SHP2与MEK1双靶点抑制剂及其制备方法与应用
文献发布时间:2023-06-19 19:30:30
技术领域
本发明属于药物化学领域,具体涉及SHP2与MEK1双靶点抑制剂及其制备方法与应用。
背景技术
SHP2是体内广泛存在的非受体型蛋白酪氨酸磷酸酶,具有两个N末端Src同源性2结构域(N-SH2和C-SH2)、催化结构域(PTP)和C末端尾部。这两个SH2结构域控制SHP2的亚细胞定位和功能调节。功能上,SHP2是连接多个细胞内致癌信号通路的关键枢纽,作为血小板源性生长因子(PDGF)、表皮生长因子(EGF)、成纤维细胞因子(FGF)、白细胞介素-3(IL-3)、白血病抑制因子(LIF)及α-干扰素(INF-α)等生长因子的下游信号分子,SHP2参与RAS/MARK通路、PI3K/AKT通路、JAK/STAT通路、JNK通路等在内的多条信号通路。研究显示,SHP2突变或过度活化将会导致骨髓增生异常综合征、B细胞急性淋巴细胞白血病及固体瘤(肺癌、结肠癌、神经母细胞瘤、黑色素瘤、肝癌)的发生。
RAS-RAF-MEK-ERK通路参与调节细胞生长过程,包括细胞增殖、侵袭和血管生成等,其中MEK是一种磷酸化丝裂原活化蛋白激酶的激酶,RAF被激活后C端与MEK结合并使其催化区的丝氨酸磷酸化激活MEK,级联激活ERK信号。MEK1/2是调节ERK活性的关键靶点,靶向抑制MEK1/2蛋白可特异性地抑制ERK信号输出,进而抑制癌细胞增殖。
研究发现,SHP2抑制剂与MEK1抑制剂联合给药具有较强的协同作用,如RMC-4630(SHP2抑制剂)与Cobimetinib(MEK1抑制剂)已于2019年联合使用治疗实体瘤。
本发明将SHP2抑制剂与MEK1抑制剂通过药效团拼接策略,设计和制备出一类全新的SHP2与MEK1双靶点抑制剂,为后期开发RAS突变引起的癌症提供可能的新型药物治疗解决方案。
发明内容
本发明需解决的技术问题之一是SHP2抑制剂和MEK1抑制剂的临床疗效差的问题。因此提供一种新型含SHP2与MEK1双靶点抑制剂,为后续抗癌药物开发提供可能。
解决上述技术问题的方案如下:
如本发明所述一种如下式I所示的芳基螺环类化合物或其药学上可接受的盐、异构体、代谢产物、前药、溶剂合物或水合物,其结构如下:
L为键或-NH-O-(CH
R
或者,R
上述化合物的药学上可接受的盐为通式I的酸加成盐,其中用于成盐的酸为:氯化氢、溴化氢、硫酸、碳酸、草酸、柠檬酸、琥珀酸、酒石酸、磷酸、乳酸、丙酮酸、乙酸、马来酸、甲磺酸、苯磺酸、富马酸、对甲苯磺酸或阿魏酸。
优选地,所述的化合物为以下任一化合物:
/>
上述的芳基螺环类化合物的制备方法,所述化合物的合成路线如下:
其中,M为键或-CO-NH-O-(CH
R
或者,R
具体合成步骤如下:
(1)化合物II-1与II-2经缩合反应得到化合物II-3;
(2)化合物II-3脱除保护基得到化合物I。
其中,步骤(1)中缩合反应所采用的溶剂包括但不限于:苯、甲苯、乙醇、甲醇、1,4-二氧六环、四氢呋喃、丙酮、乙腈、乙酸乙酯、正己烷、二氯甲烷、氯仿、N,N-二甲基甲酰胺、二甲亚砜或者用这些溶剂任选组成的混合溶剂,优选二甲亚砜;所采用的碱包括但不限于:N,N-二异丙基乙胺、4-甲基吗啡啉、三乙胺,优选4-甲基吗啡啉;
步骤(2)中脱除保护基反应所采用的溶剂包括但不限于:苯、甲苯、乙醇、甲醇、1,4-二氧六环、四氢呋喃、丙酮、乙腈、乙酸乙酯、正己烷、二氯甲烷、氯仿、N,N-二甲基甲酰胺、二甲亚砜或者用这些溶剂任选组成的混合溶剂,优选乙酸乙酯;所采用的试剂包括但不限于:盐酸的乙酸乙酯溶液、二氧六环的乙酸乙酯溶液,优选盐酸的乙酸乙酯溶液。
