一种炔丙醚基取代TPT3型非天然碱基三磷酸及其合成方法和应用
文献发布时间:2023-06-19 19:28:50
技术领域
本发明属于非天然碱基的合成技术领域,具体涉及一种炔丙醚基取代TPT3型非天然碱基三磷酸及其合成方法和应用。
背景技术
碘代TPT3型碱基在实验过程中显示出不稳定性,该化合物在进行硫代反应时更是显示出了极低的转化率及产率。非天然碱基的后修饰研究在生物活性研究中占据了十分重要的地位,合成一种含有后修饰位点的非天然碱基具有特别的意义。
设计合成结构多样性的非天然碱基核苷酸是构筑功能核酸和开展基因密码子扩增研究的化学基础。但是传统的非天然碱基核苷酸的化学合成路线冗长,涉及到条件苛刻的糖基化反应、磷酸化反应和焦磷酸化反应。近年来,基于后功能化修饰的合成策略为复杂生物分子的研究提供一种简洁、高效的化学工具。
发明内容
本发明解决的技术问题是提供了一种炔丙醚基取代TPT3型非天然碱基三磷酸及其合成方法和应用,首次以后功能化修饰合成策略为工具,设计合成了包含炔丙基醚的TPT3型核苷三磷酸关键中间体,进而通过click反应和酰胺化反应对其衍生化,并利用酶促反应动力学、PCR扩增筛选获得了系列衍生物具有良好体外复制效率。
本发明为解决上述技术问题采用如下技术方案,一种炔丙醚基取代TPT3型非天然碱基三磷酸,其特征在于其结构式为:
本发明所述的炔丙醚基取代TPT3型非天然碱基三磷酸的制备方法,其特征在于具体步骤为:
步骤S1:以碘代TPT3型非天然碱基为反应原料,重蒸甲苯为溶剂,劳森试剂为硫代试剂,氮气保护下于120℃回流反应制得含硫代羰基的碘代TPT3型非天然碱基b,合成过程中对应的反应方程式如下:
步骤S2:以含硫代羰基的碘代TPT3型非天然碱基b和炔丙基醚为反应原料,重蒸DMF为溶剂,碘化亚铜与四(三苯基膦)钯共同催化,三乙胺提供碱性环境,在氮气保护下于65℃油浴条件下反应制得化合物c,合成过程中对应的反应方程式如下:
步骤S3:以化合物c和甲醇钠为反应原料,甲醇为溶剂,于室温脱去羟基保护基制得化合物d,合成过程中对应的反应方程式如下:
步骤S4:以化合物d、三氯氧磷和焦磷酸盐为反应原料,磷酸三甲酯为溶剂,三氯氧磷作为单磷酸化试剂,在氮气保护下于-15℃冰盐浴反应,焦磷酸盐用N,N-二甲基甲酰胺作溶剂溶解后加入进一步三磷酸化制得目标产物炔丙醚基取代TPT3型非天然碱基三磷酸d,合成过程中对应的反应方程式如下:
进一步限定,步骤S4中所述焦磷酸盐为三(四丁基胺)氢焦磷酸。
本发明所述的炔丙醚基取代TPT3型非天然碱基三磷酸的制备方法,其特征在于合成路线如下:
本发明所述的炔丙醚基取代TPT3型非天然碱基三磷酸在核酸标记和DNA数据存储中的应用。
本发明与现有技术相比具有以下优点和有益效果:本发明提供了碘代TPT3型非天然碱基的羰基氧转硫试验方案;含硫代羰基的碘代TPT3型非天然碱基的偶联反应优化条件;以及一种稳定的含炔丙基醚修饰的TPT3型非天然碱基三磷酸的合成。优化后的反应方案产率更高,含有端基炔的修饰位点的三磷酸的合成可以方便地实现非天然碱基三磷酸的结构多样化修饰,并利用酶促反应动力学、PCR扩增筛选获得了系列修饰衍生物具有良好体外复制效率,能够进一步在核酸标记和DNA数据存储中应用。
附图说明
图1是偶联反应的条件筛选图;
图2是偶联反应的温度筛选图;
