一种用于原位合成核酸的固相载体的制备方法与应用
文献发布时间:2023-06-19 19:28:50
技术领域
本发明属于寡核苷酸原位合成技术领域,更具体的,本发明涉及用于寡核苷酸原位合成的固相载体的制备技术。
背景技术
目前,商业化的寡核苷酸合成仪为柱式合成仪,此种仪器合成方法是将商品化的固相无机颗粒载体,例如CPG载体,填充于反应柱中,然后进行自动化合成。随着对原位合成核酸序列的需要,此种寡核苷酸的柱式合成仪的缺点也越来越凸显,寡核苷酸柱式合成仪主要存在两方面问题:一是由于填充柱的局限性导致了合成仪的通量较小,单位时间内合成寡核苷酸序列种类比较少,已经不能满足目前的市场需求;另一方面是由于现有固相载体CPG制备技术的缺陷导致。
现阶段商品化的CPG载体主要有三部分组成:一是固相基底材料多孔玻璃;二是用于核酸起始步骤合成的带有羟基保护基的可裂解连接物,可以是带有羟基保护基的单体或其它通用结构载体等;三是用于连接固相基底和可裂解连接物的接头,如:丁二酸酐等。由于目前商品化的CPG仅在多孔玻璃上实现,受多孔玻璃本身对核酸的吸附作用影响,利用现有的商品化CPG在核酸合成过程中,存在核酸合成链越长,核酸合成能力降低的缺陷虽然现专利CN110511973A中公开了一种用于核酸原位合成的固相载体及其制备方法,其所制备的载体的载量虽然相比于市面上CPG载体的载量较高,但是其载量仍然需要进一步的提高。
发明内容
针对上述问题本发明提供了一种新型疏水性官能团与功能化官能团混合接枝方式连接固相基底材料和可裂解连接物,通过对试剂用量的优化能够有效提高CPG载体载量以及核酸产物的纯度,且本发明的新连接方法不限于仅用多孔玻璃作为固相载体的制备基底材料,其固相基底材料可以为多孔玻璃、玻片,也可以为聚苯乙烯微球、磁性颗粒、陶瓷、有机玻璃等材质作为固相基底。另外本发明还可应用于未来基于片基固相基底研发的高通量核酸合成仪的开发应用。
本发明的目的在于克服现有原位合成核酸技术中,固相载体制备技术存在核酸合成链越长,核酸合成能力降低、合成产物纯度降低的缺陷。
为了实现上述发明目的,本发明提供以下技术方案:
本发明提供了一种新型用于核酸原位合成的固相载体制备技术,利用一种新的疏水性官能团与功能化官能团混合接枝方式,可连接多种固相基底和用于核酸起始步骤合成的可裂解连接物,可以有效选择多种固相基底材料,进而可选择性的优化提高核酸原位合成的效率和核酸产物的纯度。
本发明用于核酸原位合成固相载体的制备路线包括:活化固相基底材料,疏水性官能团混合接枝,将带有保护基的可裂解连接物连接至接枝的固相载体上
优选的,所述活化固相基底材料是将固相基底材料羟基化。
优选的,所述固相基底材料为玻璃、石英、陶瓷、聚苯乙烯以及硅材质的片基或微球,多孔微珠以及磁性Fe
优选的,所述固相基底材料的形状不受特别限定。
优选的,所述固相基底材料的形状是片基、微球状和多孔微珠中的一种或多种。
优选的,所述羟基化的方法包括食人鱼溶液,浓碱溶液,等离子清洗活化。
