一种方酰胺类离子型有机催化剂及其合成方法和应用
文献发布时间:2023-06-19 19:30:30
技术领域
本发明属于催化剂及应用技术领域,涉及一种方酰胺类离子型有机催化剂及其合成方法和应用。
背景技术
有机/无金属催化剂相对于有机金属催化剂而言,具有生物毒性低、结构稳定、溶解性好、易于纯化、水氧耐受性好、原料易得、合成简便以及绿色环保等优点,使得有机/无金属催化体系在调控聚合物的微观链结构、立体结构、序列结构乃至拓扑结构等方面均取得了卓越成就,在聚合反应中,有机/无金属催化剂所体现出的催化活性与选择性,可比肩甚至超越有机金属催化剂,现阶段有机/无金属催化聚合已成为绿色、可持续发展化学和材料研究领域的重要组成部分,尤其是在对高分子材料中金属残留物有严格要求的微电子和生物医药等领域,有机/无金属催化聚合发挥着不可替代的作用。
有机/无金属催化剂主要包括有机碱类、有机(质子或路易斯)酸类和氢键给受体双官能催化剂类。其中,有机碱类催化剂使用最为广泛,主要包括N-杂环卡宾、脒类、胍类、磷腈碱、有机磷类化合物等中性(不带电)有机碱,可通过发挥其碱性或亲核性对底物(如引发剂)、单体等进行活化。在聚合反应体系中,有机碱类催化剂的催化活性往往依赖于其碱性的强弱,但对于中性有机碱来说,越强的碱性往往要求其具有更为复杂的共轭结构,而复杂的共轭结构会导致催化剂活性调节的范围和灵活度受限、催化剂合成工艺繁琐、成本增加、实用性降低,中性有机碱的这些局限性极大限制了其在工业上的应用。
文献Zhuolun Jiang,etal.,Ionic Organocatalyst with a Urea Anion andTetra-n-utyl Ammonium Cation for Rapid,Selective,and Versatile Ring-OpeningPolymerization of Lactide.ACS Macro Lett.2019,8(7),759-765,通过四正丁基氢氧化铵与N,N-二取代硫脲/脲间简单的脱水反应,获得了一系列氢键给受体双官能离子型有机催化剂,对丙交酯的开环聚合的催化,具有显著的高效性和可控性。硫脲/脲与季铵碱通过脱水反应进行离子化后,阴离子部分以氢键作用活化引发剂和增长链末端的羟基,这是催化活性的主要来源;而剩余的NH基团可通过氢键作用活化单体,从而实现对单体和引发剂的双重、协同活化,得以同时提高催化效率和选择性。相比于中性有机碱,离子型有机催化剂的结构和制备方法更简单,而且其催化活性可通过两个N原子上丰富的取代基结构进行广泛而灵活的调节。然而,双官能协同催化作用的充分发挥需要离子化后剩余的NH基团仍具有较强的氢键给体作用,这要求催化剂前体具有足够的酸性,但是脲的本征酸性较低,因而往往需要以连有多个强吸电子基团(如F原子和三氟甲基等)的苯环为N-取代基;硫脲的本征酸性虽高于脲,但仍需要连有一定量的吸电子基团(如Cl、F原子和三氟甲基等)的苯环为N-取代基才可具有足够的氢键给体效应,且硫原子导致催化剂的生物毒性不可忽视。有研究表明,具有吸电子共轭效应的苯环取代基(如硝基、氰基、羰基等)虽可提高硫脲和脲的酸性,却因本身也是强氢键受体而导致催化活性受损甚至完全丧失,从而降低聚合反应中产物的产率以及纯度。
发明内容
针对现有离子型有机催化剂存在的活性低、结构复杂以及成本高的技术问题,本发明公开一种方酰胺类离子型有机催化剂及其合成方法和应用,催化剂结构简单,合成路线容易操作,成本低,在开环聚合反应和交替共聚反应中具有很好的催化活性,能提高共聚物纯度和产率。
为了实现上述目的,本发明采用的技术方案是:
一种方酰胺类离子型有机催化剂,结构式如下式所示:
式中;
Y表示N或P;
R
R
一种方酰胺类离子型有机催化剂的合成方法,包括以下步骤:
将化合物A、化合物B和有机溶剂四氢呋喃混匀后,在压力0.01mbar~1mbar的真空条件下反应2h~4h,然后在70℃~80℃进行脱水反应,得到固体产物,即为方酰胺类离子型有机催化剂;
所述化合物A的结构式如式(1)所示:
上式中,R
所述化合物B的结构式如式(2)所示:
上式中,Y表示N或P;R
进一步的,所述化合物A和化合物B的摩尔比为(1~10):1,所述化合物B和有机溶剂的用量比为0.5mmol:15ml;所述有机溶剂为四氢呋喃、2-甲基四氢呋喃、甲醇、乙醇、正丙醇、异丙醇、甲苯或乙腈。
一种方酰胺类离子型有机催化剂在环酯单体或环状碳酸酯单体的开环聚合反应中的应用。
一种方酰胺类离子型有机催化剂在环氧化合物与异硫氰酸酯化合物交替共聚反应中的应用。
一种方酰胺类离子型有机催化剂在环氧化合物与苯酐交替共聚反应中的应用。
进一步的,所述应用时的反应条件为:温度为20℃~60℃,时间5min~68h。
进一步的,所述环酯单体为ε-己内酯、δ-戊内酯、外消旋丙交酯、左旋丙交酯、右旋丙交酯或C1~C12的δ-烷基戊内酯;所述环状碳酸酯单体为三亚甲基碳酸酯。
进一步的,所述环氧化合物为环氧乙烷、C1~C20的直链烷基环氧乙烷、C1~C16的直链烷基缩水甘油醚、异丙基缩水甘油醚、叔丁基缩水甘油醚、2-乙基己基缩水甘油醚、氧化苯乙烯、苯基缩水甘油醚、苄基缩水甘油醚、烯丙基缩水甘油醚、炔丙基缩水甘油醚、甲基丙烯酸缩水甘油酯、环氧环己烷、4-乙烯基环氧环己烷或氧化柠檬烯。
进一步的,所述异硫氰酸酯化合物为异硫氰酸甲酯、直链烷基异硫氰酸酯、脂环族异硫氰酸酯、异硫氰酸异丙酯、异硫氰酸仲丁酯、异硫氰酸异丁酯、异硫氰酸苄酯、异硫氰酸苯酯、邻/间/对甲苯异硫氰酸酯、苯甲酰异硫氰酸酯、异硫氰酸氯代乙酯、环己基甲基异硫氰酸酯和烯丙基异硫氰酸酯中的至少一种;所述直链烷基的含碳原子数为2~20,脂环族异硫氰酸酯中的脂环的含碳原子数为3~12。
本发明的有益效果是:
1、本发明的催化剂采用方酰胺衍生物作为原料,相比于常用的经典氢键给体类有机小分子,方酰胺在含有相同的N-取代基时具有更高的酸性,可避免在催化剂分子结构中另外引入S和卤素(主要是Cl和F)原子,仅通过方酰胺自身的C、H、O、N原子构建催化剂,就能获得足够的氢键强度和协同催化作用,从而简化催化剂结构以及合成路径,降低成本,进一步提高有机催化剂的生物安全性和环境友好性。
2、本发明中,方酰胺衍生物与四烃基取代的氢氧化铵/磷进行脱水反应,由于方酰胺自身的结构,能够容易的合成出结构丰富、性质稳定的方酰胺类离子型有机催化体系,
3、本发明中,方酰胺类离子型有机催化剂可以灵活地改变两个N-取代基和四烃基铵/磷反离子的结构,以及催化剂离子部分和中性部分的比例来调节催化活性,以适应不同聚合反应的需要。此外,方酰胺类离子型有机催化剂在阴离子与被保留的NH基团的作用下,能实现对单体和引发剂的双重活化,极大程度上地保证了聚合反应可控高效地进行,从而解决现有单组分有机强碱型催化剂活性调节的范围和灵活度受限的问题。
4、本发明中,方酰胺类离子型有机催化剂与不同的引发剂(如官能化引发剂、多官能引发剂、大分子引发剂)和单体搭配使用,高效可控地进行环酯单体和环状碳酸酯单体开环聚合,制备分子量可控、结构明确而丰富的聚合物(具有端基功能化、侧基功能化、嵌段、多嵌段、星状和接枝等结构特征),尤其是以聚酯/聚碳酸酯以及以聚酯/聚碳酸酯为主要成分的共聚物,应用灵活方便。
5、本发明中,方酰胺类离子型有机催化剂在环氧化合物与异硫氰酸酯的交替共聚中,能够有效地抑制乃至消除异硫氰酸酯三聚、环氧与异硫氰酸酯二聚等副反应,提高共聚物纯度和产率,使环氧与异硫氰酸酯化合物进行严格的交替共聚反应,完全地避免了环氧的均聚和聚醚链段的生成。
6、本发明中,方酰胺类离子型有机催化剂能够在较低的温度(如室温)下实现环氧化合物与苯酐的交替共聚,使这一反应摆脱对高温和高压条件的需求,极大提升了操作的简便性、灵活性和安全性。
附图说明
图1为Sq1A3催化丙交酯开环聚合获得的粗产物SEC曲线;
图2为Sq1A3催化丙交酯开环聚合获得的粗产物
图3为Sq5A3催化δ-戊内酯开环聚合获得的粗产物SEC曲线;
图4为Sq5A3催化δ-戊内酯开环聚合获得的粗产物
图5为Sq5A2催化ε-己内酯开环聚合获得的粗产物SEC曲线;