本发明提供了一种芳基螺环类化合物的药物组合物,所述药物组合物为由所述芳基螺环类化合物或其药学上可接受的盐、消旋体、旋光异构体或溶剂化合物作为活性成分和药学上可接受的载体或辅料。化合物可以添加药学上可接受的载体制成常见的药用制剂,如片剂、胶囊、糖浆、悬浮剂、注射剂,可以加入香料、甜味剂、液体或固体填料或稀释剂等常用药用辅料。本发明提供了一种如式I所示化合物或其药学上可接受的盐、异构体、代谢产物、前药、溶剂合物、水合物或一种药物组合物在制备SHP2和/或MEK1抑制剂中的用途。
本发明提供了一种如式I所示化合物或其药学上可接受的盐、异构体、代谢产物、前药、溶剂合物、水合物或一种药物组合物在制备用于预防和/或治疗癌症的药物中的用途。
本发明所述的用途,其特征在于,所述的癌症为结肠癌、直肠癌、肺癌、肝癌、胃癌。
有益效果:
与现有技术相比,本发明具有如下优点:
1、本发明提供一类全新骨架的双靶点小分子抑制剂,结构新颖,可同时靶向SHP2与MEK1,产生抑制活性,该活性良好,解决了单独使用单靶点抑制剂临床疗效差的问题,且相较于药物联合使用策略无需多次给药,患者依从性好。
2、以典型化合物I-3为例,进行分子对接,结果分析如下:
(1)对MEK1蛋白对接结果进行分析
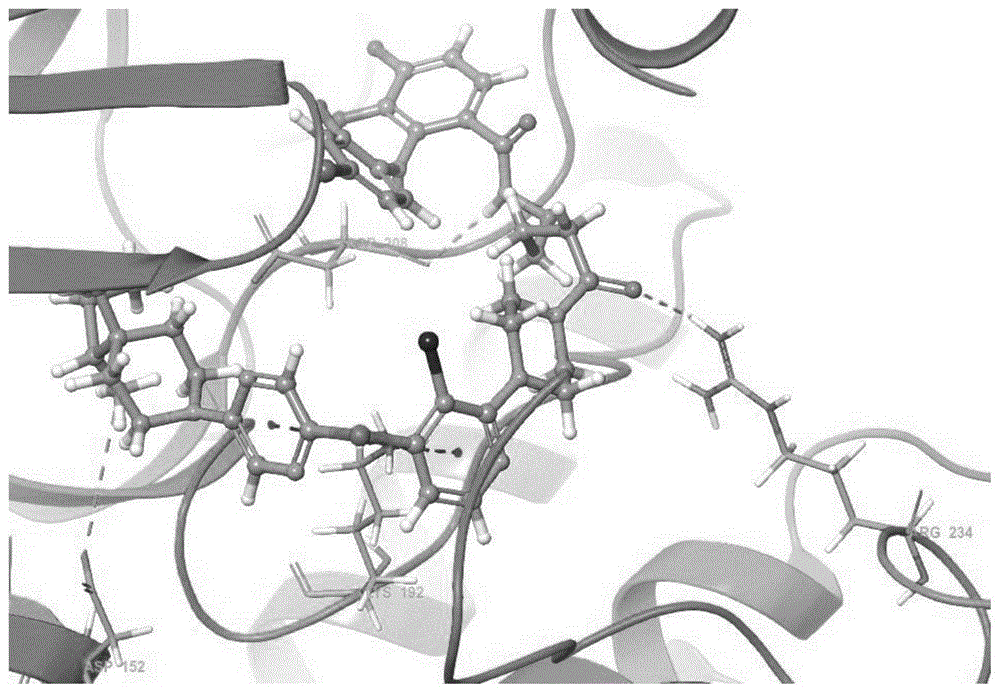
以化合物I-3为例分析其与MEK1蛋白的相互作用方式:作用模式3D图(图1),作用模式2D图(图2)
根据对接结果,化合物I-3的吡嗪环与吡啶环通过与Lys192的质子化氨基形成π-阳离子相互作用;螺环上的质子化氨基与Asp152末端羧基形成盐键;与哌嗪环相连的酰胺键羰基O原子与Arg234末端质子化NH形成氢键相互作用;双F取代苯环的酰胺键NH与Asp208末端羧基形成氢键相互作用。
(2)对SHP2蛋白对接结果进行分析
以化合物I-3为例分析其与SHP2蛋白的相互作用方式:作用模式3D图(图3),作用模式2D图(图4)
根据对接结果,化合物I-3螺环上的质子化氨基与Glu232末端的两个羧基分别形成氢键与盐键相互作用;双F取代苯环的酰胺键羰基O原子与Gln495末端酰胺的NH
再次说明拥有本发明的骨架的化合物可以同时与MEK1蛋白和SHP2蛋白发生作用。
3、体外SHP2和MEK1酶水平活性测试显示:
以上数据显示,本发明多个实施例化合物对SHP2及MEK1都具有良好的酶水平抑制作用。
4、体外抗肿瘤实验结果再次证实本发明所述的化合物不仅可以同时靶向同时与MEK1蛋白和SHP2,同时抗肿瘤活性比阳性对照药物(SHP099和ARRY-162)更好抗肿瘤活性,再次说明本发明优越性,取得意料不到技术效果。,
具体如下:化合物对结直肠癌细胞株DLD-1细胞的抑制活性如下:
以上数据显示,本发明实施例化合物对DLD-1细胞的增殖具有良好的抑制作用。相比较单独使用SHP2抑制剂(SHP099)、MEK1抑制剂(ARRY-162)而言,本发明实施例具备更好的体外抗增殖活性。
附图说明
图1化合物I-3与MEK1蛋白的作用模式3D图(PDB ID:3V04);
图2化合物I-3与MEK1蛋白的作用模式2D图(PDB ID:3V04);
图3化合物I-3与SHP2蛋白的作用模式3D图(PDB ID:5EHR);