图3是复制性能曲线。
具体实施方式
以下通过实施例对本发明的上述内容做进一步详细说明,但不应该将此理解为本发明上述主题的范围仅限于以下的实施例,凡基于本发明上述内容实现的技术均属于本发明的范围。
实施例1
规避碘代不稳定性问题及硫代条件优化探究
从上述研究中对比能够得出,在不同的条件下,产物分解的速度并不相同,通过优化实验条件在最大的产率下得到产物。
(1)反应溶剂的筛选
根据查阅文献,不同的反应溶剂可能对反应产生影响,我们首先对反应溶剂进行了筛选。氮气保护的条件下,选用了甲苯、四氢呋喃、二氯作为溶剂分别在其沸点条件下进行回流1h,检测分析。
(2)反应温度的优化
鉴于该化合物不稳定性,我们对反应温度进行了优化,尝试降低反应温度,分别在60℃、90℃、120℃,但是该化合物在60℃、90℃的条件下并未表现出好的转化率,在同样的时间条件下三种温度下的转化率分别为5%、8%、18%。故而选用120℃作为接下来实验的反应温度。
(3)反应时间的优化
我们选用甲苯作为溶剂在120℃及氮气保护的条件下对不同时间下的反应体系进行分析,通过对比产物的比例确认最佳反应时间,分别选用了30min、1h、1.5h、2h、3h、5h六个时间点,通过TLC检测分析数据(表1)。随这反应时间的延长,产物的比率在逐步但是同时生成了更多的副产物,考虑到最终在分离过程中的损耗,我们综合考虑采用了2h的反应时间。
表1不同时间反应体系中各组分所占比例
产物与副产物生成方程式
(4)柱层析分离优化
反应处理是及其重要的一步,产物在硅胶上的不稳定性提醒我们该化合物的纯化是一个需要重点攻克的难关。
首先对不同的上样方式进行了对比,采用反应体系直接上样与拌硅胶后干法上样两种方法进行对比。分别平行取(20mg,0.03mmol)原料和(14.6mg,0.036mmol)劳森试剂,真空干燥2h后在氮气保护下加入甲苯于120℃条件下回流2h后分离。
得到产物后称重,直接上样与干法上样分别得到5.3mg和2.5mg,产率分别为27.4%与12.9%。直接上样减少了样品与硅胶接触的时间,同时甲苯也在产物与杂质的分离过程中起到了正向作用。
不同颗粒度硅胶及避光条件对收率的影响
平行4份(20mg,0.03mmol)碘代TPT3型非天然碱基和(14.6mg,0.036mmol)劳森试剂,真空干燥2h后在氮气保护下加入甲苯于120℃回流2h后,直接上样,分别在避光-200目硅胶,避光-400目硅胶,不避光-200目硅胶,不避光-400目硅胶四种条件下分离得到产物的产率分别为29.3%、26.2%、26.5%、23.3%。
碘代的TPT3型化合物的不稳定性与其光敏性以及键的不稳定性有密切的关系,其对保存环境也有着较差的耐受性。我们在合成过程中通过对比研究,在不同的反应条件以及分离条件对其进行优化,最终得出,甲苯作为反应溶剂,反应时间为2h时,转化率达到35%。分离纯化过程中采用200目硅胶,湿法上样,在避光条件下进行分离,能够以29.3%的产率得到含有硫代羰基的碘代TPT3型非天然碱基。
实施例2
偶联反应条件的优化
(1)反应溶剂的筛选
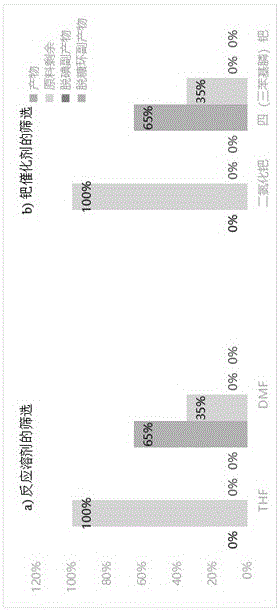
