一组戴索唑类化合物及其制备方法和应用
文献发布时间:2023-06-19 19:30:30
技术领域
本发明涉及一组戴索唑类化合物,以及该类戴索唑化合物产生菌株、制备方法和其在制备抗肿瘤药物中的应用。属于微生物技术及其制品与医药应用领域。
背景技术
戴索唑类化合物(disorazols)是由纤维堆囊菌(Soranguiumcellulosum)产生的一类大环双内酯化合物,因其结构新颖活性独特而备受关注,目前约有30种衍生物被报道。该类化合物对多种肿瘤细胞系具有强效的细胞毒活性,半数抑制浓度大多纳摩尔至皮摩尔水平。进一步对其作用机理进行研究发现,该类化合物主要是通过抑制细胞微管蛋白的组装而发挥作用,与现在临床上使用的化疗药物长春花碱类接近,这也在一定程度上增加了其成药的可能性。
尽管目前已报到的戴索唑类化合物在体外显示了较强的抗肿瘤活性,但是在体内试验的效果大都不尽如人意,除了disorazol Z衍生后的抗体偶联药物AEZS-125到了临床二期,其余disorazols均未能进入临床试验,其中A1可能由于过高的毒性和体内不稳定未能开发为临床药物,因此有必要设计合成新的戴索唑类衍生物,为抗肿瘤药物开发提供化合物资源。申请人通过对重组菌E264ΔBAC::attB/3P-disDH1
发明内容
针对现有抗肿瘤药物急需开发的现状,本发明的目的在于提供一组戴索唑类化合物,以及该戴索唑类化合物的产生菌株、制备方法,和其在制备抗肿瘤药物中的应用。
本发明所述的一组戴索唑类化合物,其特征在于:所述戴索唑类化合物是命名为戴索唑F4的化合物1,其化学构式如式Ⅰ所示;或是命名为戴索唑F5的化合物2,其化学构式如式Ⅱ所示:
上述戴索唑类化合物的制备方法,其特征在于:戴索唑F4的化合物1是由工程菌株泰国伯克氏菌E264ΔBAC::attB/3P-disDH1
其中:发酵条件是所述工程菌株泰国伯克氏菌在M9培养基中,30℃,200rpm发酵,3天后加入灭菌后的大孔树脂,继续培养2天,发酵结束;其中所述M9培养基的配方是:葡萄糖10g/L,磷酸氢二钠6.8g/L;磷酸二氢钠3.0g/L;氯化钠1g/L;硫酸铵1.0g/L;七水硫酸镁0.5g/L;柠檬酸钠0.5g/L。
本发明所述戴索唑类化合物在制备抗肿瘤药物中的应用。
其中:所述戴索唑类化合物是命名为戴索唑F4的化合物1;或是命名为戴索唑F5的化合物2。
本发明提供了一种含戴索唑类化合物的抗肿瘤药物,其特征在于:所述药物含有治疗有效量的命名为戴索唑F4的化合物1或命名为戴索唑F5的化合物2和药学上可接受的载体。
本发明提供了一组生产上述戴索唑类化合物的工程菌株,其特征在于:所述生产戴索唑F4的工程菌株是基因工程菌泰国伯克氏菌E264ΔBAC::attB/3P-disDH1
其中:所述工程菌株E264ΔBAC::attB/3P-disDH1
其中:所述工程菌株E264ΔBAC::attB/3P-disDH7
上述泰国伯克氏菌E264ΔBAC::attB/3P-disDH1
本发明提供了一组戴索唑类化合物(戴索唑F4的化合物1或戴索唑F5的化合物2),所述化合物由重组菌E264ΔBAC::attB/3P-disDH1
附图说明
图1:戴索唑F4的化合物1在氘代甲醇中的核磁图谱。
其中:(A)1H谱;(B)13C谱;(C)DEPT谱;(D)HSQC谱;(E)1H-1H COSY谱;(F)HMBC谱。
图2:戴索唑F5的化合物2在氘代甲醇中的核磁图谱。
其中:(A)1H谱;(B)13C谱;(C)DEPT谱;(D)HSQC谱;(E)1H-1H COSY谱;(F)HMBC谱。
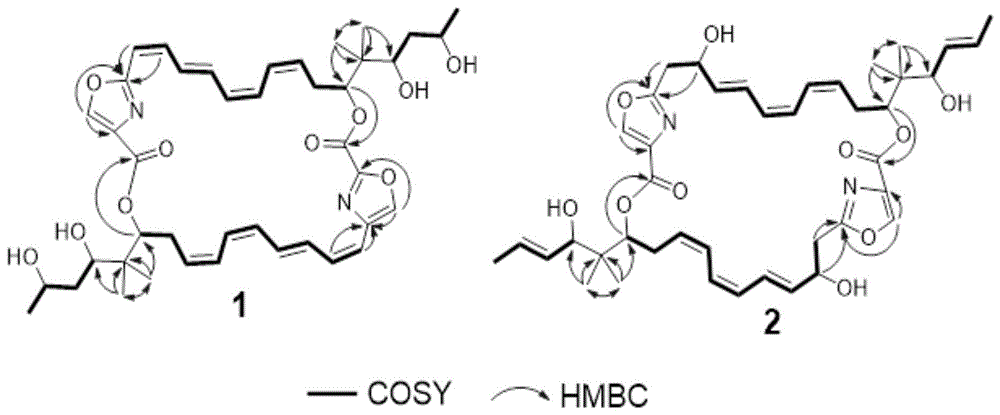
图3:戴索唑F4的化合物1及戴索唑F5的化合物2关键HMBC和
具体实施方式
下面结合附图和具体实施例对本发明内容进行详细说明。