图4化合物I-3与SHP2蛋白的作用模式2D图(PDB ID:5EHR)。
具体实施方式
为了更好的理解本发明,通过以下实施例来进一步阐明本发明,但是本发明的内容不仅仅局限于以下实施例。
SHP2抑制剂(SHP099)、MEK1抑制剂(ARRY-162)分别购自上海源叶生物科技有限公司、上海麦克林生化科技有限公司。
实施例1
合成路线:
化合物II-1-1的合成
将2-氟-3-氯-4-碘吡啶(2.60g,10.00mmol)、哌嗪-1-甲酸-2-甲基丙-2-基酯(2.10g,11.00mmol,1.1eq)、碳酸钾(2.80g,20.00mmol,2.0eq)溶于10.0mL的二甲基亚砜中,50℃下搅拌12小时,抽滤后滤液用乙酸乙酯稀释,水洗后无水硫酸钠干燥、浓缩,柱色谱纯化得化合物II-1-1,3.40g白色固体,收率80%。ESI-MS(m/z):424.0[M+H]
化合物II-1-2的合成
将化合物II-1-1(3.40g,8.00mmol)、5-氯吡嗪-2-硫钠(1.60g,9.70mmol,1.2eq)、三(二亚苄基丙酮)二钯(146.5mg,0.16mmol,0.02eq)、4,5-双二苯基膦-9,9-二甲基氧杂蒽(228.8mg,0.48mmol,0.06eq)置于120mL封管中,氮气置换三次后将N,N-二异丙基乙胺(2.80mL,16.10mmol,2.0eq)及20.0mL的1,4-二氧六环加入,95℃下反应12小时,停止反应,浓缩,柱色谱纯化得化合物II-1-2,2.55g淡黄色固体,收率72%。ESI-MS(m/z):442.1[M+H]
化合物II-1-3的合成
将化合物II-1-2(2.50g,5.70mmol)、(3S,4S)-4-{[(R)-(2-甲基丙-2-基)(氧亚基)-4-硫基]氨基}-3-甲基-8-氮杂-2-氧杂螺[4.5]癸烷(1.70g,6.20mmol,1.1eq)、N,N-二异丙基乙胺(3.50mL,20.10mmol,3.5eq)溶于20.0mL的N-甲基吡咯烷酮中,95℃下反应12小时,乙酸乙酯稀释后饱和食盐水洗涤,无水硫酸钠干燥、浓缩,柱色谱纯化得化合物II-1-3,2.42g白色固体,收率63%。ESI-MS(m/z):680.2[M+H]
化合物II-1的合成
将化合物II-1-3(2.40g,3.60mmol)溶于60mL的二氯甲烷中,室温下滴加20.0mL三氟乙酸并搅拌6小时,蒸除溶剂得化合物II-1,1.85g淡黄色固体,收率90%。ESI-MS(m/z):580.2[M+H]
实施例2
合成路线:
化合物I-1-1的合成
将2-氟-4-碘苯胺(5.00g,21.10mmol)、2,3,4-三氟苯甲酸(3.70g,21.10mmol,1.0eq)溶于15.0mL的四氢呋喃中,再将氨基锂(1.60g,67.50mmol,3.2eq)配制为15.0mL的四氢呋喃溶液并在0℃下缓慢滴入上述反应液,滴毕加热至50℃搅拌12小时。0℃下缓慢滴加3M盐酸调节pH至1,乙酸乙酯萃取后浓缩、干燥,二氯甲烷/甲醇重结晶得化合物I-1-1,6.60g棕色固体,收率79%。ESI-MS(m/z):393.9[M+H]
化合物I-1-2的合成