优选的,所述疏水性官能团混合接枝使用的是疏水性试剂与氨基硅烷偶联剂,配比为1~1:10。
优选的,所述疏水性试剂包括氟硅烷偶联剂,烷基硅烷偶联剂,苯基硅烷偶联剂。
优选的,所述疏水性官能团包括-CH
优选的,所述固相基底材料为片基时,相对于单位面积所需的混合接枝试剂浓度为0.1%-30%,更优选浓度为1%-5%。
优选的,所述固相基底材料为微球或微珠时,大小为1μm-1000μm,更优选大小为50μm-150μm。
优选的,所述固相基底材料为多孔微珠时,多孔微珠的孔径大小为
优选的,所述固相基底材料为多孔微珠时,相对于多孔微珠的用量来说,所述接枝试剂用量为100μmol/g-100mmol/g,更优选的用量为500μmol/g-5mmol/g。
优选的,所述可裂解连接物包括核苷羟基连接物,具有以下结构式:
上式中R3表示的基团包括碱基和通过保护基来保护氨基的碱基;
R4表示的基团包括氢原子、羟基、卤素原子、具有1-20个碳原子的烷氧基和被叔丁基二甲基硅烷基保护的羟基。
优选的,所述碱基包括核酸碱基,所述保护基包括氨基乙酰基、异丁酰基、苯甲酰基。
另一方面,本发明还提供了一种高效原位合成核酸的反应体系。通过对上述方法制备的固相载体表面载量进行优化,得到一套可以高效原位合成核酸的反应体系。
所述固相载体的可裂解连接物的负载量为1-150μmol/g;优选1-100μmol/g。
另一方面,本发明还提供了一种寡核苷酸合成的固相载体的应用,所述固相载体用于核酸合成起始步骤,在接头修饰载体上负载有可裂解连接物,该可裂解连接物是作为核酸合成反应的起点化合物,其在加热及碱性等条件下可裂解,用来从接头修饰载体上切割合成的核酸。
所述固相载体的应用是在缩合剂的存在下通过脱水缩合反应将可裂解的连接物与固相载体共价键合。
优选的,所述脱水缩合反应在对反应惰性的溶剂中进行,所述包括卤化烃溶剂、芳香族溶剂、脂族烃类溶剂,腈溶剂。
优选的,所述溶剂为芳香族溶剂吡啶。
优选的,所述缩合剂包括二环己基碳二亚胺(DCC)、4-二甲氨基吡啶(DMAP)、二异丙基碳二亚胺(DIC)、N-乙基-N'-3-二甲基氨基丙基碳二亚胺或其盐酸盐(EDC·HCl)。
更优选的,所述缩合剂为二环己基碳二亚胺(DCC)和4-二甲氨基吡啶(DMAP)。
本发明的有益效果:
(1)本发明通过对固相基底表面采用疏水官能团与亲水官能团的混合改性,并且改进固相基底与连接单体之间的新方法的接枝,进而可以选择多种材质作为固相基底材料,从而可选择性的优化提高核酸原位合成效率和纯度。
(2)本发明所用的接头试剂为疏水性修饰试剂和功能化修饰试剂,通过对固相基底材料的表面官能团混合修饰,从而降低了固相基底材料表面分子间的电子干扰效应以及降低化学反应的空间位阻效应,从而通过对试剂用量的优化达到了优于商品化CPG载体的核酸合成效果。
附图说明
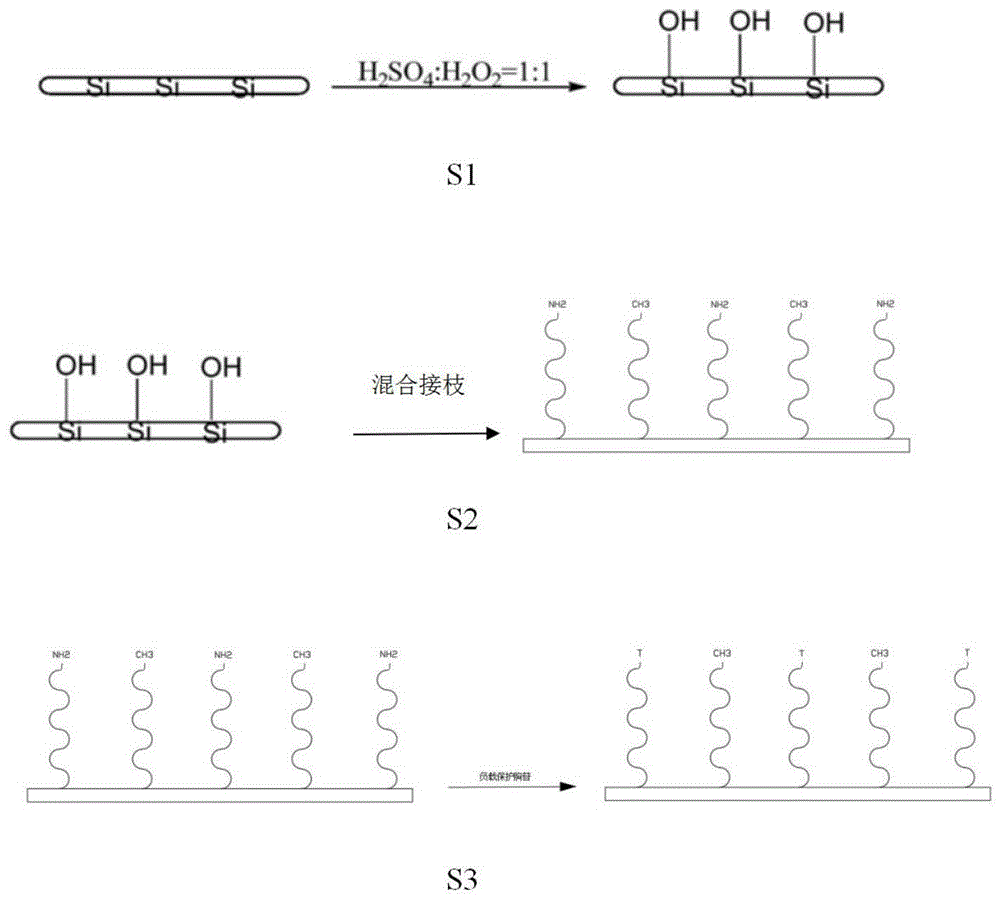