如下所述例子仅是本发明的较佳实施方式而已,应该说明的是,下述说明仅仅是为了解释本发明,而不应视为限制本发明的范围,凡是依据本发明的技术实质对实施方式所做的任何简单修改,等同变化与修饰,均属于本发明技术方案的范围内。
实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。其他实验方法及试剂或仪器如无特殊说明,均为相关领域常规方法与市售常规产品。质粒p15A-amp-ccdB获取见文献((Wang,H.;Li,Z.;Jia,R.,et al.Nat.Protoc.2016,11,1175-1190),质粒p15A-genta-int-attP-3P-dis获取见文献(Wang Z.et al.Front.Microbiol.13:1073243.doi:10.3389/fmicb.2022.1073243),菌株泰国伯克氏菌E264(ATCC 700388)可购自DSMZ,突变株E264ΔBAC::attB获取见文献(Wang Z.et al.Front.Microbiol.13:1073243.doi:10.3389/fmicb.2022.1073243),菌株E.coli GB2005、E.coli GB05-redgyrA462获取见文献(Fu,J.;Bian,X.;Hu,S.,et al.Nat.Biotechnol.2012,30,440-446),E.coli WM3064购自北京科瑞思搏生物科技有限公司。
实施例1:泰国伯克氏菌E264ΔBAC::attB/3P-disDH1
1.1由质粒p15A-genta-int-attP-3P-dis出发构建质粒p15A-genta-int-attP-3P-disDH1
(1)以质粒p15A-amp-ccdB为模板,以DH1
DH1
CGGCGGACCGGGCTGAACGCCCCTCGATCCCCGTGGACCGCCTGATCGCCGATGTTTAAACTAAATACATTCAAATATG
DH1
(2)从平板上挑取GB05-redgyrA462至1mL的LB培养基中(1.5mL EP管),37℃,950rpm培养过夜。
(3)取40uL过夜培养的GB05-redgyrA462接种至1.3mL的LB液体培养基中,37℃,950rpm,培养2h。
(4)将GB05-redgyrA462置于离心机中,9000rpm离心30秒,去掉上清;
(5)用1ml无菌去离子水将菌体重悬,再次9000rpm离心30秒,去掉上清;
(6)重复步骤5,并用约50μL去离子水将菌体重悬;
(7)将质粒p15A-genta-int-attP-3P-dis约200ng加入菌体;
(8)用移液器将混合液转移至1mm的电转杯中
(9)1250V电击后加入1ml LB培养基,将菌体充分悬浮后转移至1.5ml EP管中,37℃,950rpm复苏60min;
(10)复苏完后9000rpm离心1min,去掉上清,并用残留的培养基将菌体重新悬浮后涂布于含有Genta(庆大霉素)的LB平板上;
注:步骤2-10是将质粒电转进大肠杆菌GB05-redgyrA462中,此菌株含有诱导型red重组系统,并且GyrA亚基的462位氨基酸由精氨酸突变为半胱氨酸,导致E.coli细胞产生CcdB(CcdB是一种毒蛋白)抗性。
(11)挑取部分长出的克隆,接种至1mL的含有Genta的LB培养基中(1.5mL EP管),37℃,950rpm培养过夜。
(12)取40uL过夜培养的GB05-redgyrA462/p15A-genta-int-attP-3P-dis接种至1.3mL的LB液体培养基中,30℃,950rpm,培养2h。
(13)向步骤12中的GB05-redgyrA462/p15A-genta-int-attP-3P-dis中加入20μL阿拉伯糖(10%v/w)诱导液,并转至37℃,950rpm诱导40min;
(14)将培养物置于离心机中,9000rpm离心30秒,去掉上清;
(15)用1mL无菌去离子水将菌体重悬,再次9000rpm离心30秒,去掉上清;