反应溶剂对反应体系有着极大的影响,常见的偶联反应体系为THF或DMF,我们选用这两种溶剂做不同的反应体系。平行取含硫代羰基的碘代TPT3型非天然碱基(20mg)于25mL圆底烧瓶中,按顺序加入四(三苯基膦)钯、碘化亚铜、三乙胺、炔丙基醚(该反应要及其注意无水无氧的控制)。随后在35℃油浴条件下搅拌反应。取反应体系监测,THF反应体系没有产物生成全部为原料剩余(图1中a),故而选用DMF反应体系进一步探究其他影响因素。
(2)钯催化剂的筛选
我们在两种不同的钯催化剂的催化下平行取含硫代羰基的碘代TPT3型非天然碱基(20mg),按顺序加入不同的催化剂(四(三苯基膦)钯或者二氯化钯和碘化亚铜)、三乙胺、炔丙基醚化合物与炔丙基醚在氮气保护下严格除水后于35℃条件下进行偶联反应。通过TLC检测,二氯化钯催化作用下并无产物点生成。四(三苯基膦)钯催化剂的条件下有产物(65%)生成,同时有仍有原料剩余(35%),低反应温度下反应不完全但副产物生成减少(图1中b)。
(3)反应温度的调节
反应温度对反应速率有较大的影响,我们在四(三苯基膦)钯的催化下,分别在35℃、50℃、65℃、75℃的条件下进行了催化反应,在65℃的条件下进行的反应在3h时能够反应完全,有5%的脱碘副产物的生成,温度较低的情况下有原料的剩余(图2,表2)。
表2偶联反应条件优化
实施例3
不同的末端炔烃以及不同卤素取代底物进行Sonogashira偶联反应
方案a:取含硫代羰基的碘代TPT3型非天然碱基(60mg,0.09mmol)于25mL圆底烧瓶中,真空条件下干燥后,按照顺序加入碘化亚铜(34.4mg,0.18mmol),四(三苯基膦)钯(20.8mg,0.018mmol),放置于干燥罐中继续干燥2h,随后加入DMF(N,N-二甲基甲酰胺)以及氮气保护(该反应要及其注意无水无氧的控制)。将三乙胺和炔丙基醚分别溶于250μL DMF中然后按照顺序注入反应体系。随后在65℃油浴条件下搅拌反应,3h检测一次,约3h反应完全。柱层析分离得到产物30mg,产率为58.1%。
方案b:取反应物、催化剂等,具体操作步骤同上,将反应物炔丙基醚换为三甲基硅基乙炔溶于250μL DMF中然后注入反应体系。随后在65℃油浴条件下搅拌反应3h检测。
方案c:取含硫代羰基溴代TPT3非天然碱基作为反应物(20mg,0.08mmol)于25mL圆底烧瓶中,真空条件下干燥后,按照顺序加入碘化亚铜,四(三苯基膦)钯,放置于干燥罐中继续干燥2h,随后加入DMF(N,N-二甲基甲酰胺)以及氮气保护(该反应要及其注意无水无氧的控制)。将三乙胺和炔丙基醚分别溶于250μLDMF中然后按照顺序注入反应体系。随后在65℃油浴条件下搅拌反应,检测反应,并无产物生成。
偶联反应底物筛选方程式
硫对钯催化剂的毒化作用使含有硫羰基的碘代化合物的偶联过程需要注意的细节较多,首先对副产物生成的条件探究过程中发现,在水以及有氧气的的情况下副产物生成,在反应过程中严格控制反应条件。通过筛选不同的反应溶剂,反应温度,反应时间。我们最终选用了以DMF为反应溶剂,四(三苯基膦)钯作为催化剂,反应温度为65℃,反应3h,能够以65%的产率得到碘代TPT3保护核苷与炔丙基醚的反应产物。
底物活性影响反应的进行,实验中设计了三种带有端基炔的产物,但是方案b的产物稳定性较差。方案c中采用的底物是同实验室其他工作人员设计合成的非天然碱基,考虑到其他可能性我们也对该化合物的后修饰进行了尝试,但反应并不成功。从反应的可进行性以及产物的稳定性考虑,选用方案a在进一步的三磷酸底物合成过程中使用。