图6为Sq5A2催化ε-己内酯开环聚合获得的粗产物
图7为Sq1A3催化丙交酯开环聚合获得的纯化产物SEC曲线;
图8为Sq1A3催化丙交酯开环聚合获得的纯化产物
图9为Sq1A3催化丙交酯开环聚合获得的纯化产物MALDI-TOF谱图;
图10为Sq5A3催化环氧丙烷与异硫氰酸苯酯交替共聚获得的纯化产物SEC曲线;
图11为Sq5A3催化环氧丙烷与异硫氰酸苯酯交替共聚获得的纯化产物
图12为Sq5A3催化环氧丙烷与异硫酸氰异丙酯交替共聚获得的粗产物SEC曲线;
图13为Sq5A3催化环氧丙烷与异硫酸氰异丙酯交替共聚获得的粗产物
图14为Sq5A3催化环氧丙烷与苯酐交替共聚获得的粗产物SEC曲线;
图15为Sq5A3催化环氧丙烷与苯酐交替共聚获得的粗产物
图16为Sq10A3催化环氧丙烷与苯酐交替共聚获得的粗产物SEC曲线;
图17为Sq10A3催化环氧丙烷与苯酐交替共聚获得的粗产物
图18为Sq5A3催化氧化苯乙烯与苯酐交替共聚获得的粗产物SEC曲线;
图19为Sq5A3催化氧化苯乙烯与苯酐交替共聚获得的粗产物
具体实施方式
下面结合实施例对本发明作进一步详细的描述,但本发明的实施方式不限于此。
本发明将方酰胺与季铵/磷碱通过脱水反应制备成离子型有机催化剂,再应用于环酯或环状碳酸酯单体开环聚合反应、环氧化合物与苯酐的交替共聚,以及环氧化合物与异硫氰酸酯交替共聚。
本发明提供的方酰胺类离子型有机催化剂,结构式如下式所示:
式中;
Y表示N或P;
R
R
本发明方酰胺类离子型有机催化剂的合成方法,包括以下步骤:
将化合物A、化合物B和有机溶剂四氢呋喃混匀后,在压力0.01mbar~1mbar的真空条件下反应2h~4h,然后在70℃~80℃进行脱水反应,得到固体产物,即为方酰胺类离子型有机催化剂;
化合物A的结构式如式(1)所示。
上式中,R
化合物B的结构式如式(2)所示。
上式中,Y表示N或P;R
本发明有机溶剂为四氢呋喃、2-甲基四氢呋喃、甲醇、乙醇、正丙醇、异丙醇、甲苯或乙腈。优选的,有机溶剂为四氢呋喃。
本发明中,化合物A和化合物B的摩尔比为(1~10):1,化合物B和有机溶剂的用量比为0.05mmol:15ml。
合成方法的反应式表达如下所示。
本发明制备的方酰胺类离子型有机催化剂,具有很好的催化活性,合成路线简单,催化剂结构简单,成本低,且催化活性好,能很好的用于单体开环聚合反应以及交替共聚反应。
优选的,本发明的方酰胺类离子型有机催化剂用于环酯单体或环状碳酸酯单体的开环聚合反应。
进一步优选的,环酯单体为ε-己内酯(ε-CL)、δ-戊内酯(δ-VL)、外消旋丙交酯(LA)、左旋丙交酯(LLA)、右旋丙交酯(DLA)、烷基碳原子数为1~12的δ-烷基戊内酯(5-alkyl-VL)。环状碳酸酯单体为三亚甲基碳酸酯(TMC)。具体结构式如下。
优选的,本发明的方酰胺类离子型有机催化剂用于环氧化合物与异硫氰酸酯化合物的交替共聚反应,以及用于环氧化合物与苯酐的交替共聚反应。
优选的,环氧化合物为(1)环氧乙烷、(2)C1至C20的直链烷基环氧乙烷、(3)氧化苯乙烯、(4)环氧环己烷、(5)4-乙烯基环氧环己烷、(6)氧化柠檬烯、(7)C1至C16的直链烷基缩水甘油醚、(8)异丙基缩水甘油醚、(9)叔丁基缩水甘油醚、(10)2-乙基己基缩水甘油醚、(11)苯基缩水甘油醚、(12)苄基缩水甘油醚、(13)烯丙基缩水甘油醚、(14)炔丙基缩水甘油醚和(15)甲基丙烯酸缩水甘油酯中的至少一种。结构如下图所示。
优选的,异硫氰酸酯化合物为:(1)异硫氰酸甲酯、(2)直链烷基异硫氰酸酯,其中直链烷基含碳原子数为2~20、(3)脂环族异硫氰酸酯,其中脂环含碳原子数为3至12、(4)异硫氰酸异丙酯、(5)异硫氰酸仲丁酯、(6)异硫氰酸异丁酯、(7)异硫氰酸苄酯、(8)异硫氰酸苯酯、(9)邻/间/对甲苯异硫氰酸酯、(10)苯甲酰异硫氰酸酯、(11)异硫氰酸氯代乙酯、(12)环己基甲基异硫氰酸酯和(13)烯丙基异硫氰酸酯中的至少一种。具体结构式如下。