(16)重复步骤15,并用约50μL去离子水将菌体重悬;
(17)将步骤1中的PCR产物300ng加入菌体;
(18)用移液器将混合液转移至1mm的电转杯中
(19)1350V电击后加入1mL LB培养基,将菌体充分悬浮后转移至1.5ml EP管中,37℃,950rpm复苏60min;
(20)复苏完后9000rpm离心1min,去掉上清,并用残留的培养基将菌体重新悬浮后涂布于含有Amp(氨苄青霉素)的LB平板上;
(21)挑取部分长出的克隆,酶切鉴定,选取酶切正确的质粒p15A-genta-int-attP-3P-disDH1
注:步骤11-21是在大肠杆菌GB05-redgyrA462中,诱导red重组酶,使加入的PCR片段与质粒p15A-genta-int-attP-3P-dis进行重组,插入amp-ccdB并引入突变序列,以Amp作为正向筛选标记。
(22)从平板上挑取GB05-redgyrA462至1mL的LB培养基中(1.5mL EP管),37℃,950rpm培养过夜。
(23)取40μL过夜培养的GB05-redgyrA462接种至1.3mL的LB液体培养基中,37℃,950rpm,培养2h。
(24)将GB05-redgyrA462置于离心机中,9000rpm离心30秒,去掉上清;
(25)用1mL无菌去离子水将菌体重悬,再次9000rpm离心30秒,去掉上清;
(26)重复步骤25,并用约50μL去离子水将菌体重悬;
(27)取步骤21中质粒p15A-genta-int-attP-3P-disDH1
(28)用移液器将混合液转移至1mm的电转杯中
(29)1250V电击后加入1ml LB培养基,将菌体充分悬浮后转移至1.5mL EP管中,37℃,950rpm复苏60min;
(30)复苏完后9000rpm离心1min,去掉上清,并用残留的培养基将菌体重新悬浮后涂布于含有Amp的LB平板上;
(31)挑取克隆,Amp液体培养,提取质粒,取一部分进行酶切鉴定并测序。
注:步骤22-31是将质粒p15A-genta-int-attP-3P-disDH1
(32)将重转化的质粒p15A-genta-int-attP-3P-disDH1
(33)酶切后的质粒用除盐膜除盐40分钟
(34)酶切并除盐后的质粒用DNA聚合酶T4处理,所用程序依次为25℃,30分钟,70℃,30分钟,50℃,30分钟,4℃永久保存。
(35)从平板上挑取GB2005至1mL的LB培养基中(1.5mL EP管),37℃,950rpm培养过夜。
(36)取40uL过夜培养的GB2005接种至1.3mL的LB液体培养基中,37℃,950rpm,培养2h。
(37)将GB2005置于离心机中,9000rpm离心30秒,去掉上清;
(38)用1mL无菌去离子水将菌体重悬,再次9000rpm离心30秒,去掉上清;
(39)重复步骤38,并用约50μL去离子水将菌体重悬;
(40)取步骤34中酶切并除盐后的质粒约200ng加入菌体;
(41)用移液器将混合液转移至1mm的电转杯中
(42)1250V电击后加入1ml LB培养基,将菌体充分悬浮后转移至1.5ml EP管中,37℃,950rpm复苏60min;
(43)复苏完后9000rpm离心1min,去掉上清,并用残留的培养基将菌体重新悬浮后涂布于含有Genta的LB平板上;
(44)挑取克隆至Genta的LB中培养,提取质粒,酶切验证并测序,得到质粒p15A-genta-int-attP-3P-disDH1
注:步骤32-44是用限制性内切酶PmeI切掉amp-ccdB,并暴露同源臂,利用T4DNA聚合酶处理同源臂,产生5’端单链,通过退火自身环化成双链,得到聚酮合酶模块1中的脱水酶结构域DH1位点失活突变的的质粒p15A-genta-int-attP-3P-disDH1
1.2由质粒p15A-genta-int-attP-3P-dis出发构建质粒p15A-genta-int-attP-3P-disDH7
