氧化异阿朴菲生物碱与联吡啶衍生物的环金属有机铑配合物及其合成方法和应用
文献发布时间:2023-06-19 19:30:30
技术领域
本发明涉及氧化异阿朴菲生物碱与联吡啶衍生物的环金属有机铑配合物及其合成方法和应用,属于医药技术领域。
背景技术
自从发现顺铂具有抗癌活性以来,金属抗肿瘤药物的研究和开发引起了人们的广泛关注(Rosenberg,B.;et al.Nature,1965,205:698-699.)。顺铂的临床应用受限最主要原因是耐药性及毒副性。虽然其它耐药性或毒性相对改善的第二代和第三代铂类抗肿瘤药相继在临床应用,但未彻底解决其毒副性和耐药性问题。因此,新型金属药物研究具有迫切需求性。非铂类金属化合物,如钌、铑、金等金属配合物具有较大的潜能。近年研究表明,部分新型环金属类过渡金属化合物优势明显,稳定性、膜透性及细胞毒活性等都具有更大的潜能。近年,天然活性配体调控金属抗癌化合物的生物活性已成为一种新策略。综上所述,用天然活性配体调控过渡金属环金属化合物结构与生物活性是一种新颖且潜能更大的科学研究策略。
氧化异阿朴菲是一种特殊的生物碱化合物,广泛存在于天然产物中,具有一定的生物活性。目前,氧化异阿朴菲的金属配合物及其各种生物活性已被广泛研究,如本申请人之前的研究成果(公布号为CN103421048A的发明专利申请)中,采用6-羟基氧化异阿朴菲和二氯·二(二甲基亚砜)合铂溶解于极性溶剂中进行配位反应得到一氯·二甲基亚砜·6-羟基氧化异阿朴菲合铂(II),其结构如式(A)所示:
发明内容
本发明要解决的技术问题是提供一系列结构新颖且抗肿瘤活性显著的氧化异阿朴菲生物碱与联吡啶衍生物的环金属有机铑配合物及其合成方法和应用。
为解决上述技术问题,本发明采用以下技术方案:
本发明所述的氧化异阿朴菲生物碱与联吡啶衍生物的环金属有机铑配合物为具有下述通式(I)所示结构的配合物或其药学上可接受的盐:
其中,R
优选的,通式(I)中,R
本发明所述氧化异阿朴菲生物碱与联吡啶衍生物的环金属有机铑配合物的合成方法,主要包括以下步骤:取下述式(Ⅱ)所示化合物置于极性溶剂中,加入银盐于加热或不加热条件下进行除氯反应,所得反应料液过滤,收集滤液;向滤液中加入N^N配体于加热或不加热条件下进行配位反应,反应结束后,向其中加入饱和六氟磷酸钾水溶液或饱和六氟磷酸钠水溶液,静置,有沉淀析出,收集沉淀,即得到相应的目标配合物粗品;其中,
所述的N^N配体为2,2-联吡啶、4,4-二甲氧基-2,2-联吡啶、4,4-二叔丁基-2,2-联吡啶、5,5-二甲基-2,2-联吡啶或4,4-二苯基-2,2-联吡啶;
上述合成方法中,所述的极性溶剂具体可以是选自N,N-二甲基甲酰胺、二甲基亚砜、乙二醇、乙二醇甲醚和水中的一种或两种以上的组合。极性溶剂的用量可以根据需要进行确定,以能够充分溶解所参加反应的原料为宜。具体的,以0.1mmol的式(II)所示化合物为基准计算,全部原料所用极性溶剂的总用量通常为5~60mL,优选为5~15mL。