进一步地,聚合反应采用溶液聚合或本体聚合,即催化剂直接参与聚合反应,或是将催化剂配成溶液参与反应。
溶液聚合时,采用的溶剂为甲苯、丙酮、四氢呋喃、1,4-二氧六环、二氯甲烷、N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、二甲基亚砜、γ-丁内酯、丙烯碳酸酯和乙腈中的一种或两种以上的混合。溶液中催化剂浓度为0.04mol/L~0.2mol/L。
进一步地,聚合反应是,根据反应需要还可选用加入醇类引发剂,离子型有机催化剂和醇类引发剂加入的摩尔比为(0.01~10):1。
优选的,醇类引发剂为:(1)甲醇或碳原子数为2~18的直链烷基醇、(2)异丙醇、(3)2-丁醇、(4)叔丁醇、(5)烷基碳原子数为1~10的1-苯基直链烷基醇、(6)烯丙基醇或饱和碳原子数为2~10的直链端烯1-醇、(7)1-萘甲醇、(8)顺丁烯二醇、(9)乙二醇、(10)1,4-丁二醇、(11)对苯二甲醇、(12)1,1,1-三羟甲基丙烷、(13)甘油、(14)季戊四醇、(15)双季戊四醇、(16)山梨醇、(17)三季戊四醇、(18)数均分子量为400~20000g/mol的聚乙二醇或聚乙二醇单甲醚。具体结构式如下。
进一步地,催化剂的反应条件是温度为20~60℃,反应时间为5min~68h。
下面以几组具体的实施方式说明本发明提供的催化剂、合成方法以及在开环聚合反应和交替共聚反应中的应用。
需要说明的是,以下实施例中环酯单体或环状碳酸酯单体的转化率和聚合物结构特征由BrukerAV600 NMR波谱仪进行测试,CDCl
聚酯或聚碳酸酯的分子量及分子量分散度由体积排除色谱(SEC)测得,仪器采用美国安捷伦(Agilent)1260 Infinity型号的体积排除色谱仪,流动相为四氢呋喃,柱温35℃,流速1mL/min;以一系列聚苯乙烯标准样品做校准曲线。
聚酯的质谱峰使用Bruker AutoflexШ Smartbeam MALDL-TOF质谱仪进行测试。样品溶于THF配成10mg/mL的溶液,再将其与浓度为10mg/mL的三氟乙酸钠的THF溶液以体积比5/1混合。再将此混合溶液与2,5-二羟基苯甲酸的THF溶液(20mg/mL)以1/10的体积比混合,取0.4μL混合液滴加在测试板上进行测试,通过阳离子反射模式得到样品的质谱峰。
以下实施例中所述份数均为摩尔份。
其他未进行特别说明的均为本领域常规操作。
实施例1
本实施例为1,4-二苯基方酰胺四丁基铵催化剂(Sq1A3)的合成。
催化剂Sq1A3的合成方法如下:
将1,4-二苯基方酰胺(0.648mmol)、四丁基氢氧化铵(0.54mmol)与15mL四氢呋喃(THF)加入50mL的Schlenk瓶中,在40℃下搅拌4h。然后在0.1~0.01mbar的真空下缓慢抽走四氢呋喃(THF),将剩余固体在真空条件下加热至80℃保持2h。将生成的固体溶解在2.7mL的二甲基亚砜(DMSO)中,配成0.2mol/L催化剂溶液。
实施例2
本实施例为1,4-二(4-对甲氧基苯基)方酰胺四丁基铵催化剂(Sq2A3)的合成。
催化剂Sq2A3的合成方法如下:
将1,4-二(4-对甲氧基苯基)方酰胺(0.648mmol)、四丁基氢氧化铵(0.54mmol)与15mL四氢呋喃(THF)混合均匀。其他操作与实施例1中合成相同。
实施例3
本实施例为1,4-二环己基方酰胺四丁基铵催化剂(Sq5A3)的合成。
催化剂Sq5A3的合成方法如下:
将1,4-二环己基方酰胺(0.648mmol)、四丁基氢氧化铵(0.54mmol)与15mL四氢呋喃(THF)混合均匀。其他操作与实施例1中合成相同。
实施例4
本实施例为1,4-二环己基方酰胺四丁基磷催化剂(Sq5P1)的合成。
催化剂Sq5P1的合成方法如下:
将1,4-二环己基方酰胺(0.648mmol)、四丁基氢氧化磷(0.54mmol)与15mL四氢呋喃(THF)混合均匀,其他操作与实施例1中合成相同。
实施例5
本实施例为1,4-二环己基方酰胺四甲基胺催化剂(Sq5A1)的合成。