(1)以质粒p15A-amp-ccdB为模板,以DH7
DH7
ACACGTCGACGTTCAGAGAGCAGCGGTTCGCCACGACCTTCACCGGCGAGGAGAT CCTCCTCTCGGACGTTTAAACTAAATACATTCAAATATGTATC
DH7
CTCCAGGTAAGCCGTGCCCGGCAGCAGGGCGCGGCCTCGGATCCGGAAGTCCGAG AGGAGGATCTCCTCGCCGGTGAAGTTTAAACAGCCCGCTCATTAG
(2)从平板上挑取GB05-redgyrA462至1mL的LB培养基中(1.5mL EP管),37℃,950rpm培养过夜。
(3)取40uL过夜培养的GB05-redgyrA462接种至1.3mL的LB液体培养基中,37℃,950rpm,培养2h。
(4)将GB05-redgyrA462置于离心机中,9000rpm离心30秒,去掉上清;
(5)用1ml无菌去离子水将菌体重悬,再次9000rpm离心30秒,去掉上清;
(6)重复步骤5,并用约50μL去离子水将菌体重悬;
(7)将质粒p15A-genta-int-attP-3P-dis约200ng加入菌体;
(8)用移液器将混合液转移至1mm的电转杯中
(9)1250V电击后加入1ml LB培养基,将菌体充分悬浮后转移至1.5ml EP管中,37℃,950rpm复苏60min;
(10)复苏完后9000rpm离心1min,去掉上清,并用残留的培养基将菌体重新悬浮后涂布于含有Genta的LB平板上;
注:步骤2-10是将质粒电转进大肠杆菌GB05-redgyrA462中,此菌株含有诱导型red重组系统,并且GyrA亚基的462位氨基酸由精氨酸突变为半胱氨酸,导致E.coli细胞产生CcdB(CcdB是一种毒蛋白)抗性。
(11)挑取部分长出的克隆,接种至1mL的含有Genta的LB培养基中(1.5mL EP管),37℃,950rpm培养过夜。
(12)取40uL过夜培养的GB05-redgyrA462/p15A-genta-int-attP-3P-dis接种至1.3mL的LB液体培养基中,30℃,950rpm,培养2h。
(13)向步骤12中的GB05-redgyrA462/p15A-genta-int-attP-3P-dis中加入20μL阿拉伯糖(10%v/w)诱导液,并转至37℃,950rpm诱导40min;
(14)将培养物置于离心机中,9000rpm离心30秒,去掉上清;
(15)用1mL无菌去离子水将菌体重悬,再次9000rpm离心30秒,去掉上清;
(16)重复步骤15,并用约50μL去离子水将菌体重悬;
(17)将步骤1中的PCR产物300ng加入菌体;
(18)用移液器将混合液转移至1mm的电转杯中;
(19)1350V电击后加入1mL LB培养基,将菌体充分悬浮后转移至1.5ml EP管中,37℃,950rpm复苏60min;
(20)复苏完后9000rpm离心1min,去掉上清,并用残留的培养基将菌体重新悬浮后涂布于含有Amp的LB平板上;
(21)挑取部分长出的克隆,酶切鉴定,选取酶切正确的质粒p15A-genta-int-attP-3P-disDH7
注:步骤11-21是在大肠杆菌GB05-redgyrA462中,诱导red重组酶,使加入的PCR片段与质粒p15A-genta-int-attP-3P-dis进行重组,插入amp-ccdB并引入突变序列,以Amp作为正向筛选标记。
(22)从平板上挑取GB05-redgyrA462至1mL的LB培养基中(1.5mL EP管),37℃,950rpm培养过夜。
(23)取40μL过夜培养的GB05-redgyrA462接种至1.3mL的LB液体培养基中,37℃,950rpm,培养2h。
(24)将GB05-redgyrA462置于离心机中,9000rpm离心30秒,去掉上清;