将化合物I-1-1(66.8mg,0.17mmol)、化合物II-1(100mg,0.17mmol,1.0eq)、1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(38.3mg,0.20mmol,1.2eq)、1-羟基苯并三唑(35.4mg,0.26mmol,1.5eq)溶于1.00mL的二甲基亚砜中,滴加4-甲基吗啡啉(56μL,0.50mmol,3.0eq),室温搅拌12小时,乙酸乙酯稀释反应液,饱和食盐水洗涤,无水硫酸钠干燥、浓缩,柱色谱纯化得化合物I-1-2,白色固体90.0mg,收率55%。ESI-MS(m/z):955.1[M+H]
化合物I-1的合成
将化合物I-1-2(90.0mg,0.094mmol)溶于1.00mL的乙酸乙酯,滴加2M盐酸的乙酸乙酯溶液(2.00mL),室温搅拌6小时,蒸除溶剂后加水溶解,饱和碳酸氢钠调节pH至8-9,二氯甲烷:异丙醇(3:1)萃取,合并有机层,无水硫酸钠干燥、浓缩得化合物I-1,77.3mg白色固体,收率97%。ESI-MS(m/z):851.1[M+H]
实施例3
合成路线:
化合物I-2-1的合成:
将化合物N-羟基邻苯二甲酰亚胺(3.00g,18.40mmol)、溴乙酸甲酯(5.59g,36.80mmol,2.0eq)、碳酸钾(7.60g,55.20mmol,3.0eq)溶于30.0mL的N,N-二甲基甲酰胺中,70℃下反应过夜。TLC板监测至原料转化完全,饱和食盐水洗涤,无水硫酸钠干燥,浓缩,柱色谱纯化得化合物I-2-1,3.42g白色固体,收率79%。ESI-MS(m/z):236.0[M+H]
化合物I-2-2的合成
将化合物I-2-1(2.94g,12.50mmol)溶于60.0mL的二氯甲烷中,0℃下缓慢滴加水合肼(1.00mL,18.80mmol,1.5eq),0℃搅拌4小时后抽滤,二氯甲烷洗涤滤饼,滤液浓缩得化合物I-2-2,774.7mg无色液体,收率59%。ESI-MS(m/z):106.0[M+H]
化合物I-2-3的合成
将化合物I-1-1(392.9mg,1.00mmol)、化合物I-2-2(105.0mg,1.00mmol,1.0eq)、1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(230.0mg,1.20mmol,1.2eq)、1-羟基苯并三唑(204.2mg,1.50mmol,1.5eq)溶于二甲基亚砜中,滴加4-甲基吗啡啉(0.33mL,3.00mmol,3.0eq),室温搅拌12小时,乙酸乙酯稀释反应液,饱和食盐水洗涤,无水硫酸钠干燥、浓缩,柱色谱纯化得化合物I-2-3,297.4mg白色固体,收率62%。ESI-MS(m/z):480.9[M+H]
化合物I-2-4的合成
将化合物I-2-3(288.0mg,0.60mmol)、氢氧化锂(57.6mg,2.40mmol,4.0eq)溶于3.00mL的甲醇中,室温搅拌6小时,蒸除溶剂后加水溶解,1M盐酸调节pH至7,抽滤、干燥,得化合物I-2-4,248.5mg白色固体,收率89%。ESI-MS(m/z):466.9[M+H]
化合物I-2-5的合成
将化合物I-2-4(79.2mg,0.17mmol)、化合物II-1(100mg,0.17mmol,1.0eq)、1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(38.3mg,0.20mmol,1.2eq)、1-羟基苯并三唑(35.4mg,0.26mmol,1.5eq)溶于1.00mL的二甲基亚砜中,滴加4-甲基吗啡啉(56μL,0.50mmol,3.0eq),室温搅拌12小时,乙酸乙酯稀释反应液,饱和食盐水洗涤,无水硫酸钠干燥、浓缩,柱色谱纯化得化合物I-2-5,100.0mg白色固体,收率57%。ESI-MS(m/z):1028.1[M+H]