上述合成方法中,所述的银盐优选为六氟磷酸银、三氟甲磺酸银、氧化银或硝酸银,优选为六氟磷酸银或氧化银。通过添加银盐,利用其中的银离子与式(II)所示化合物中的氯离子反应,使式(II)所示化合物中氯离子与铑离子的连接键打断,为后续N^N配体与铑离子进行配位作铺垫。所述银盐与式(II)所示化合物的摩尔比优选为2:1~5:1。从提高反应速率的角度而言,除氯反应优选是在加热条件下进行,进一步优选是在大于或等于40℃条件下进行,更优选是在50℃至极性溶剂的沸点温度之间进行。
上述合成方法中,配位反应在加热条件下进行相对于在不加热条件下进行可以提高反应速率,进一步优选是在大于或等于40℃条件下进行,更优选是在60℃至极性溶剂的沸点温度之间进行。反应是否完全采用薄层层析(TLC)跟踪检测。所述N^N配体的加入量优选为式(II)所示化合物摩尔用量的2~5倍。
本申请中,当N^N配体为2,2-联吡啶时,通式(I)所示配合物中的R
当N^N配体为4,4-二甲氧基-2,2-联吡啶时,通式(I)所示配合物中的R
当N^N配体为4,4-二叔丁基-2,2-联吡啶时,通式(I)所示配合物中的R
当N^N配体为5,5-二甲基-2,2-联吡啶时,通式(I)所示配合物中的R
当N^N配体为4,4-二苯基-2,2-联吡啶时,通式(I)所示配合物中的R
为了提高目标配合物的产率,本发明所述合成方法中的除氯反应和配位反应优选是在气体保护(如氮气、氩气等)下进行。除氯反应进一步优选在避光条件下进行,更优选是在避光且气体保护(如氮气、氩气等)下进行。
在配位反应结束后,通过加入饱和六氟磷酸钾水溶液或饱和六氟磷酸钠水溶液使目标配合物以沉淀的形式析出,所述饱和六氟磷酸钾水溶液或饱和六氟磷酸钠水溶液通常是以体积上相对过量于反应后所得溶液的量进行加入,优选饱和六氟磷酸钾水溶液或饱和六氟磷酸钠水溶液的加入量为反应后所得溶液体积的1.5~2倍。
上述方法合成得到的是目标配合物的粗品,可采用现有常规的纯化方法对其进行纯化以提高目标配合物的纯度。在本申请中,具体可以采用硅胶柱层析以获得纯化后的目标配合物,柱层析时以二氯甲烷与甲醇或乙醇组成的混合溶剂为洗脱剂,其中二氯甲烷与甲醇或者是二氯甲烷与乙醇的体积比优选为500:1~10:1,更优选为200:1~10:1。也可以采用重结晶的方法达到进一步纯化的目的,在重结晶时所用的溶剂可以是选自二氯甲烷、丙酮和氯仿中的任意一种,也可以是上述任意一种与乙醇或石油醚或甲醇的组合。
本发明所述合成方法中涉及的原料式(Ⅱ)所示化合物的名称为氯双(1-氮杂苯并蒽酮)铑二聚体,其可参考现有文献(Lowry M S,Hudson W R,Pasca l R A,etal.Accelerated Luminophore Discovery through Combinatorial Sy nthesis[J].Journal of the American Chemical Society,2004,126(43):14129-14135.Li Y,LiuB,Lu X R,et al.Cyclometalated iridium(III)N-heterocyc lic carbene complexesas potential mitochondrial anticancer and photodynami c agents[J].DaltonTrans,2017,46(34):11363-11371.Ma D L,Liu L J,Leung K H,et al.AntagonizingSTAT3 dimerization with a rhodium(III)co mplex[J].Angewandte Chemie,2014,126(35):9332-9336.)进行制备,也可自行设计合成路线进行制备,优选按下述方法进行制备:
取无水三氯化铑或氯铑酸盐和氧化异阿朴菲置于由2-甲氧基乙醇和水组成的混合溶剂中,在气体保护(如氮气、氩气等)及加热条件下进行反应,反应结束后,除去溶剂,干燥,即得。
在上述式(Ⅱ)所示化合物的制备方法中,所述的氯铑酸盐优选为氯铑酸钠或氯铑酸钾,氯铑酸盐和氧化异阿朴菲的摩尔比优选为1:1~1:4。在混合溶剂的组成中,2-甲氧基乙醇和水的体积比优选为3:1。所述反应优选在100~120℃条件下进行。
本发明还包括上述氧化异阿朴菲生物碱与联吡啶衍生物的环金属有机铑配合物或其药学上可接受的盐在制备抗肿瘤药物中的应用。
本发明进一步包括一种药物组合物,该药物组合物中含有治疗上有效剂量的上述氧化异阿朴菲生物碱与联吡啶衍生物的环金属有机铑配合物或其药学上可接受的盐。
与现有技术相比,本发明提供了一系列结构新颖的氧化异阿朴菲生物碱与联吡啶衍生物的环金属有机铑配合物及其合成方法,申请人的试验结果表明,这些配合物对某些肿瘤细胞株如人三阴乳腺癌MDA-MB-231细胞具有显著的增殖抑制活性,同时还具有较高的生理稳定性,有望开发成抗肿瘤药物。
附图说明
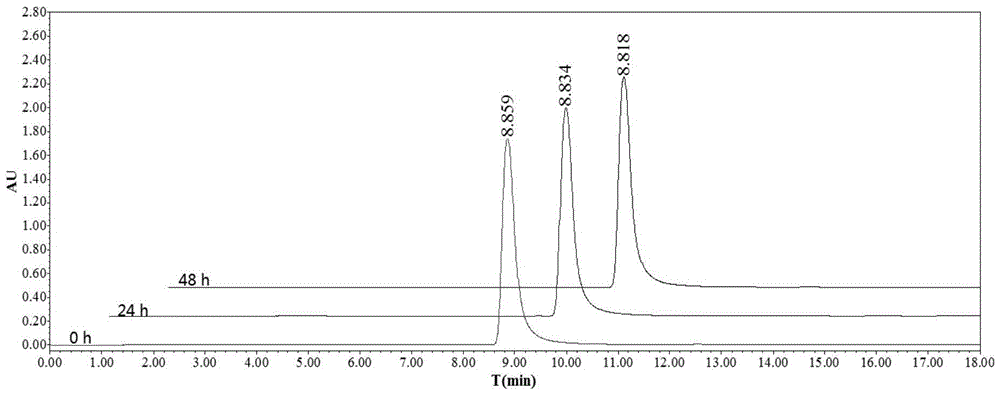
图1为按本发明实施例1所述方法制得的Rh-1在生理缓冲溶液条件下作用0h、24h及48h的HPLC检测结果图。
图2为按本发明实施例5所述方法制得的Rh-2在生理缓冲溶液条件下作用0h、24h及48h的HPLC检测结果图。
图3为按本发明实施例8所述方法制得的Rh-3在生理缓冲溶液条件下作用0h、24h及48h的HPLC检测结果图。
图4为按本发明实施例11所述方法制得的Rh-4在生理缓冲溶液条件下作用0h、24h及48h的HPLC检测结果图。
图5为按本发明实施例14所述方法制得的Rh-5在生理缓冲溶液条件下作用0h、24h及48h的HPLC检测结果图。
具体实施方式
为了更好的解释本发明的技术方案,下面结合实施例对本发明作进一步详细的描述,但本发明的实施方式不限于此。
以下的实施例中涉及的式(Ⅱ)所示化合物根据下述合成路线及制备方法制备得到:
具体的制备方法为:
称取0.5g(2.39mmol)RhCl
实施例1:目标配合物Rh-1的合成
取0.1g(0.067mmol)式(Ⅱ)所示化合物和0.034g(0.134mmol)六氟磷酸银置于容器中,加入8mL乙二醇,将容器移至油浴中,在氮气保护下加热至110℃反应1小时,过滤,收集滤液;向滤液中加入0.034g(0.217mmol)2,2-联吡啶,在氮气保护下,加热至110℃回流反应3小时,冷却至室温,向反应所得料液中加入饱和六氟磷酸钾水溶液,室温静置析出沉淀,收集沉淀,得到粗产品,烘干后上硅胶柱层析分离纯化(洗脱溶剂二氯甲烷/甲醇=200:1,体积比),得到黄色固体产物,产率为60%。
对本实施例所得产物进行表征:
1)核磁谱分析,所得波谱数据如下:
核磁氢谱:
核磁碳谱:
2)电喷雾质谱:M=719.0954 found:m/z 719.0975[M-PF
因此,可确定本实施例所得黄色固体产物为目标配合物Rh-1,分子式为C
实施例2:目标配合物Rh-1的合成
重复实施例1,不同的是:用二甲基亚砜代替乙二醇。产率56%。
对本实施例所得产物进行核磁氢谱、核磁碳谱和电喷雾质谱分析,确定本实施例所得产物为目标配合物Rh-1。
实施例3:目标配合物Rh-1的合成
重复实施例1,不同的是:配位反应改在60℃条件下进行。产率45%。
对本实施例所得产物进行核磁氢谱、核磁碳谱和电喷雾质谱分析,确定本实施例所得产物为目标配合物Rh-1。
实施例4:目标配合物Rh-1的合成
重复实施例1,不同的是:配位反应的时间改为12h。产率58%。
对本实施例所得产物进行核磁氢谱、核磁碳谱和电喷雾质谱分析,确定本实施例所得产物为目标配合物Rh-1。
实施例5:目标配合物Rh-2的合成
取0.1g(0.067mmol)式(Ⅱ)所示化合物和0.034g(0.134mmol)六氟磷酸银置于容器中,加入8mL乙二醇,将容器移至油浴中,在氮气保护下加热至110℃反应1小时,过滤,收集滤液;向滤液中加入0.032g(0.149mmol)4,4-二甲氧基-2,2-联吡啶,在氮气保护下,加热至110℃回流反应3小时,冷却至室温,向反应所得料液中加入饱和六氟磷酸钾水溶液,室温静置析出沉淀,收集沉淀,得到粗产品,烘干后上硅胶柱层析分离纯化(洗脱溶剂二氯甲烷/甲醇=200:1,体积比),得到黄色固体产物,产率为50%。
对本实施例所得产物进行表征:
1)核磁氢谱分析,所得波谱数据如下:
核磁氢谱:
核磁碳谱:
2)电喷雾质谱:M=779.1166found:m/z 779.1185[M-PF
因此,可确定本实施例所得黄色固体产物为目标配合物Rh-2,分子式为C
实施例6:目标配合物Rh-2的合成
重复实施例5,不同的是:用乙二醇甲醚代替乙二醇。产率45%。
对本实施例所得产物进行核磁氢谱、核磁碳谱和电喷雾质谱分析,确定本实施例所得产物为目标配合物Rh-2。
实施例7:目标配合物Rh-2的合成
重复实施例5,不同的是:配位反应改在常温条件下进行,反应的时间改为24h。产率42%。
对本实施例所得产物进行核磁氢谱、核磁碳谱和电喷雾质谱分析,确定本实施例所得产物为目标配合物Rh-2。
实施例8:目标配合物Rh-3的合成
取0.1g(0.067mmol)式(Ⅱ)所示化合物和0.034g(0.134mmol)六氟磷酸银置于容器中,加入8mL乙二醇,将容器移至油浴中,在氮气保护下加热至110℃反应1小时,过滤,收集滤液;向滤液中加入0.04g(0.149mmol)4,4-二叔丁基-2,2-联吡啶,在氮气保护下,加热至110℃回流反应3小时,冷却至室温,向反应所得料液中加入饱和六氟磷酸钾水溶液,室温静置析出沉淀,收集沉淀,得到粗产品,烘干后上硅胶柱层析分离纯化(洗脱溶剂二氯甲烷/甲醇=200:1,体积比),得到黄色固体产物,产率为65%。
对本实施例所得产物进行表征:
1)核磁氢谱分析,所得波谱数据如下:
核磁氢谱:
核磁碳谱:
2)电喷雾质谱:M=831.2206found:m/z 831.2225[M-PF
因此,可确定本实施例所得黄色固体产物为目标配合物Rh-3,分子式为C