图1:S1活化固相基底材料;S2活性官能团与疏水性官能团的混合接枝产物;S3将带有保护基的可裂解连接物连接至接枝的固相载体上。
图2:根据本发明自制CPG载体合成的核酸产物的质谱图和相应的纯度数据。
图3:商品化的CPG载体合成的核酸产物的质谱图和相应的纯度数据。
具体实施方式
本发明提供了一种新型用于核酸原位合成的固相载体制备技术,利用一种新的疏水性官能团与功能化官能团混合接枝方式,可连接多种固相基底和用于核酸起始步骤合成的可裂解连接物,可以有效选择多种固相基底材料,进而可选择性的优化提高核酸原位合成的效率和核酸产物的纯度。
在本发明中,若无特殊说明,所有的原料组分均为本领域技术人员熟知的市售商品。
本发明用于原位合成核酸的固相基底材料性状不受特别限定,可以是片基、微球状、多孔微珠等。
本发明用于原位合成核酸固相基底材料的羟基化修饰方法不受限定可使用食人鱼溶液,浓碱溶液,等离子清洗活化等方案。
本发明所用混合接枝试剂不受限定,可以是氟硅烷偶联剂,烷基硅烷偶联剂,苯基硅烷偶联剂等疏水性试剂与氨基硅烷偶联剂混合配比。
本发明用于原位合成核酸的固相基底材料为片基时,相对于单位面积所需的混合接枝试剂浓度为0.1%-30%,优选1%-5%。当混合接枝试剂浓度过低时,基片表面活性官能团较少,产物量较低;当混合接枝试剂浓度过高时,其表面活性官能团较多,可连接分子密度过高,受表面空间位阻效应和电子效应的影响使得产物纯度降低,合成效率下降。
本发明所述混合接枝试剂,疏水性试剂与氨基硅烷偶联剂的配比为1:1~10,其中当混合接枝试剂配比为1:2~3为最佳,得到基片表面疏水官能团与活性官能团的分散度较好,进行后续修饰合成试验后,产物质量高。
本发明用于原位合成核酸的固相基底材料为微球或微珠时,优选利用多孔微珠,多孔微珠的大小为1μm-1000μm,优选50μm-150μm;多孔微珠的孔径大小为
本发明所用的接头试剂为疏水性修饰试剂和功能化修饰试剂,通过对固相基底材料的表面官能团混合修饰,从而降低了固相基底材料表面分子间的电子干扰效应以及降低化学反应的空间位阻效应,从而通过对试剂用量的优化达到了优于商品化CPG载体的核酸合成效果。
相对于多孔微珠的用量来说,本发明所用的接头修饰试剂用量为100μmol/g-100mmol/g,优选为500μmol/g-5mmol/g。当接头修饰试剂用量低于这一范围时,表面修饰效率不高,载量过低;当接头修饰试剂用量高于这一范围时,表面核酸合成能力降低,产物纯度降低。
本发明用于核酸合成起始步骤,在接头修饰载体上负载有可裂解连接物,该可裂解连接物是作为核酸合成反应的起点化合物,其在加热及碱性等条件下可裂解,以便于从接头修饰载体上切割合成的核酸。
本发明用于核酸合成的固相载体的可裂解连接物的例子,包括不限于如下所示的核苷羟基连接物,可以使用结合有各种修饰基的核苷连接物和不包含核苷的普通连接物。
上式中R3表示碱基,可以采用核酸碱基(例如:腺嘌呤、鸟嘌呤、胞嘧啶、胸腺嘧啶、尿嘧啶等),并且上述核酸碱基可以通过乙酰基、异丁酰基、苯甲酰基等的保护基来保护氨基。