化合物I-2的合成
将化合物I-2-5(100.0mg,0.088mmol)溶于1.00mL的乙酸乙酯,滴加2M盐酸的乙酸乙酯溶液(2.00mL),室温搅拌6小时,蒸除溶剂后加水溶解,饱和碳酸氢钠调节pH至8-9,二氯甲烷:异丙醇(3:1)萃取,合并有机层,无水硫酸钠干燥、浓缩得化合物I-2,34.9mg白色固体,收率43%。ESI-MS(m/z):924.2[M+H]
实施例4
参照化合物I-2的合成方法,将溴乙酸甲酯替换为4-溴丁酸甲酯,可制得化合物I-3。ESI-MS(m/z):952.2[M+H]
实施例5
参照化合物I-2的合成方法,将溴乙酸甲酯替换为5-溴戊酸甲酯,可制得化合物I-4。ESI-MS(m/z):966.2[M+H]
实施例6
参照化合物I-2的合成方法,将溴乙酸甲酯替换为6-溴己酸甲酯,可制得化合物I-5。ESI-MS(m/z):980.2[M+H]
实施例7
参照化合物I-2的合成方法,将溴乙酸甲酯替换为7-溴庚酸甲酯,可制得化合物I-6。ESI-MS(m/z):994.2[M+H]
实施例8
合成路线:
化合物I-7-1的合成
将6-氨基-7-氟-3-甲基苯并[d]咪唑-5-甲酸甲酯(1.00g,4.80mmol)、1-溴-2-氟-4-碘苯(1.70g,5.80mmol,1.2eq)、三(二亚苄基丙酮)二钯(44.0mg,0.048mmol,0.01eq)、4,5-双二苯基膦-9,9-二甲基氧杂蒽(23.0mg,0.096mmol,0.02eq)、碳酸铯(3.10g,9.60mmol,2.0eq)置于120mL封管中,氮气置换三次后将20.0mL的甲苯加入,100℃下反应12小时,停止反应,浓缩,柱色谱纯化得化合物I-7-1,539.1mg白色固体,收率29%。ESI-MS(m/z):396.0[M+H]
化合物I-7-2的合成
将化合物I-7-1(530.0mg,1.30mmol)、氢氧化锂(64.4mg,2.70mmol,2.0eq)溶于甲醇中,室温搅拌6小时,蒸除溶剂后加水溶解,1M盐酸调节pH至7,抽滤、干燥,得化合物I-7-2,356.1mg白色固体,收率70%。ESI-MS(m/z):381.9[M+H]
化合物I-7的合成
参照化合物I-3的合成方法将化合物I-1-1替换为化合物I-7-2,可制得化合物I-7。ESI-MS(m/z):940.2[M+H]
实施例9
参照化合物I-7的合成方法,将1-溴-2-氟-4-碘苯替换为1-溴-2-氯-4-碘苯,可制得化合物I-8。ESI-MS(m/z):956.2[M+H]
实施例10
分子对接分析
对实施例中化合物利用
1.分子对接过程
(1)分别将MEK1蛋白(PDB ID:3V04)、SHP2蛋白(PDB ID:5EHR)导入
(2)将实施例中化合物导入
(3)对准备好的蛋白分别选择Receptor Grid Generation模块限定对接范围。
(4)对MEK1蛋白/SHP2蛋白选择Ligand Docking模块进行分子对接,对接受体为上述限定对接范围的蛋白,对接配体为已完成配体分子准备的实施例化合物,对接精度选择XP
2.分子对接结果分析
(1)对MEK1蛋白对接结果进行分析
以化合物I-3为例分析其与MEK1蛋白的相互作用方式:作用模式3D图(图1),作用模式2D图(图2)