催化剂Sq5A1的合成方法如下:
将1,4-二环己基方酰胺(0.648mmol)、四甲基氢氧化铵(0.54mmol)与15mL四氢呋喃(THF)混合均匀,其他操作与实施例1中相同。
实施例6
本实施例为1,4-二环己基方酰胺苄基三甲基胺催化剂(Sq5A2)的合成。
催化剂Sq5A2的合成方法如下:
将1,4-二环己基方酰胺(0.648mmol)、苄基三甲基铵氢氧化铵(0.54mmol)与15mL四氢呋喃(THF)混合均匀,其他操作与实施例1中合成相同。
实施例7
本实施例为1,4-(2,5-二对甲氧基苯基)方酰胺四丁基胺催化剂(Sq3A3)的合成。
催化剂Sq3A3的合成方法如下:
将1,4-(2,5-二对甲氧基苯基)方酰胺(0.648mmol)、四丁基氢氧化铵(0.54mmol)与15mL四氢呋喃(THF)混合均匀,其他操作与实施例1中合成相同。
实施例8
本实施例为1,4-(3,4,5-三对甲氧基苯基)方酰胺四丁基胺催化剂(Sq4A3)的合成。
催化剂Sq4A3的合成方法如下:
将1,4-(3,4,5-三对甲氧基苯基)方酰胺(0.648mmol)、四丁基氢氧化铵(0.54mmol)与15mL四氢呋喃(THF)混合均匀,其他操作与实施例1中合成相同。
实施例9
本实施例为1,4-二叔丁基方酰胺四丁基铵催化剂(Sq10A3)的合成。
催化剂Sq10A3的合成方法如下:
将1,4-叔丁基方酰胺(0.648mmol)、四丁基氢氧化铵(0.54mmol)与15mL四氢呋喃(THF)混合均匀,其他操作与实施例1中合成相同。
实施例10
本实施例为1,4-三环[3,3,1,1(3,7)]癸烷基方酰胺四丁基铵催化剂(Sq11A3)的合成。
催化剂Sq11A3的合成方法如下:
将1,4-三环[3,3,1,1(3,7)]癸烷基方酰胺(0.648mmol)、四丁基氢氧化铵(0.54mmol)与15mL四氢呋喃(THF)混合均匀,其他操作与实施例1中合成相同。
上述实施例1~实施例10列出了10组不同结构的方酰胺类离子型有机催化剂,但是本发明提供的方酰胺类离子型有机催化剂结构不限于此;此外,合成过程中,化合物的用量比,合成参数均在本发明前述的范围内任意选择,均能得到方酰胺类离子型有机催化剂。
下面以实施例1~实施例10的催化剂的应用,进一步阐明催化剂的催化活性。
实施例11
本实施例为1,4-二苯基方酰胺四丁基铵催化剂(Sq1A3)催化的丙交酯(LA)开环聚合。
本实施例中,开环聚合的反应式如下。
催化剂(Sq1A3)催化丙交酯(LA)开环聚合制备聚丙交酯(PLA)的方法如下。
(1)将2.88gLA溶解在26.5mL的二氯甲烷(DCM)的溶剂中,配制成0.75mol/L的LA溶液,将0.3160g1-萘甲醇(NtA)溶解在40mL的DCM溶液中配制成0.05mol/L的NtA溶液。
(2)在惰性气体的氛围下将75份的LA(1.0mL,0.75mmol)和1份的NtA(200μL,0.01mmol)和1份的催化剂Sq1A3(50μL,0.01mmol)加入到干燥好的容器中,在室温下反应5min后,加入0.5mL乙酸终止反应,取少量粗产物进行SEC和
本实施例中,催化剂Sq1A3催化丙交酯开环聚合制备的粗产物的SEC曲线和
参见图1和图2中,SEC和
实施例12
本实施例为1,4-二苯基方酰胺四丁基铵催化剂(Sq1A3)催化的γ-丁内酯(BL)开环聚合。
将催化剂Sq1A3溶解在2.7mLγ-丁内酯(BL)中,其他操作步骤与实施例11相同。在室温下反应5min后,加入0.5mL乙酸终止反应,取少量粗产物进行SEC和
BL的转化率为81%,计算可得理论数均分子量(M
实施例13
本实施例为1,4-二(4-对甲氧基苯基)方酰胺四丁基铵催化剂(Sq2A3)催化的LA开环聚合。
催化剂(Sq2A3)催化LA开环聚合制备聚PLA的方法与实施例11相同。
根据SEC和
实施例14
本实施例为1,4-二环己基方酰胺四丁基铵催化剂(Sq5A3)催化的LA开环聚合。
