一种与鸡核糖体RNA结合的特异性探针组及应用
文献发布时间:2024-01-17 01:16:56
技术领域
本发明涉及一种与鸡核糖体RNA结合的特异性探针组及应用,属于生物技术领域。
背景技术
转录组测序是目前除全基因组测序外广泛应用于生物学和遗传学的一种高效实验方法,通过转录组测序可了解生物体的基因表达情况,进而发现影响发育、生殖或者疾病的关键基因。随着测序技术的不断发展,各种非编码RNA,如长非编码RNA(lncRNA)、微小非编码RNA(miRNA)和环状非编码RNA(circRNA)被确定在生物的发育中发挥重要功能。目前通过mRNA建库手段可同时在真核生物总RNA中检测出mRNA和含有poly A结构的lncRNA的表达量,但研究表明真核生物中核糖体RNA(rRNA)分为细胞质核糖体RNA和线粒体核糖体RNA,二者共占总RNA的80%左右,这两种类型的RNA表达丰度高且高度保守,因此rRNA的存在导致大多数测序的数据是无效信息。尽管转录组测序价格逐渐降低,但通过提高测序数据量获得完整有效信息的方法仍会造成浪费,因此mRNA测序时需预先去除总RNA中的rRNA。
目前主要有三种去除核糖体RNA的方法,第一种方法使用oligo-DT磁珠富集带有Poly A结构的mRNA,间接实现去除rRNA的效果;第二种方法通过设计链霉亲和素标记与rRNA互补的探针,结合后通过链霉亲和素磁珠去除rRNA;第三种方法设计大量rRNA反义探针使其与rRNA结合形成rRNA-DNA双链,通过RNase H酶降解rRNA。
第一种方法是目前主流的转录组测序方法,市场上有多种beads试剂盒产品,如诺唯赞公司的VAHTS mRNA Capture Beads和翌圣公司的Oligo dT-Coated Magnetic Beads,该方法虽能有效去除rRNA,但会导致总RNA中不含有poly A结构的非编码RNA和质量较差的mRNA丢失。第二种方法最为稳定,但其操作复杂且链霉亲和素标记的探针与磁珠价格昂贵,不适用于大量样本的处理。第三种方法设计rRNA序列反义探针体系,通过梯度降温使探针与靶RNA结合形成靶RNA-DNA探针杂交链,通过RNase H酶消化杂交链中的靶RNA,最后借助DNase I酶清除基因组DNA和DNA探针。该方法操作便捷,但由于各物种之间rRNA序列一致性有限,因此商业试剂盒大多通过第三种方法设计针对人或小鼠的探针进行rRNA清除,申请人通过检索发现中国专利申请202010056986.X公开了一种去除动物总RNA中的核糖体RNA的方法,其针对所有物种,在使用后只能把所有物种核糖体RNA相似的区域清除,不能去除掉物种特异性的核糖体RNA区域并且无法做到仅针对鸡这一品种。目前,市面上尚未见可高效去除鸡核糖体RNA的方法,极大限制了鸡这一物种的科学研究。
发明内容
本发明的目的是针对现有技术存在的缺陷,提出与鸡核糖体RNA结合的特异性探针组,并将该特异性探针组用于有效去除鸡总RNA中的核糖体RNA。
本发明通过以下技术方案解决技术问题:首先提供与鸡核糖体RNA结合的特异性探针组,由145条反义探针组成,其序列为SEQ ID NO.1-SEQ ID NO.145。
本发明通过以下技术方案进一步解决技术问题,与鸡核糖体RNA结合的特异性探针组的应用,包括以下步骤:
第一步、针对鸡的细胞质核糖体RNA和线粒体核糖体RNA序列设计特异性反义探针,提取鸡的总RNA,通过梯度降温方法使探针与鸡的rRNA结合形成靶RNA-DNA探针杂交双链;
第二步、采用RNase H酶清除RNA-DNA双链中的靶RNA;
第三步、采用DNase I酶清除基因组DNA和探针;
第四步、采用磁珠回收纯化后的RNA。
以上方法的第一步中的杂交体系为杂交Buffer 3μL、探针1μL、总RNA 1μg,反应总体积15μL,反应条件为105℃热盖,95℃ 2min、95℃-22℃ 0.1℃/s,22℃ 5min、至4℃结束。
所述第二步中RNase H酶处理的反应体系如下:RNase H Buffer 3μL、RNase H 2μL、上步产物15μL、总体积20μL;反应条件为37℃,30min。
所述第三步中DNase I酶处理的反应体系如下:DNase I Buffer 27.5μL、DNase I2.5μL、上一步产物20μL、总体积50μL;反应条件为37℃,30min。
