一种α-萘酮衍生物二聚体类化合物及其制备方法与用途
文献发布时间:2023-06-19 19:30:30
技术领域
本发明属于生物医药技术领域,尤其涉及一种α-萘酮衍生物二聚体类化合物及其制备方法与用途。
背景技术
α-萘酮二聚体衍生物主要来自微生物的代谢产物,通常从枝孢属(Cladosporium)真菌中分离得到,其结构特点是以α-萘酮衍生物作为结构单元,大部分通过8~4’碳碳键,少部分为碳氧键将两个α-萘酮衍生物聚合为二聚体,连接方式通常为一分子α-萘酮衍生物的苯环和另一分子α-萘酮衍生物的苯环连接或一分子α-萘酮衍生物的苯环与另一分子α-萘酮衍生物的环己酮环连接。α-萘酮二聚体衍生物具有广泛的生物活性,如抗癌、抗菌等,因此具有用于药物或作为药物前体开发的潜力,相关论文报道包括:NasiniG等发表的“Secondarymould metabolitesofCladosporiumtenuissimum,ahyperparasiteofrustfungi”,YamazakiH等发表的“Halogenatedcladosporolsproducedbythesodiumhalide-supplementedfermentationofthe plant-associatedfungusCladosporiumsp.TMPU1621”,等等。
随着科研人员致力于研究和开发新的活性药物,α-萘酮二聚体衍生物因其广泛的生物活性越来越受到科学界的关注,但现有的α-萘酮二聚体衍生物大部分通过8~4’碳碳键,少部分为碳氧键,将两个α-萘酮聚合,即一分子α-萘酮衍生物的苯环和另一分子α-萘酮衍生物的苯环连接或一分子α-萘酮衍生物的苯环与另一分子α-萘酮衍生物的环己酮环连接。尚未发现通过一分子α-萘酮衍生物的环己酮环和另一分子α-萘酮衍生物的环己酮环连接方式聚合的α-萘酮二聚体衍生物。因此,提供一种新聚合方式、具有抑癌活性的α-萘酮衍生物二聚体类化合物具有重要意义。
发明内容
为解决现有技术中存在的问题,本发明提供一种α-萘酮衍生物二聚体类化合物及其制备方法与用途,该化合物是发明人从腐生真菌Neohelicosporiumgriseum的发酵产物中提取、分离和纯化得到,经鉴定具有罕见的3~2’碳碳键聚合方式(一分子α-萘酮衍生物的环己酮环和另一分子α-萘酮衍生物的环己酮环连接),并经进一步的检测具有显著的抑制卵巢癌细胞株的活性,有望在制备防治卵巢癌的药物中发挥重要作用。
本发明所述的腐生真菌Neohelicosporiumgriseum,是由贵州理工学院真菌资源研究室分离获得并已进行了保藏,保藏单位:贵州省农业科学院保藏中心,保藏日:2019年11月20日,保藏登记号为GZAAS22-2002。
本发明的目的将通过下面的详细描述来进一步说明。
本发明提供一种α-萘酮衍生物二聚体类化合物,其结构如式Ⅰ或式Ⅱ所示:
标示碳序号的式Ⅰ如下所示:
相应地,本发明还提供所述的α-萘酮衍生物二聚体类化合物的制备方法,包括如下步骤:A)菌种活化:取出保藏的腐生真菌Neohelicosporiumgriseum,接种,静置培养后,备用;B)发酵种子液制备:将步骤A)活化后的菌落切碎后,接种于灭菌后的液体培养基中培养8~12d;
C)接种:采用固体发酵方式,每个发酵袋装入大米50-100份、水50-100份,高压蒸汽灭菌后接种步骤B)所得发酵种子液,静置培养90-110d后得到发酵培养基;