实施例4
取β构型糖基化产物(1g,1.98mmol)于圆底烧瓶中,加氮气保护后,重蒸二氯甲烷溶解,然后取氯化亚碘溶解于二氯甲烷中注入反应瓶中,室温条件下反应6h后,用饱和硫代硫酸钠淬灭反应后,二氯甲烷与饱和食盐水萃取,取有机相旋干后柱层析分离,得到纯净的米黄色产物a 800mg,产率为64.5%。
实施例5
称取化合物碘代产物a(100mg,0.16mmol),劳森试剂(77.2mg,0.19mmol)于圆底烧瓶中加回流管后真空干燥,加氮气保护,保证反应体系无水无氧,有利于反应的正向进行。最后将反应瓶置于120℃油浴锅中,搅拌回流两小时,直接上样过柱。得淡黄色纯净产物b23mg,产率为22.3%。
实施例6
取化合物b(60mg,0.09mmol)于25mL圆底烧瓶中,真空条件下干燥后,按照顺序加入碘化亚铜(34.4mg,0.18mmol),四(三苯基膦)钯(20.8mg,0.018mmol),放置于干燥罐中继续干燥2h,随后加入DMF(N,N-二甲基甲酰胺)以及氮气保护(该反应要及其注意无水无氧的控制)。将三乙胺和炔丙基醚分别溶于250μL DMF中然后按照顺序注入反应体系。随后在65℃油浴条件下搅拌反应,3h检测一次,约3h反应完全。柱层析分离得到产物c 30mg,产率为58.1%。
实施例7
取化合物c(30mg,0.049mmol),加5mL甲醇搅拌,随后取5M的甲醇钠溶液滴加至反应体系,搅拌约2h反应完全。柱层析分离后的黄色产物d 15mg,产率为83.3%。
实施例8
将化合物d(10mg,0.026mmol),质子海绵(12mg,0.056mmol),三(四丁基铵)氢焦磷酸(160mg,0.38mmol)以及实验中所需注射器、磁子等实验用品提前干燥。次日取磁子,质子海绵加入提前准备好的加有化合物d的10mL圆底烧瓶中,加橡胶翻口塞后氮气保护。随后加入干燥的磷酸三甲酯作为溶剂将脱保护产物溶解,接下来把反应瓶置于提前准备好的冰盐浴中(温度已经达到-15℃以下),待反应瓶中温度平衡后,加入5μL三氯氧磷,严格控制反应温度3h后,将完全溶解于DMF中的三(四丁基铵)氢焦磷酸注入反应体系,随即注入叔丁胺,然后在45min的时间内缓慢升温至0℃,然后注入300μL的TEAB(pH 7.5)淬灭反应,然后加入纯水在-20℃的条件下保存。最后用葡聚糖凝胶柱粗分离后得到三磷酸产物,最后采用反向的C18半制备柱进行分离,得到干净的三磷酸产物。最后用紫外分光光度计单点定量法定量,得e产物1.9mg,产率为11.6%。
实施例9
以该三磷酸进一步进行酶促反应的掺入效率与延伸效率,掺入效率能够达到92%,延伸效率能够达到94%。在聚合酶体系下其复制忠实性能够达到98%(图3)。该化合物在末端带有的炔基与其较高的复制忠实性,给在DNA水平的进一步修饰提供了更多可能。
以上实施例描述了本发明的基本原理、主要特征及优点,本行业的技术人员应该了解,本发明不受上述实施例的限制,上述实施例和说明书中描述的只是说明本发明的原理,在不脱离本发明原理的范围下,本发明还会有各种变化和改进,这些变化和改进均落入本发明保护的范围内。
- 一类非天然碱基三磷酸及其合成方法和应用
- 一种合成在炔端含有不同取代基的炔丙胺衍生物的方法