实施例9:目标配合物Rh-3的合成
重复实施例8,不同的是:用N,N-二甲基甲酰胺代替乙二醇。产率60%。
对本实施例所得产物进行核磁氢谱、核磁碳谱和电喷雾质谱分析,确定本实施例所得产物为目标配合物Rh-3。
实施例10:目标配合物Rh-3的合成
重复实施例8,不同的是:配位反应的时间改为12h。产率62%。
对本实施例所得产物进行核磁氢谱、核磁碳谱和电喷雾质谱分析,确定本实施例所得产物为目标配合物Rh-3。
实施例11:目标配合物Rh-4的合成
取0.1g(0.067mmol)式(Ⅱ)所示化合物和0.034g(0.134mmol)六氟磷酸银置于容器中,加入8mL乙二醇,将容器移至油浴中,在氮气保护下加热至110℃反应1小时,过滤,收集滤液;向滤液中加入0.028g(0.151mmol)5,5-二甲基-2,2-联吡啶,在氮气保护下,加热至110℃回流反应3小时,冷却至室温,向反应所得料液中加入饱和六氟磷酸钾水溶液,室温静置析出沉淀,收集沉淀,得到粗产品,烘干后上硅胶柱层析分离纯化(洗脱溶剂二氯甲烷/甲醇=200:1,体积比),得到黄色固体产物,产率为60%。
对本实施例所得产物进行表征:
1)核磁氢谱分析,所得波谱数据如下:
核磁氢谱:
核磁碳谱:
2)电喷雾质谱:M=747.1267found:m/z 747.1295[M-PF
因此,可确定本实施例所得黄色固体产物为目标配合物Rh-4,分子式为C
实施例12:目标配合物Rh-4的合成
重复实施例11,不同的是:反应用水代替乙二醇。产率56%。
对本实施例所得产物进行核磁氢谱、核磁碳谱和电喷雾质谱分析,确定本实施例所得产物为目标配合物Rh-4。
实施例13:目标配合物Rh-4的合成
重复实施例12,不同的是:除氯反应中用三氟甲磺酸银代替六氟磷酸银;配位反应改在40℃条件下进行。产率48%。
对本实施例所得产物进行核磁氢谱、核磁碳谱和电喷雾质谱分析,确定本实施例所得产物为目标配合物Rh-4。
实施例14:目标配合物Rh-5的合成
取0.1g(0.067mmol)式(Ⅱ)所示化合物和0.034g(0.134mmol)六氟磷酸银置于容器中,加入8mL乙二醇,将容器移至油浴中,在氮气保护且避光条件下加热至110℃反应1小时,过滤,收集滤液;向滤液中加入0.046g(0.146mmol)4,4-二苯基-2,2-联吡啶,在氮气保护下,加热至110℃回流反应3小时,冷却至室温,向反应所得料液中加入饱和六氟磷酸钾水溶液,室温静置析出沉淀,收集沉淀,得到粗产品,烘干后上硅胶柱层析分离纯化(洗脱溶剂二氯甲烷/甲醇=200:1,体积比),得到黄色固体产物,产率为55%。
对本实施例所得产物进行表征:
1)核磁氢谱分析,所得波谱数据如下:
核磁氢谱:
核磁碳谱:
2)电喷雾质谱:M=871.1580found:m/z 871.1606[M-PF
因此,可确定本实施例所得黄色固体产物为目标配合物Rh-5,分子式为C
实施例15:目标配合物Rh-5的合成
重复实施例14,不同的是:除氯反应中用氧化银代替六氟磷酸银,除氯反应在氮气保护且避光条件下进行;配位反应在80℃条件下进行,用饱和六氟磷酸钠水溶液代替饱和六氟磷酸钾水溶液。产率48%。
对本实施例所得产物进行核磁氢谱、核磁碳谱和电喷雾质谱分析,确定本实施例所得产物为目标配合物Rh-5。