D)萃取:用乙酸乙酯/甲醇混合溶剂浸泡步骤C)所得发酵培养基,搅拌,离心,收集上清液于旋转蒸发仪中回收溶剂,得到粗浸膏;
E)分离纯化:将步骤D)所得粗浸膏经层析分离,得到如式Ⅰ或式Ⅱ所示的α-萘酮衍生物二聚体类化合物;所述层析分离方法包括硅胶柱层析法、凝胶柱层析法和半制备高效液相色谱法。
优选地,所述液体培养基由如下质量份数的组分制得:麦芽糖18-22份、谷氨酸钠8-12份、磷酸二氢钾0.4-0.6份、硫酸镁0.2-0.4份、葡萄糖9-11份、酵母粉2.5-3.5份、甘露醇18-20份、水950-1050份。
优选地,所述乙酸乙酯/甲醇混合溶剂由乙酸乙酯和甲醇按8-12:1的体积比组成。
优选地,所述步骤E)包括:用二氯甲烷/甲醇混合溶剂溶解所述步骤D)所得粗浸膏,与硅胶按1:1.4-1.6的质量比混合均匀,待溶剂挥发后得到河沙状样品,该样品作为一次上柱样品,备用;称取硅胶粉装入分离柱中,让硅胶粉缓缓下沉直至不再下沉时,加入所述一次上柱样品,分别采用石油醚/乙酸乙酯进行梯度洗脱,每个梯度洗脱2-3个柱体积,每480-520mL洗脱溶剂为一份进行收集,每份洗脱样品经薄层层析点板,使用石油醚乙酸乙酯按100:0~0:100进行梯度洗脱,每个梯度洗脱2-3个柱体积,每480-520mL洗脱溶剂为一份进行收集,每份洗脱样品经薄层层析点板,使用石油醚乙酸乙酯体系展开,TLC分析,合并斑点颜色和R
优选地,所述二氯甲烷/甲醇混合溶剂由二氯甲烷和甲醇按0.9-1.1:0.9-1.1的体积比组成。
此外,本发明还提供α-萘酮衍生物二聚体类化合物在制备防治卵巢癌的药物中的应用。
此外,本发明还提供一种药物组合物,包含权利要求1所述的α-萘酮衍生物二聚体类化合物、辅料;所述α-萘酮衍生物二聚体类化合物的质量百分含量为0.1%–99%。
与现有技术相比,本发明的有益效果包括:(1)本发明在对腐生真菌Neohelicosporium griseum的发酵产物进行深入分析的基础上,提供的α-萘酮衍生物二聚体类化合物(化合物1、化合物2)结构新颖,经质谱等结构鉴定,具有罕见的3~2’碳碳键聚合方式(一分子α-萘酮衍生物的环己酮环和另一分子α-萘酮衍生物的环己酮环连接),不同于现有α-萘酮二聚体衍生物的聚合方式(一分子α-萘酮衍生物的苯环和另一分子α-萘酮衍生物的苯环连接或一分子α-萘酮衍生物的苯环与另一分子α-萘酮衍生物的环己酮环连接)。(2)本发明提供的α-萘酮衍生物二聚体类化合物对人卵巢癌细胞株A2780具有显著的选择性抑制作用,而对PC-3等其他肿瘤细胞株的抑制活性较弱,有望在制备抗卵巢癌药物中发挥作用。
附图说明
图1是本发明化合物1的高分辨质谱图。
图2是本发明化合物2的高分辨质谱图。
图3是本发明化合物1的一维核磁共振氢谱图。
图4是本发明化合物1的一维核磁共振碳谱图。
图5是本发明化合物1的二维核磁共振氢氢相关谱图(COSY)。
图6是本发明化合物1的二维核磁共振碳氢相关谱图(HSQC)。
图7是本发明化合物1的二维核磁共振碳氢相关谱图(HMBC)。
图8是本发明化合物1的核磁共振碳谱图(NOE)。
图9是本发明化合物2的一维核磁共振氢谱图。
图10是本发明化合物2的一维核磁共振碳谱图。
图11是本发明化合物2的二维核磁共振氢氢相关谱图(COSY)。
图12是本发明化合物2的二维核磁共振碳氢相关谱图(HSQC)。