上式中R4可以采用氢原子、羟基、卤素原子(例如氟原子、氯原子、溴原子等)、具有1-20个碳原子的烷氧基(例如甲氧基等)、被叔丁基二甲基硅烷基等保护的羟基和其类似基团。
本发明用于寡核苷酸合成的固相载体的可裂解连接物的负载量为1-150μmol/g,优选1-100μmol/g,当连接物的负载量少于1μmol/g时,可合成的核苷酸的量变得很小;当连接物的负载量超过150μmol/g时,寡核苷酸的合成能力,与不含长链有机载体的用于寡核苷酸的合成的固相载体相比,没有显示出不同。
在用于本发明寡核苷酸合成的固相载体上负载可裂解的连接物的结合方法为脱水缩合反应,在缩合剂的存在下通过脱水缩合反应而共价键合。
该脱水缩合反应在对反应惰性的溶剂中进行,只要反应能进行,该溶剂没有特别限定,可以为卤化烃溶剂、芳香族溶剂、脂族烃类溶剂,腈溶剂等。其中优选芳香族溶剂吡啶。
缩合剂的例子包括二环己基碳二亚胺(DCC)、4-二甲氨基吡啶(DMAP)、二异丙基碳二亚胺(DIC)、N-乙基-N'-3-二甲基氨基丙基碳二亚胺或其盐酸盐(EDC·HCl)等,其中优选二环己基碳二亚胺(DCC)和4-二甲氨基吡啶(DMAP)。
通过上述反应可以获得本发明用于原位合成核酸的固相载体。
利用已知方法对本发明制备的固相载体进行载量验证。
下面将结合本发明中的实施例,对本发明中的技术方案进行清楚、完整地描述。显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
实施例1
自制CPG修饰:
1、羟基化处理:取0.3g空白CPG置于2mL离心管中,向其中加入1.5mL浓硫酸:双氧水=7:3的混合溶液,然后将其置于恒温混匀仪上,50℃恒温混匀反应1h,反应完成后用去离子水多次离心清洗,真空干燥备用;
2、官能团混合接枝:向上述处理后的离心管中分别加入25μl甲基硅烷偶联剂,75μL氨基硅烷偶联剂,1700μL无水乙醇。将其置于恒温混匀仪50℃恒温混匀反应24h,反应完成后用去离子水多次离心清洗,真空干燥备用;
3、功能化修饰:向上述离心管中加入100mg带羧基保护胸苷,1.5mL吡啶,置于恒温混匀仪上,50℃恒温混匀反应24h,反应完成后采用乙腈进行多次离心清洗,真空干燥即可;
4、利用紫外分光光度计进行DMT脱保护检测。
实施例2
利用玻片作为固相基底进行制备。
1、羟基化处理:取两张石英玻片置于加工好的反应器皿中,向其中加入20mL浓硫酸:双氧水=7:3的反应溶液,在70℃超声条件下反应3h,去离子水清洗,氮气吹干备用;
2、官能团混合接枝:向上述处理完的石英玻片中分别加入0.25mL甲基硅烷偶联剂,0.75mL氨基硅烷偶联剂,19mL无水乙醇,在50℃超声条件下进行反应24h,反应完成后用去离子水清洗,氮气吹干,并置于110℃烘箱中过夜固化,得到活性氨基化玻片;