催化剂(Sq5A3)催化LA开环聚合制备PLA的方法与实施例11的方法相同。
根据SEC和
实施例15
本实施例为1,4-二环己基方酰胺四丁基铵催化剂(Sq5A3)催化的δ-戊内酯(δ-VL)开环聚合。
本实施例中,δ-戊内酯(δ-VL)开环聚合的反应式如下所示。
在惰性气体的氛围下将100份的δ-VL(0.9mL,9mmol)和1份的苯甲醇(90μL,0.09mmol)和1份的催化剂Sq5A3(0.45mL,0.09mmol)加入到干燥好的容器中,在室温下反应5min后,加入0.5mL乙酸终止反应,取少量粗产物进行SEC和
催化剂Sq5A3催化δ-戊内酯开环聚合制备的粗产物的SEC曲线和
参见图3和图4,根据SEC和
实施例16
本实施例为1,4-二环己基方酰胺四丁基铵催化剂(Sq5A3)催化的ε-己内酯(ε-CL)开环聚合。
本实施例中,ε-己内酯(ε-CL)开环聚合反应式如下。
在惰性气体的氛围下将100份的ε-CL(1.0mL,9mmol)和1份的苯甲醇(90μL,0.09mmol)和1份的催化剂Sq5A3(0.45mL,0.09mmol)加入到干燥好的容器中,在室温下反应2h后,加入0.5mL乙酸终止反应,取少量粗产物进行SEC和
本实施例中,根据SEC和
实施例17
本实施例为1,4-二环己基方酰胺四丁基磷催化剂(Sq5P1)催化的ε-CL开环聚合。
催化剂(Sq5P1)催化ε-己内酯(ε-CL)开环聚合制备聚己内酯(PCL)的方法与实施例16相同,在室温下反应2h后,加入0.5mL乙酸终止反应,取少量粗产物进行SEC和
根据SEC和
实施例18
本实施例为1,4-二环己基方酰胺四甲基胺催化剂(Sq5A1)催化的ε-CL开环聚合。
催化剂(Sq5A1)催化ε-CL开环聚合制备PCL的方法与实施例16相同。在室温下反应2h后,加入0.5mL乙酸终止反应,取少量粗产物进行SEC和
根据SEC和
实施例19
本实施例为1,4-二环己基方酰胺苄基三甲基胺催化剂(Sq5A2)催化的ε-CL开环聚合。
催化剂(Sq5A2)催化ε-CL开环聚合制备PCL的方法与实施例16相同,在室温下反应2h后,加入0.5mL乙酸终止反应,取少量粗产物进行SEC和
催化剂Sq5A2催化ε-己内酯开环聚合制备的粗产物的SEC曲线和
参见图5和图6,根据SEC和
实施例20
本实施例为1,4-二环己基方酰胺四丁基铵催化剂(Sq5A3)催化的δ-葵内酯(δ-DL)开环聚合。
本实施例δ-葵内酯(δ-DL)开环聚合的反应式如下。
/>
在惰性气体的氛围下将100份的δ-DL(1.6mL,9mmol)和1份的苯甲醇(90μL,0.09mmol)和1份的催化剂Sq5A3(0.45mL,0.09mmol)加入到干燥好的容器中,在室温下反应15min后,加入0.5mL乙酸终止反应,取少量粗产物进行SEC和
根据SEC和
实施例21
本实施例为1,4-(2,5-二对甲氧基苯基)方酰胺四丁基胺催化剂(Sq3A3)催化的左旋丙交酯(LLA)开环聚合。
本实施例中,左旋丙交酯(LLA)开环聚合反应式如下。
催化剂(Sq3A3)催化左旋丙交酯(LLA)开环聚合制备聚左旋丙交酯(PLLA)的方法如下。
在惰性气体的氛围下将200份的LLA(2.7mL,2.0mmol)和1份的NtA(200μL,0.01mmol)和1份的催化剂Sq3A3(50μL,0.01mmol)加入到干燥好的容器中,在室温下反应15min后,加入0.5mL乙酸终止反应,取少量粗产物进行SEC和
根据SEC和
实施例22
本实施例为1,4-(3,4,5-三对甲氧基苯基)方酰胺四丁基胺催化剂(Sq4A3)催化的LLA开环聚合。
本实施例中,LLA开环聚合的反应式如下。
本实施例中,催化剂(Sq4A3)催化LLA开环聚合制备PLLA的方法如下。
本实施例与实施例11操作相同,将丙交酯换成左旋丙交酯,将引发剂换成1,1,1-三羟基丙烷,在室温下反应2min后,加入0.5mL乙酸终止反应,取少量粗产物进行SEC和
根据SEC和
实施例23