根据对接结果,化合物I-3的吡嗪环与吡啶环通过与Lys192的质子化氨基形成π-阳离子相互作用;螺环上的质子化氨基与Asp152末端羧基形成盐键;与哌嗪环相连的酰胺键羰基O原子与Arg234末端质子化NH形成氢键相互作用;双F取代苯环的酰胺键NH与Asp208末端羧基形成氢键相互作用。
(2)对SHP2蛋白对接结果进行分析
以化合物I-3为例分析其与SHP2蛋白的相互作用方式:作用模式3D图(图3),作用模式2D图(图4)
根据对接结果,化合物I-3螺环上的质子化氨基与Glu232末端的两个羧基分别形成氢键与盐键相互作用;双F取代苯环的酰胺键羰基O原子与Gln495末端酰胺的NH
实施例11
体外SHP2和MEK1酶水平活性测试
1.体外SHP2酶水平活性测试
对上述实施例中化合物的SHP2酶水平活性进行测试,具体操作如下:
1.1化合物配制
化合物溶解在100%DMSO中,配制成10mM储存液,于-20度冰箱避光保存。
1.2SHP2反应过程
(1)配制1×Kinase Buffer。
(2)化合物浓度梯度的配制:受试化合物测试起始浓度为10μM,4倍稀释,8个浓度,单孔测试。在384孔板中稀释成100倍终浓度的100%DMSO溶液,使用分液器Echo550向目的板384-well plate转移250nL 100倍终浓度的化合物。正对照加入250nL的DMSO,负对照加入250nL 1mM的SHP099。
(3)用1×ReactionBuffer配制5倍终浓度的激活肽溶液,分别加入5μL到反应板中,1000rpm离心1分钟。
(4)用1×ReactionBuffer配制2.5倍终浓度的酶溶液,分别加入10μL到反应板中,1000rpm离心1分钟,室温孵育60分钟。
(5)用1×ReactionBuffer配制2.5倍终浓度的底物溶液,分别加入10μL到反应板中,1000rpm离心1分钟孵育20分钟。
(6)用EnVision读取Ex355/Em460荧光数值。
1.3数据分析
计算公式
其中:RFU:样品的荧光值;Mean(NC):含10μM SHP099的对照孔荧光值均值;
Mean(PC):阳性对照孔荧光值均值。
拟合量效曲线
以浓度的log值作为X轴,百分比抑制率为Y轴,采用分析软件GraphPad Prism 7的log(inhibitor)vs.response-Variable slope拟合量效曲线,从而得出各个化合物对酶活性的IC
计算公式是Y=Bottom+(Top-Bottom)/(1+10^((LogIC
2.体外MEK1酶水平活性测试
对上述实施例中化合物的MEK1酶水平活性进行测试,具体操作如下:
2.1配制1×Kinase Base Buffer和Stop Buffer
(1)1×Kinase Buffer
50mM HEPES,pH 7.5
0.0015%Brij-35
(2)Stop Buffer
100mM HEPES,pH 7.5
0.015%Brij-35
0.2%Coating Reagent#3
50mM EDTA
2.2化合物配制
化合物溶解在100%DMSO中,配制成10mM储存液,于-20度冰箱避光保存。
(1)化合物浓度梯度的配制:受试化合物测试起始浓度为10μM,4倍稀释,10个浓度,单孔测试。在96孔板中稀释成50倍终浓度的100%DMSO溶液,使用移液枪向下一个96孔板转移100μL 50倍终浓度的化合物进行梯度稀释。正对照加入100μL100%DMSO。该96孔板标记为源板。