图13是本发明化合物2的二维核磁共振碳氢相关谱图(HMBC)。
图14是本发明化合物2的核磁共振碳谱图(NOE)。
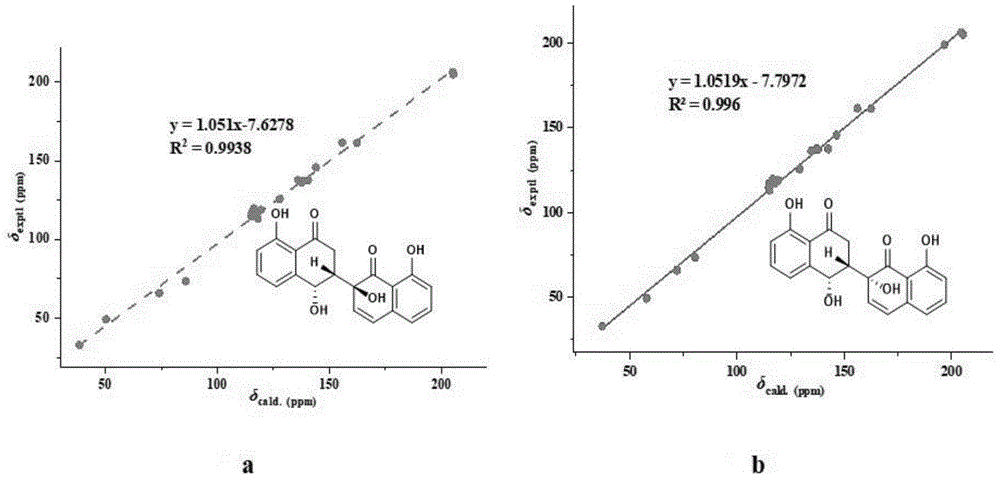
图15是化合物1实验和计算的
图16是化合物2实验和计算的13CNMR异构体化学位移之间的线性回归分析结果图;其中,16-a为实验的结果图,16-b为计算的结果图。
图17是化合物1和2实验和计算的ECD谱图。
具体实施方式
下面结合附图和实施例对本发明做进一步详细说明。本发明中,如未明确指出,比指体积比,百分含量指体积百分含量。
实施例一α-萘酮衍生物二聚体类化合物
一种α-萘酮衍生物二聚体类化合物(化合物1),其结构如式Ⅰ所示:
化合物1的制备方法,包括如下步骤:
A)菌种活化:取出保藏的腐生真菌Neohelicosporiumgriseum,接种在PDA平板培养基上,于28℃静置培养后,备用;
B)发酵种子液制备:将步骤A)活化后的菌落切碎后,接种于灭菌后的100mL液体培养基中,28℃、180r/min条件下震荡培养10d;所述液体培养基由如下组分制得:麦芽糖20g、谷氨酸钠10g、磷酸二氢钾0.5g、硫酸镁0.3g、葡萄糖10g、酵母粉3g、甘露醇20g、加水定容至1L;
C)接种:采用固体发酵方式,准备400个发酵袋,每个发酵袋装入大米50g、水55g,高压蒸汽灭菌后接种步骤B)所得发酵种子液,每袋的接种量为5mL,28℃下静置培养100d后得到发酵培养基;
D)萃取:用乙酸乙酯/甲醇混合溶剂(乙酸乙酯/甲醇的体积比为10:1)浸泡步骤C)所得发酵培养基72h,分批次搅拌,离心,收集上清液于旋转蒸发仪中回收溶剂,得到粗浸膏99.4g;E)分离纯化:用二氯甲烷/甲醇混合溶剂溶解所述步骤D)所得粗浸膏,与硅胶按1:1.5的质量比混合均匀,待溶剂挥发后得到河沙状样品,该样品作为一次上柱样品,备用;称取200-300目硅胶粉装入长为0.9m、内径为50mm的分离柱中,让硅胶粉缓缓下沉直至不再下沉时,加入所述一次上柱样品,分别采用石油醚/乙酸乙酯按100:0逐渐变至0:100进行梯度洗脱,每个梯度洗脱2-3个柱体积,每500mL洗脱溶剂为一份进行收集,每份洗脱样品经薄层层析点板,使用石油醚乙酸乙酯体系(石油醚:乙酸乙酯=50:1、石油醚:乙酸乙酯=20:1石油醚:乙酸乙酯=10:1、石油醚:乙酸乙酯=8:2、石油醚:乙酸乙酯=7:3、石油醚:乙酸乙酯=6:4、石油醚:乙酸乙酯=5:5)展开,TLC分析,合并斑点颜色和R
实施例二α-萘酮衍生物二聚体类化合物
一种α-萘酮衍生物二聚体类化合物(化合物2),其结构如式Ⅱ所示:
化合物2的制备方法,包括如下步骤:
A)菌种活化:取出保藏的腐生真菌Neohelicosporiumgriseum,接种在PDA平板培养基上,于28℃静置培养后,备用;
B)发酵种子液制备:将步骤A)活化后的菌落切碎后,接种于灭菌后的100mL液体培养基中,28℃、180r/min条件下震荡培养10d;所述液体培养基由如下组分制得:麦芽糖20g、谷氨酸钠10g、磷酸二氢钾0.5g、硫酸镁0.3g、葡萄糖10g、酵母粉3g、甘露醇20g、加水定容至1L;
C)接种:采用固体发酵方式,准备400个发酵袋,每个发酵袋装入大米50g、水55g,高压蒸汽灭菌后接种步骤B)所得发酵种子液,每袋的接种量为5mL,28℃下静置培养100d后得到发酵培养基;