本实施例为1,4-二环己基方酰胺四丁基铵催化剂(Sq5A3)催化的ε-CL与左旋丙交酯(LLA)开环共聚。
本实施例中,左旋丙交酯(LLA)开环共聚反应式如下。
本实施例中,催化剂(Sq5A3)催化的ε-CL与左旋丙交酯(LLA)开环共聚方法如下。
在惰性气体的氛围下将100份的ε-CL(1.0mL,9mmol)和1份的NtA(90μL,0.09mmol)和1份的催化剂Sq5A3(0.45mL,0.09mmol)加入到干燥好的容器中,在室温下反应1h后,取少量粗产物进行SEC和
根据SEC和
然后加入75份的LLA(1.0mL,0.75mmol)在室温下反应1min后,加入0.5mL乙酸终止反应,取少量粗产物进行SEC和
根据SEC和
实施例24
本实施例为1,4-二苯基方酰胺四丁基铵催化剂(Sq1A3)催化的LA开环聚合。
在惰性气体的氛围下将35份的LA(0.47mL,0.35mmol)和1份的NtA(200μL,0.01mmol)和1份的催化剂Sq1A3(50μL,0.01mmol)加入到干燥好的容器中,在室温下反应5min后,加入0.5mL乙酸终止反应,取少量粗产物进行SEC和
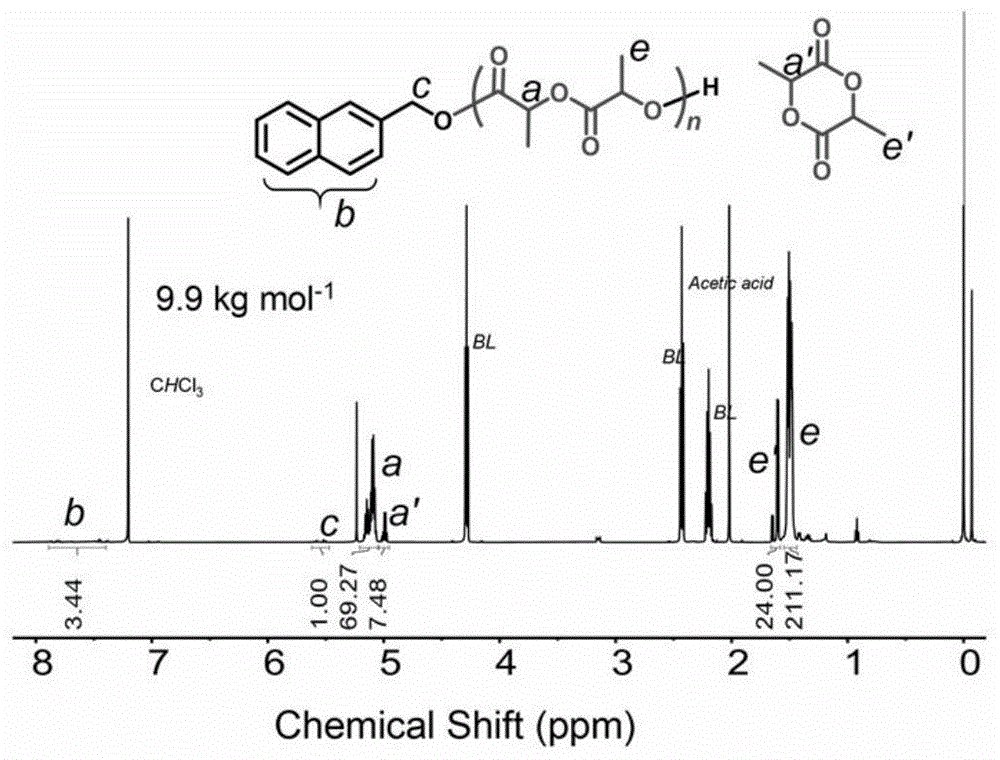
本实施例中,催化剂Sq1A3催化丙交酯开环聚合制备的纯化产物的SEC曲线、
参见图7~图9,根据SEC和
实施例25
本实施例为1,4-二环己基方酰胺四丁基铵催化剂(Sq5A3)催化的环氧丙烷与异硫氰酸苯酯交替共聚。
本实施例中,环氧丙烷与异硫氰酸苯酯交替共聚的反应式如下。
本实施例中,催化剂Sq5A3催化环氧丙烷与异硫氰酸苯酯交替共聚的方法如下。
催化剂Sq5A3不溶于任何溶剂,直接在惰性条件的氛围下将100份异硫氰酸苯酯(1.6mL,13.4mmol),150份环氧丙烷(1.4mL,20mmol),1份的顺式丁烯二醇(268μL,0.134mmol)加入Schlenk瓶中。在室温下反应26h后,加入0.5mL乙酸终止反应,取少量粗产物进行SEC和
Sq5A3催化环氧丙烷与异硫氰酸苯酯交替共聚制备的纯化产物的SEC曲线和
参见图10和图11,根据SEC和
实施例26
本实施例为1,4-二(4-对甲氧基苯基)方酰胺四丁基铵催化剂(Sq2A3)催化的环氧丙烷与异硫氰酸苯酯交替共聚。
本实施例中,将离子型催化剂Sq2A3溶解于DMSO中,配成0.2mol/L催化剂溶液。然后在惰性条件的氛围下将30份异硫氰酸苯酯(0.8mL,6.7mmol),45份环氧丙烷(0.7mL,10mmol),1份的顺式丁烯二醇(450μL,0.225mmol)以及0.1份的Sq5A3(0.0225mmol)加入到干燥好的容器中,在室温下反应36h后,加入0.5mL乙酸终止反应,取少量粗产物进行SEC和
根据SEC和
实施例27
本实施例为1,4-二环己基方酰胺四丁基铵催化剂(Sq5A3)催化的环氧丙烷与异硫氰酸异丙酯交替共聚。
本实施例中,交替共聚反应式如下。
本实施例中,催化剂(Sq5A3)催化的环氧丙烷与异硫氰酸异丙酯交替共聚反应过程与实施例26操作相同,将催化剂Sq2A3换成Sq5A3,将异硫氰酸苯酯换成异硫氰酸异丙酯,在室温下反应48h后,加入0.5mL乙酸终止反应,取少量粗产物进行SEC和
Sq5A3催化环氧丙烷与异硫酸氰异丙酯交替共聚制备的粗产物的SEC曲线和
参见图12和图13,根据SEC和
实施例28
本实施例为1,4-二环己基方酰胺四丁基铵催化剂(Sq5A3)催化的环氧丙烷与异硫氰酸异乙酯交替共聚。
本实施例中,交替共聚的反应式如下。
本实施例中,催化剂Sq5A3催化的交替共聚反应与实施例26的操作相同,将催化剂Sq2A3换成Sq5A3,将异硫氰酸苯酯换成异硫氰酸异乙酯,在室温下反应96h后,加入0.5mL乙酸终止反应,取少量粗产物进行SEC和
根据SEC和
实施例29
本实施例为1,4-二苯基方酰胺四丁基铵催化剂(Sq1A3)催化的环氧丙烷与异硫氰酸苯酯交替共聚。