(2)将10μL化合物从源板转移到一个新的96孔板作为中间板,中间板每孔加入90μL 1xKinase Buffer,并在摇床上搅拌10分钟。
(3)从96孔中间板每孔取5μL转移到384孔板,一式两份。
2.3激酶反应
(1)配制2.5倍酶溶液
在1x kinase base buffer中加入激酶。
(2)配制2.5倍肽溶液
在1x kinase base buffe中加入FAM标记肽和ATP。
(3)检测板已含5μL的10%DMSO溶液。
(4)将2.5倍酶溶液转移到检测板上,在384孔检测板的每孔中加入2.5倍酶溶液10μL。
(5)室温孵育10分钟。
(6)将2.5倍肽溶液转移到检测板上,在384孔检测板的每孔中加入2.5倍肽溶液10μL。
(7)激酶反应和停止
28℃恒温培养下反应,终止反应时加入25μL的Stop Buffer。
(8)Caliper收集数据。
2.4数据分析
曲线拟合
(1)从Caliper中复制相关数据。
(2)将数据转换为抑制率
%inhibition=(max-conversion)/(max-min)*100
其中max:DMSO对照组数值;min:最低数值
(3)将数据导入XLFit(Version 5.4.0.8),得到IC
计算公式是Y=Bottom+(Top-Bottom)/(1+(IC
具体结果如表所示:
以上数据显示,本发明多个实施例化合物对SHP2及MEK1都具有良好的酶水平抑制作用。
实施例12
化合物体外抗增殖活性测试
1.实验步骤
(1)PBS溶液进行高压灭菌,置于冰箱4℃保存。
(2)称量胰蛋白酶和胰酶消化液,加入超纯水充分溶解,用微孔过滤器过滤得液体,置于冰箱-20℃保存。
(3)分别称取培养基粉和NaHCO
(4)将人结直肠癌细胞DLD-1从液氮罐中取出,立刻置于37℃恒温水浴锅中,摇晃使其融化,再将细胞倒入培养瓶中,加入培养液(含10%胎牛血清)稀释。将稀释后的培养基转入离心管中,1000r/min离心5分钟,舍弃上清液,再加入新鲜的培养基吹打混匀,移入培养瓶中培置于5%CO
(5)取对数期细胞,倒掉旧培养基,加入胰蛋白酶溶液消化3分钟,加入含10%胎牛血清的新鲜培养基终止消化,将溶液转移至离心管,1000r/min离心5分钟,舍弃上清液。加入培养基将其配制成细胞悬浊液,进行细胞计数。计数完成后,按照每孔500-1000个细胞浓度将细胞植于96孔板中。将铺好细胞的96孔板置于37℃、5%CO
2.数据处理
绘制曲线并计算药物对细胞的抑制率及IC
抑制率=[(对照组平均OD值-实验组平均OD值)/(对照组平均OD值-空白对照组平均OD值)]X 100%
3.实验结果
化合物对结直肠癌细胞株DLD-1细胞的抑制活性如下:
以上数据显示,本发明实施例化合物对DLD-1细胞的增殖具有良好的抑制作用。相比较单独使用SHP2抑制剂(SHP099)、MEK1抑制剂(ARRY-162)而言,本发明实施例具备更好的体外抗增殖活性。
- 一类HDAC/ALK双靶点抑制剂及其制备方法与应用
- EGFR和HER2双靶点酪氨酸激酶抑制剂及制备方法和用途
- 一种具有IDO1/TDO双靶点的小分子化合物及其制备方法与应用
- 吲哚胺2,3-双加氧酶抑制剂化合物及其制备方法和用途
- N-酰基苯磺酰胺异羟肟酸类Bcl-2、HDAC双靶点抑制剂及制备方法和应用
- BChE和HDAC双靶点抑制剂及其制备方法和应用