Leecho与经过限制性内切酶Nde I和Xhol I双酶切后的质粒pRSG,使用一步克隆法在ExnaseⅡ作用下进行连接,得到重组质粒pRSG-α-Leecho(图7);
将重组质粒pRSG-α-Leecho转化至感受态大肠杆菌DH5α中,涂布含有100μg/mL氨苄青霉素的LB固体培养基上,37℃恒温培养12~16h得到初步阳性克隆;分别挑取初步阳性克隆于5mL含有100μg/mL氨苄青霉素的LB液体培养基中,37℃、200rpm培养过夜,提取质粒,经限制性内切酶Nde I和Xhol I双酶切质粒,根据电泳结果判断具有序列表SEQ ID NO:6-7的α-Leecho质粒为重组质粒pRSG-α-Leecho,具有该质粒的菌落为阳性克隆菌株。
(2)将验证成功后的重组质粒pRSG-α-Leecho进行质粒抽提,采用化学转化法转化到筛选得到的马克斯克鲁维酵母单倍体HP11中,将复苏转化液涂布到含有G418(100μg/mL)的YPD琼脂平板,37℃,静置培养12h。从平板上挑取长出来的单菌落观察重组菌菌株菌落形态与菌落PCR验证阳性菌株,即为具有透明质酸降解酶表达质粒的马克斯克鲁维酵母单倍体工程菌。
实施例6工程菌产酶发酵考察
将具有透明质酸降解酶表达质粒的马克斯克鲁维酵母单倍体工程菌的甘油管种子接种于抗性种子培养基(葡萄糖20g/L,酵母粉10g/L,蛋白胨20g/L),于30℃,220rpm过夜培养,转接到装有50mL发酵培养基的250mL三角瓶中(葡萄糖20g/L,酵母粉10g/L,蛋白胨20g/L),接种量2%v/v,于30℃,220rpm,培养72h。摇瓶发酵结束后,对发酵液中的蛋白含量及酶活进行检测。根据蛋白浓度标准曲线计算发酵液中蛋白含量,发酵液中的胞外蛋白含量为0.297mg/mL,根据酶活计算公式,可计算出酶活2643±21U/mL(图8)。酶活定义如下:给定条件下,每分钟降解透明质酸形成1μmol不饱和双键所需要的酶量。通过基因工程改造,初步实现了日本医蛭透明质酸降解酶在马克斯克鲁维酵母单倍体HP11中的胞外表达,后续可通过信号肽优化,培养基优化等进一步提高透明质酸降解酶的产量。
本发明提供了一种马克斯克鲁维单倍体酵母及其构建方法与应用的思路及方法,具体实现该技术方案的方法和途径很多,以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。本实施例中未明确的各组成部分均可用现有技术加以实现。
- 一种乙酸和木糖共利用的马克斯克鲁维酵母菌株及筛选方法
- 一种抑制马克斯克鲁维酵母目的基因的表达的载体
- 一种马克斯克鲁维酵母培养物及其制备方法和应用
- 一种马克斯克鲁维酵母启动子及其制备方法与应用