(25)用1mL无菌去离子水将菌体重悬,再次9000rpm离心30秒,去掉上清;
(26)重复步骤25,并用约50μL去离子水将菌体重悬;
(27)取步骤21中质粒p15A-genta-int-attP-3P-disDH7
(28)用移液器将混合液转移至1mm的电转杯中
(29)1250V电击后加入1ml LB培养基,将菌体充分悬浮后转移至1.5mL EP管中,37℃,950rpm复苏60min;
(30)复苏完后9000rpm离心1min,去掉上清,并用残留的培养基将菌体重新悬浮后涂布于含有Amp的LB平板上;
(31)挑取克隆,Amp液体培养,提取质粒,取一部分进行酶切鉴定并测序。
注:步骤22-31是将质粒p15A-genta-int-attP-3P-disDH7
(32)将重转化的质粒p15A-genta-int-attP-3P-disDH7
(33)酶切后的质粒用除盐膜除盐40分钟
(34)酶切并除盐后的质粒用DNA聚合酶T4处理,所用程序依次为25℃,30分钟,70℃,30分钟,50℃,30分钟,4℃永久保存。
(35)从平板上挑取GB2005至1mL的LB培养基中(1.5mL EP管),37℃,950rpm培养过夜。
(36)取40uL过夜培养的GB2005接种至1.3mL的LB液体培养基中,37℃,950rpm,培养2h。
(37)将GB2005置于离心机中,9000rpm离心30秒,去掉上清;
(38)用1mL无菌去离子水将菌体重悬,再次9000rpm离心30秒,去掉上清;
(39)重复步骤38,并用约50μL去离子水将菌体重悬;
(40)取步骤34中酶切并除盐后的质粒约200ng加入菌体;
(41)用移液器将混合液转移至1mm的电转杯中
(42)1250V电击后加入1ml LB培养基,将菌体充分悬浮后转移至1.5ml EP管中,37℃,950rpm复苏60min;
(43)复苏完后9000rpm离心1min,去掉上清,并用残留的培养基将菌体重新悬浮后涂布于含有Genta的LB平板上;
(44)挑取克隆至Genta的LB中培养,提取质粒,酶切验证并测序,得到质粒p15A-genta-int-attP-3P-disDH7
注:步骤32-44是用限制性内切酶PmeI切掉amp-ccdB,并暴露同源臂,利用T4DNA聚合酶处理同源臂,产生5’端单链,通过退火自身环化成双链,得到聚酮合酶模块7中脱水酶结构域DH7位点失活突变的的质粒p15A-genta-int-attP-3P-disDH7
1.3将质粒p15A-genta-int-attP-3P-disDH1
在本部分实验中,将表达质粒p15A-genta-int-attP-3P-disDH1
(1)挑取接合转移辅助大肠杆菌WM3064,接种至含有1mM DAP(二氨基庚二酸)的LB中,37℃过夜培养;
(2)取40μL过夜培养的WM3064接种至含有1mM DAP的1.3mL的LB液体培养基中,30℃,950rpm,培养2h,然后转至37℃,950rpm培养40min;
(3)将WM3064置于离心机中,9000rpm离心30秒,去掉上清;
(4)用1ml无菌去离子水将菌体重悬,再次9000rpm离心30秒,去掉上清;
(5)重复步骤4,并用约50μL去离子水将菌体重悬;
(6)将质粒p15A-genta-int-attP-3P-disDH1
(7)1250V电击后加入含有1mM DAP的1ml LB培养基,将菌体充分悬浮后转移至1.5ml EP管中,37℃,900rpm复苏60min;
(8)复苏完后9000rpm离心30秒,去掉上清,并用残留的培养基将菌体重新悬浮后涂布于含有1mM DAP和5μg/mL Genta的LB平板,置于37℃过夜培养。
(9)在操作(8)的同时,将泰国伯克氏菌E264ΔBAC::attB划线接种至LB平板,同样置于37℃过夜培养。
(10)步骤(8)长出的克隆为携带质粒p15A-genta-int-attP-3P-disDH1