3、功能化修饰:将混合官能团接枝玻片置于玻片反应器皿中,向其中加入500mg丁二酸干、20mL吡啶,室温下超声处理24h,乙腈清洗,然后向其中加入500mg保护胸苷、500mgDCC以及20mL吡啶,室温下超声处理48h,得到负载保护胸苷的石英芯片
4、利用紫外分光光度计进行DMT脱保护检测。
实施例3
利用硅片作为固相基底进行制备:
1、羟基化处理:取20张硅片置于等离子清洗机(CPC10-018)中进行表面羟基化,等离子清洗机设定参数为功率300W,时间3min;
2、官能团混合接枝:将上述处理完的硅片置于pp反应器皿中,向其中分别加入2mL甲基硅烷偶联剂,8mL氨基硅烷偶联剂,200mL无水乙醇,在50℃超声条件下进行反应24h,反应完成后用去离子水清洗,氮气吹干,并置于110℃烘箱中过夜固化,得到活性氨基化玻片;
3、功能化修饰:将混合官能团接枝玻片置于玻片反应器皿中,向其中加入1g丁二酸干、200mL吡啶,室温下超声处理24h,乙腈清洗,然后向其中加入1g保护胸苷、1g DCC以及200mL吡啶,室温下超声处理48h,得到负载保护胸苷的硅基芯片;
4、利用紫外分光光度计进行DMT脱保护检测。
实施例4
利用磁性颗粒Fe
1、Fe
2、Fe
3、Fe
4、向上述处理后的离心管中其中加入50mg丁二酸干,1.5mL吡啶,室温下置于恒温混匀仪混匀反应24h,乙腈清洗,然后向其中加入136mg保护胸苷,100mg DCC和1.5mL吡啶,室温下混匀反应48h,得到负载保护胸苷的Fe
5、利用紫外分光光度计进行DMT脱保护检测。
实施例5
利用紫外分光光度计(TFN UV6800S)对实施例1、实施例2、实施例3以及实施例4进行DMT脱保护检测。
a、取实施例1制备好的CPG载体10mg置于2mL离心管中,向其中加入商品化的TCA试剂浸泡2h进行脱DMT处理,收集浸泡液置于15mL离心管中,然后用乙腈清洗3次,同样将3次清洗液收集至装有浸泡液的15mL离心管中,向其中加入1%的对甲苯磺酸乙腈溶液,定容至5mL。
b、取实施例2制备好的玻片一张置于玻片反应盒中,向其中加入商品化的TCA试剂浸泡2h进行脱DMT处理,收集浸泡液置于15mL离心管中,然后用乙腈清洗3次,同样将3次清洗液收集至装有浸泡液的15mL离心管中,向其中加入1%的对甲苯磺酸乙腈溶液,定容至5mL。
c、取实施例3制备好的硅基芯片一张置于玻片反应盒中,向其中加入商品化的TCA试剂浸泡2h进行脱DMT处理,收集浸泡液置于15mL离心管中,然后用乙腈清洗3次,同样将3次清洗液收集至装有浸泡液的15mL离心管中,向其中加入1%的对甲苯磺酸乙腈溶液,定容至5mL。
d、取实施例4制备好的磁珠10mg置于2mL离心管中,向其中加入商品化的TCA试剂浸泡2h进行脱DMT处理,收集浸泡液置于15mL离心管中,然后用乙腈清洗3次,同样将3次清洗液收集至装有浸泡液的15mL离心管中,向其中加入1%的对甲苯磺酸乙腈溶液,定容至5mL。