本实施例中,催化剂Sq1A3催化的环氧丙烷与异硫氰酸苯酯交替共聚方法与实施例25操作相同,将催化剂Sq5A3换成Sq1A3,在室温下反应36h后,加入0.5mL乙酸终止反应,取少量粗产物进行SEC和
根据SEC和
实施例30
本实施例为1,4-二环己基方酰胺四丁基铵催化剂(Sq5A3)在60℃下催化的环氧丙烷与苯酐交替共聚。
本实施例中,催化剂Sq5A3交替共聚的反应式如下。
本实施例中,催化剂Sq5A3催化的环氧丙烷与苯酐交替共聚方法如下。
在惰性条件的氛围下将50份的苯酐(1mL,1mmol),150份环氧丙烷(210μL,3mmol),1份的对苯二甲醇(40μL,0.02mmol)以及1份的催化剂Sq5A3(100μL,0.02mmol)加入到干燥好的容器中,在60℃下反应48h后,加入0.5mL乙酸终止反应,取少量粗产物进行SEC和
根据SEC和
实施例31
本实施例为1,4-二环己基方酰胺四丁基铵催化剂(Sq5A3)在室温下催化的环氧丙烷与苯酐交替共聚。
本实施例与实施例30操作相同,在室温下反应48h后,加入0.5mL乙酸终止反应,取少量粗产物进行SEC和
本实施例中,催化剂Sq5A3催化环氧丙烷与苯酐交替共聚制备的粗产物的SEC曲线和
参见图14和图15,根据SEC和
实施例32
本实施例为1,4-二叔丁基方酰胺四丁基铵催化剂(Sq10A3)催化的环氧丙烷与苯酐交替共聚。
催化剂(Sq10A3)催化环氧丙烷与苯酐交替共聚的方法如下与实施例30操作相同,在60℃下反应48h后,加入0.5mL乙酸终止反应,取少量粗产物进行SEC和
Sq10A3催化环氧丙烷与苯酐交替共聚制备的粗产物的SEC曲线和
参见图16和图17,根据SEC和
实施例33
本实施例为1,4-二环己基方酰胺四丁基铵催化剂(Sq5A3)催化的氧化苯乙烯与苯酐交替共聚。
本实施例中,交替共聚的反应式如下。
催化剂Sq5A3催化的氧化苯乙烯与苯酐交替共聚方法如下。
在惰性条件的氛围下将20份的苯酐(0.4mL,0.04mmol),740份氧化苯乙烯(1.5mL,14.8mmol),1份的对苯二甲醇(40μL,0.02mmol)以及1份催化剂Sq5A3(100μL,0.02mmol)加入到干燥好的容器中,在60℃下反应68h后,加入0.5mL乙酸终止反应,取少量粗产物进行SEC和
Sq5A3催化氧化苯乙烯与苯酐交替共聚制备的粗产物的SEC曲线和
参见图18和图19,根据SEC和
实施例34
本实施例为1,4-三环[3,3,1,1(3,7)]癸烷基方酰胺四丁基铵催化剂(Sq11A3)催化的氧化苯乙烯与苯酐交替共聚。
催化剂(Sq11A3)催化氧化苯乙烯与苯酐交替共聚的方法与实施例33操作相同,在60℃下反应68h后,加入0.5mL乙酸终止反应,取少量粗产物进行SEC和
根据SEC和
实施例35
本实施例为1,4-三环[3,3,1,1(3,7)]癸烷基方酰胺四丁基铵催化剂(Sq11A3)催化的异丙基缩水甘油醚与苯酐交替共聚。
本实施例中,交替共聚的反应式如下。
本实施例交替共聚的过程与实施例33操作相同,将氧化苯乙烯换成异丙基缩水甘油醚,在60℃下反应68h后,加入0.5mL乙酸终止反应,取少量粗产物进行SEC和
根据SEC和
从上述实施例可以看出,本发明的催化剂合成简单,成本低,催化剂结构简单,催化活性好,生物安全性好;在常温下开环聚合的转化率能快速的达到100%;在常温下交替共聚反应中,异硫氰酸酯化合物的转化率达到98%,苯酐的转化率为100%,催化剂的工业应用范围大,产物的产率高,纯度好,有很好的应用前景。
以上实施例仅为本发明较优的实施方式,仅用于解释本发明,而非限制本发明,本领域技术人员在未脱离本发明精神实质下所作的改变、替换、修饰等均应属于本发明的保护范围。
- 一种采用己内酰胺类碱性离子液体作为催化剂制备酚醛树脂的方法
- 一种生物基金属有机框架组装酯酶催化剂、制备方法及其在生物柴油合成中的应用
- 一种轴手性萘胺方酰胺类有机催化剂及其制备方法和应用
- 一种轴手性萘胺方酰胺类有机催化剂及其制备方法和应用