D)萃取:用乙酸乙酯/甲醇混合溶剂(乙酸乙酯/甲醇的体积比为10:1)浸泡步骤C)所得发酵培养基72h,分批次搅拌,离心,收集上清液于旋转蒸发仪中回收溶剂,得到粗浸膏99.7g;E)分离纯化:用二氯甲烷/甲醇混合溶剂溶解所述步骤D)所得粗浸膏,与硅胶按1:1.5的质量比混合均匀,待溶剂挥发后得到河沙状样品,该样品作为一次上柱样品,备用;称取200-300目硅胶粉装入长为0.9m、内径为50mm的分离柱中,让硅胶粉缓缓下沉直至不再下沉时,加入所述一次上柱样品,分别采用石油醚/乙酸乙酯按100:0逐渐变至0:100进行梯度洗脱,每个梯度洗脱2-3个柱体积,每500mL洗脱溶剂为一份进行收集,每份洗脱样品经薄层层析点板,使用石油醚乙酸乙酯体系(石油醚:乙酸乙酯=50:1、石油醚:乙酸乙酯=20:1石油醚:乙酸乙酯=10:1、石油醚:乙酸乙酯=8:2、石油醚:乙酸乙酯=7:3、石油醚:乙酸乙酯=6:4、石油醚:乙酸乙酯=5:5)展开,TLC分析,合并斑点颜色和R
实施例三α-萘酮衍生物二聚体类化合物的结构鉴定
通过高分辨质谱,紫外光谱,红外光谱,旋光,核磁共振等数据进行综合分析,从而确定化合物1和2的结构,结果如图1至图17所示,其理化性质如下:
化合物1:土黄色粉末;分子式C
化合物2:棕黑色固体;分子式C
表1化合物1及化合物2的1DNMR数据
实施例四α-萘酮衍生物二聚体类化合物的活性测试
为了进一步验证本发明提供的化合物的有益效果,通过对化合物1和2进行肿瘤细胞抑制进行活性测试,具体实验如下:
被测样品溶液的配制:测试样品为上述实施例一中分离得到的化合物1、上述实施例二中分离得到的化合物2;准确称取适量样品,用DMSO配制成所需浓度的溶液,供活性测试。
细胞系及细胞的继代培养:活性测试采用A2780、PC-3、MBA-MD-231细胞系;A2780、PC-3细胞用RPMI-1640培养基,MBA-MD-231细胞用DMEM培养基,分别添加10%胎牛血清(FBS)和1%青霉素-链霉素溶液(1×10
本实验采用MTT比色法检测药物对A2780、PC-3、MBA-MD-231细胞的生长抑制作用。活性测试时,将对数生长期的细胞以5×10
表2化合物1和化合物2对3种肿瘤细胞的增殖抑制活性(IC
从表2可知,本发明提供的α-萘酮衍生物二聚体类化合物对人卵巢癌细胞株A2780具有显著的选择性抑制作用,尤其地,化合物1和2对人卵巢癌细胞株A2780具有非常显著的抑制作用,化合物1对A2780的抑制作用甚至超过顺铂,而它们对PC-3等其他肿瘤细胞株的抑制活性较弱。
以上内容是结合具体的优选实施方式对本发明所作的进一步详细说明,不能认定本发明的具体实施只局限于这些说明。对于本发明所属技术领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干简单推演或替换,都应当视为属于本发明的保护范围。
- 肟基萘醌类化合物及其制备方法和用途
- 萘衍生物类荧光染料及其制备方法和用途
- 一种红树内生真菌来源的1,4-萘醌类化合物及其制备方法和在制备抗炎药物中的应用
- 噻唑烷酮螺嘧啶三酮类化合物及制备方法和用途
- 喹唑啉-2,4-二酮衍生物及其制备方法和用途
- 5,8-二取代-1,6-二氮杂萘-7-羰酰胺类化合物及其二聚体化合物,其制备方法和用途
- 5,8-二取代-1,6-二氮杂萘-7-羰酰胺类化合物及其二聚体化合物,其制备方法和用途