e、取商品化的CPG载体10mg置于2mL离心管中,向其中加入商品化的TCA试剂浸泡2h进行脱DMT处理,收集浸泡液置于15mL离心管中,然后用乙腈清洗3次,同样将3次清洗液收集至装有浸泡液的15mL离心管中,向其中加入1%的对甲苯磺酸乙腈溶液,定容至5mL。
将上述b、c、d收集回来的液体稀释400倍进行分光光度计498nm处吸光度检测,a的收集液可以直接用来检测。检测结果如下:
通过上述检测结果可知,实施例1、实施例2、实施例3以及实施例4均制备成功;通过对比a和e的吸光值可知,实施例1所制备的CPG载体要比商品化的CPG载体载量高;其中d的吸光值最高,是因为自己制备的磁珠要比CPG小,所以比表面积大载量大;b为实施例2的玻片,c为实施例3硅基芯片,不管是玻片还是硅基芯片均采用长75mm、宽25mm的规定尺寸,其表面为单分子层,相对于微粒状的CPG和磁颗粒比表面积小很多,所以吸光值较小。
实施例6
核酸合成(长度40bp):
分别称取本发明实施例1所制备的CPG载体30mg以及商品化的CPG载体30mg,填充于柱式合成仪(LK-192X)进行寡核苷酸合成。
剪切脱保护:
将合成结束的CPG置于1.5mL螺帽管内,800mL浓氨水65℃处理4h,弃去CPG,取上清液,65℃加热真空浓缩干,加入TE buffer(PH7.5)200μl,室温静置1hours,使剪切后的oligo充分溶解,尽可能的取净上部溶液,置于新2mLEP管内,弃去原管及其内的不溶物。
脱盐纯化:
向待纯化的DNA内,加入1/10DNA溶液体积(20μL)的醋酸钠(3mol/L PH5.2),充分混匀;加入220μL冰预冷的乙醇,充分混合后,置于-20℃中至少20min后,12000g离心10min,移去上清,尽可能的吸去管壁液滴,然后加入1mL 70%乙醇,12000g离心2min,移去上清,尽可能的吸去管壁液滴,室温下敞口,待其挥发干,再向其中加入100mL TE buffer溶解DNA沉淀(4℃,过夜)。
定量:
将得到的核酸产物稀释100倍用NanoDrop2000进行定量,定量结果如下:
从定量结果得知自制CPG载体载量要比商品化CPG载体载量要高,与分光光度计检测结果一致。
质检:
利用质谱仪对实施例1自制CPG载体的核酸负载量进行质检分析,得到如图2所示。
利用质谱仪对商品化CPG载体的核酸负载量进行质检分析,得到的质谱图如图3所示。
由图2和图3的质谱图和相应纯度数据可知,自制CPG载体得到的产物纯度为84.75%,商品化CPG载体得到的产物纯度为63.36%,从质谱图上亦可看出自制CPG得到的产物杂峰较低且较少。所以本发明的新型CPG连接方法优于现有商品化的CPG制备方式。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 一种原位制备离子液体杂化二氧化硅气凝胶涂层固相微萃取纤维的方法
- 一种用于核酸原位合成的固相载体及其制备方法
- 一种用于核酸原位合成的固相载体及其制备方法