实验例1:本发明所述配合物对多种肿瘤细胞株进行体外抑制活性实验:
1、细胞株与细胞培养
本实验选用细胞株:小鼠结肠癌细胞CT-26、人胃癌细胞MGC-803以及人三阴乳腺癌细胞MDA-MB-231。
所有细胞株均培养在含10%胎牛血清、100U/mL青霉素、100U/mL链霉素的RPMI-1640或DMEM培养液内,置37℃含体积浓度5% CO
2、待测化合物的配制
5个待测化合物Rh-1、Rh-2、Rh-3、Rh-4、Rh-5分别按本发明实施例1、实施例5、实施例8、实施例11、实施例14所述方法制备,所得产物用二氯甲烷重结晶2次所得,其纯度≥95%,将其DMSO储液(浓度为0.002mol/L)通过RPMI-1640或DMEM培养基依次稀释成五个浓度梯度,分别为20、10、5、2.5、1.25、0.625μmol/L,其中助溶剂DMSO终浓度≤1%。分别测试不同梯度浓度下目标配合物对各种肿瘤细胞的增殖抑制程度,用以拟合计算半数抑制浓度,即IC
3、细胞生长抑制实验(MTT法)
(1)取对数生长期的肿瘤细胞,经胰蛋白酶消化后,用含10%胎牛血清的培养液配制细胞悬液,以每孔180μL接种于96孔培养板中,使待测细胞密度至1000-10000个/孔(边缘孔用无菌PBS填充);
(2)5% CO
(3)5% CO
(4)每孔加入20μL的MTT溶液(5mg/mL),继续培养4h
(5)终止培养,小心吸去孔内培养液,每孔加入150μL DMSO充分溶解甲瓒沉淀,振荡器混匀后,在酶标仪用波长为570nm,参比波长为630nm测定各孔的光密度值;
(6)同时设置调零孔(培养基、MTT、DMSO),对照孔(细胞、相同浓度的药物溶解介质、培养液、MTT、DMSO);
(7)根据测得的光密度值(OD值),来判断活细胞数量,OD值越大,细胞活性越强。利用公式:
计算化合物对肿瘤细胞生长的抑制率。进一步通过Bliss软件对五个浓度梯度的抑制率数据进行拟合,求出产物对不同肿瘤株的半数抑制浓度(IC
表1:Rh-1~5和顺铂对3种肿瘤细胞株的IC
从体外抗肿瘤活性测试结果来看,配合物Rh-1~5均具有良好的抗癌活性,对3种肿瘤细胞株的IC
综上所述,本发明所述配合物总体表现出了一定的体外抗肿瘤活性,对癌细胞具有一定程度的毒性选择性,具有良好的潜在药用价值,有望用于各种抗肿瘤药物的制备。
实验例2:本发明所述配合物的生物稳定性实验
(1)取Rh-1(按本发明实施例1所述方法制得)的10μL DMSO储液(2×10
(2)取Rh-2(按本发明实施例5所述方法制得)的10μL DMSO储液(2×10
(3)取Rh-3(按本发明实施例8所述方法制得)的10μL DMSO储液(2×10
(4)取Rh-4(按本发明实施例11所述方法制得)的10μL DMSO储液(2×10
(5)取Rh-5(按本发明实施例14所述方法制得)的10μL DMSO储液(2×10
由图1至图5可知,本发明所述配合物分别作用24和48h后,无明显其它成分的峰出现,说明本发明所述配合物在生理缓冲溶液中是稳定的。
- 氧化异阿朴菲生物碱环金属有机铑配合物及其合成方法和应用
- 一种以氧化异阿朴啡衍生物为配体的钴(II)金属配合物及其合成方法和应用