(11)刮取菌苔,划线于不含DAP的5μg/mL Genta LB平板上。此时,大肠杆菌WM3064由于缺少DAP,因此不生长,只有质粒p15A-genta-int-attP-3P-disDH1
(12)挑取克隆PCR鉴定,确定得到突变株E264ΔBAC::attB/3P-disDH1
1.4将质粒p15A-genta-int-attP-3P-disDH7
在本部分实验中,将表达质粒p15A-genta-int-attP-3P-disDH7
(1)挑取接合转移辅助大肠杆菌WM3064,接种至含有1mM DAP(二氨基庚二酸)的LB中,37℃过夜培养;
(2)取40μL过夜培养的WM3064接种至含有1mM DAP的1.3mL的LB液体培养基中,30℃,950rpm,培养2h,然后转至37℃,950rpm培养40min;
(3)将WM3064置于离心机中,9000rpm离心30秒,去掉上清;
(4)用1ml无菌去离子水将菌体重悬,再次9000rpm离心30秒,去掉上清;
(5)重复步骤4,并用约50μL去离子水将菌体重悬;
(6)将质粒p15A-genta-int-attP-3P-disDH7
(7)1250V电击后加入含有1mM DAP的1ml LB培养基,将菌体充分悬浮后转移至1.5ml EP管中,37℃,900rpm复苏60min;
(8)复苏完后9000rpm离心30秒,去掉上清,并用残留的培养基将菌体重新悬浮后涂布于含有1mM DAP和6μg/mL Genta的LB平板,置于37℃过夜培养。
(9)在操作(8)的同时,将泰国伯克氏菌E264ΔBAC::attB划线接种至LB平板,同样置于37℃过夜培养。
(10)步骤(8)长出的克隆为携带质粒p15A-genta-int-attP-3P-disDH7
(11)刮取菌苔,划线于不含DAP的5μg/mL Genta LB平板上。此时,大肠杆菌WM3064由于缺少DAP,因此不生长,只有质粒p15A-genta-int-attP-3P-disDH7
(12)挑取克隆PCR鉴定,确定得到突变株E264ΔBAC::attB/3P-disDH7
实施例2:泰国伯克氏菌E264ΔBAC::attB/3P-disDH1
将实施例1构建的E264ΔBAC::attB/3P-disDH1
实施例3:戴索唑类化合物的分离纯化和结构鉴定
3.1目的化合物的分离纯化
将实施例2中泰国伯克氏菌E264ΔBAC::attB/3P-disDH1
将实施例2中泰国伯克氏菌E264ΔBAC::attB/3P-disDH7
3.2戴索唑类化合物的结构鉴定
进一步地对上述分离到的目的化合物进行高分辨质谱、核磁的检测。
仪器型号:核磁测试为Bruker AVNEO 600MHz核磁系统;高分辨质谱用BrukerImpact HD microTOF Q III质谱仪。
目的化合物的理化性质及波谱数据(见图1、图2)如下,判定为戴索唑类化合物。
化合物1,命名为戴索唑F4:白色粉末,分子式C
化合物2,命名为戴索唑F5:白色粉末,分子式C
其化学构式如式Ⅰ(化合物1)和式Ⅱ(化合物1)所示:
表1化合物1和2在MeOH-d
根据HRESIMS推测戴索唑F4(化合物1)的分子式为C
采用同样的方式推导了化合物2的结构(见图3)。
实施例4:戴索唑类化合物的抗肿瘤细胞毒活性测试
测试样品为上述实施例3中分离精制的化合物1和2。精密称取适量样品,用DMSO配制成所需浓度的溶液,用MTT法进行抗肿瘤细胞毒活性测定。
抗肿瘤细胞毒活性结果见表2。
其中:戴索唑F
表2.化合物1和2抗肿瘤细胞毒活性(IC
比较试验显示:戴索唑F4的细胞毒性比A1降低4个数量级,为微摩尔水平,戴索唑F5的细胞毒性比A1降低1个数量级,为纳摩尔水平。由于化合物1和2表现出的高效抗肿瘤细胞毒活性,表明所述戴索唑类化合物具有开发成为抗肿瘤药物的潜力,可能进一步开发成新型抗肿瘤药物。