本发明针对鸡核糖体RNA序列设计特异性反义探针,通过梯度降温方法使设计的探针与鸡总RNA中的rRNA结合形成杂交链,再通过RNase H酶和DNase I酶将杂交链中的rRNA、基因组DNA和探针去除,随后使用Qsep100全自动核酸蛋白分析系统进行质检,最后通过建库测序手段检测rRNA的残留情况。本发明中还添加了线粒体中核糖体RNA的互补探针,在原本的细胞质核糖体RNA的去除工作上进行了补充。
本发明的有益效果是:该方法获得的RNA产物可通过建库试剂盒同时对mRNA和部分含有poly A结构的lncRNA进行测序,减少测序数据和样品的浪费,极大地降低了RNA样品转录组测序成本,加强转录组测序技术在鸡这一物种中的应用。解决了市面上尚缺少特异性去除鸡总RNA中核糖体RNA方法的问题,本发明中的去rRNA方法虽耗时略高于其余两种方法且需要数次磁珠纯化过程,但有效避免了oligo-DT磁珠法造成的部分RNA信息丢失和链霉亲和素标记法的高成本需求,有利于转录组测序技术在鸡这一物种的应用。
附图说明
图1为尚未经过本发明探针处理的总RNA样品Qsep100仪器检测结果。
图2为经过本发明探针处理后的总RNA样品Qsep100仪器检测结果。
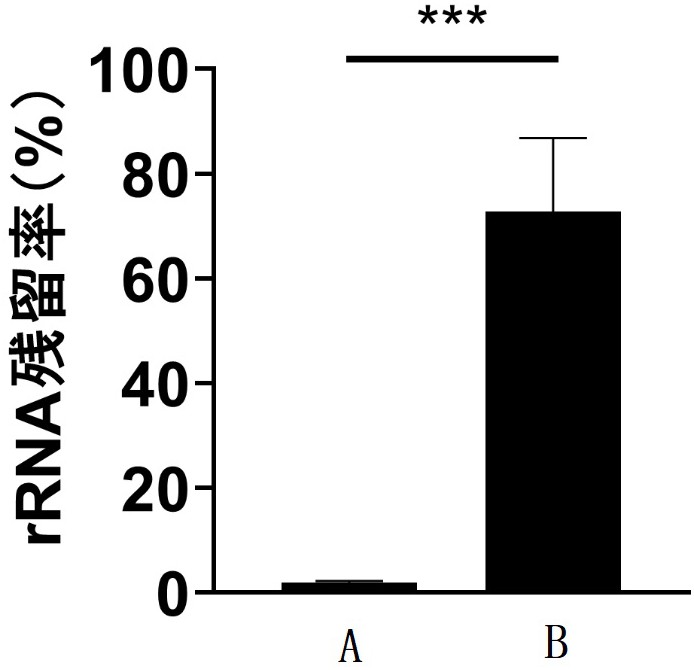
图3 为经过本发明探针A和商业化探针B处理后核糖体RNA残留率对比。
实施方式
以下实例便于更好的理解本发明,但不限定于本发明。以下实施方法中如无特别说明,均为常规方法,且下述所用试剂耗材均为试剂公司购买所得。以下实验采用三个不同样本,结果以平均值展示。
Hybridization Buffer,RNase H Buffer,RNase H,DNase I Buffer,DNase I,Hieff NGS® MaxUp Human rRNA Depletion Kit,RNA cleaner beads,Hieff NGS UltimaDual-mode RNA Library Prep Kit均来自于翌圣生物科技(上海)股份有限公司。双Barcode环化试剂盒,MGISEQ-2000RS 测序载片,cPAS条形码引物均来自于深圳华大智造科技股份有限公司。
实施例
本实施例按照以下方法去除鸡总RNA中核糖体RNA:
1.设计并制备探针
从NCBI网站上获取鸡的细胞质核糖体RNA及线粒体核糖体RNA序列,经过设计获得145条反义探针,这些探针在鸡各品种间保守,具有兼容性。
145条探针序列信息如下(5’-3’方向):
以上所有探针制备完成后,稀释混合形成探针池,保证每一条探针在探针池中的终浓度为1μM。取13胚龄鸡胚皮肤组织,采用Trizol试剂提取样本总RNA,使用Qsep100仪器总RNA的完整性,如图1所示。
2.取1μg的总RNA进行探针杂交,按以下比例配置体系。该体系中的探针分别使用本发明设计探针和商业化探针进行对比,其余条件保持一致。
将溶液混匀后瞬离至管底,放置于PCR仪上,反应程序如下所示:
3.使用RNase H酶消化,反应体系如下:
将混合溶液放置在PCR仪上,37℃,30min。
4.使用DNase I酶消化
将混合溶液震荡混匀后放置在PCR仪上,37℃ 30min。
5.RNA纯化
(5.1)将RNA Cleaner磁珠在室温下平衡半小时以上并涡旋震荡混匀,且用DEPC水配置新鲜的80%乙醇溶液于室温放置备用。
(5.2)吸取110μl的RNA Cleaner磁珠到上述反应液产物中,震荡混匀,室温孵育5min。
(5.3)将PCR管放置在磁力架上,静置5min,等待溶液澄清后弃上清。
(5.4)将PCR管始终放置在磁力架上,加入80%的乙醇溶液漂洗,30s后吸取上清弃掉。
(5.5)重复步骤(5.4),共漂洗两次。
(5.6)用移液器吸净残留液体后,室温干燥磁珠约5min。
(5.7)RNA洗脱:将PCR管从磁力架上取下,加入6μL的DEPC水,充分混匀磁珠,室温静置5min。
(5.8)将PCR管放置在磁力架上吸附5min,取4.25μL含有纯化RNA的上清液于新的200μL PCR管中,纯化后的RNA应立即进行文库构建。
6.测序文库的构建
本研究利用Hieff NGS
(6.1)RNA片段化
向步骤(5.8)的上清液中加入等体积的2x Frag/Prime Buffer,用移液器充分吹打混匀后,按照如下程序进行片段化:
(6.2)第一链cDNA的合成
配置第一链cDNA合成的反应液,体系如下:
用移液器吹打混匀,瞬离。
将PCR管放置在PCR仪上,进行第一链cDNA合成,反应程序如下:
(6.3)第二链cDNA的合成/末端修复/加A
配置第二链cDNA合成的反应液,体系如下:
用移液器吹打混匀,瞬离。
将PCR管放置在PCR仪上,进行第二链cDNA的合成,反应程序如下:
(6.4)连接接头
配置连接接头的反应液,体系如下:
用移液器吹打混匀,瞬离。
将PCR管放置于PCR仪上,进行接头连接,反应程序如下:
(6.5)连接产物纯化
将DNA Selection Beads磁珠放置室温平衡30min以上并涡旋震荡混匀,且用DEPC水配置80%的乙醇溶液于室温放置备用。
吸取30μL DNA Selection Beads磁珠至上述反应液产物中,用移液器吹打混匀,室温放置5min。
将PCR管瞬离后放置于磁力架上,静置5min,吸取上清液弃掉。
将PCR管始终保持在磁力架上,加入200μL新鲜配置的80%酒精漂洗,30s后,吸取上清弃掉。
重复上一步骤,共漂洗2次。
将PCR从磁力架上取下,加入26μL的DEPC水,用移液器吹打混匀之后,室温静置5min。将PCR管瞬离后置于磁力架上静置3min,吸取25μL上清液于PCR管中进行新一轮的纯化。
吸取20μL DNA Selection Beads磁珠至上一步反应液中,用移液器吹打混匀,室温孵育5min。
将PCR管瞬离后置于磁力架上,室温静置5min,吸取上清液弃掉。
将PCR管始终保持在磁力架上,加入200μL新鲜配置的80%酒精漂洗,30s后,吸取上清弃掉。
重复上一步骤,共漂洗2次。
用移液器吸取残留液体,开盖室温干燥磁珠。
将PCR管子从磁力架上取下,加入12μL DEPC水,吹打混匀,室温孵育5min。
将PCR管瞬离,置于磁力架上,静置5min,吸取10μL的上清液于新的PCR管中,进行下一步反应。
(6.6)文库扩增配置文库扩增的反应液,体系如下:
用移液器吹打混匀,瞬离。
将PCR管置于PCR仪中,进行文库扩增反应,反应程序如下:
(6.7)文库纯化
将DNA Selection Beads磁珠放置室温平衡30min以上并涡旋震荡混匀,且用DEPC水配置80%的乙醇溶液于室温放置备用。
吸取22.5μL DNA Selection Beads磁珠至文库扩增产物中,吹打混匀,室温孵育5min。
将PCR管瞬离后置于磁力架上,室温静置5min,吸取上清液弃掉。
将PCR管始终保持在磁力架上,加入200μL新鲜配置的80%酒精漂洗,30s后,吸取上清弃掉。
重复上一步骤,共漂洗2次。
将PCR管始终置于磁力架上,开盖干燥磁珠约5min。
将PCR管从磁力架上取下,加入11μL DEPC水,用移液器吹打混匀,室温静置5min。
将PCR管瞬离,置于磁力架上,静置5min,吸取10μL的上清液于新的PCR管中。
(6.8)RNA文库质检
本研究利用Qsep100核酸蛋白分析仪对文库进行片段大小的检测,具体操作步骤如下:
将10x的Dilution Buffer用DEPC水稀释至1x; Separation Buffer不用稀释;Alignment Marker 体积 30μL,加入 20μl矿物油油封;Size Marker体积30μL,加入20μl矿物油。
使用1x的Dilution Buffer调整文库样本最佳浓度范围为 1-2ng/μL,体积为15-20μL,加入 0.2mL PCR 管中。
P(Park)为打胶槽,W(Wash)、C(Clean)位置为清洗槽,三个孔均加纯水,S槽加入Separation Buffer。
利用S2卡夹进行检测。
利用Qubit荧光计通过荧光检测法检测样品的浓度差异,并输出每个样本的浓度数据。
7.上机测序
本研究构建的转录组文库均利用MGISEQ-2000测序仪进行测序,测序时使用MGISEQ-2000RS 高通量测序试剂套装(PE100)。
(7.1)双Barcode环化
利用双Barcode环化试剂盒进行,具体步骤如下:
文库样本混样,根据数据量的分配进行投入,每条lane的数据量相同则等量投入。
变性,此步骤是将双链的文库变性成单链的文库,反应程序如下:
反应结束后,将PCR管放置在冰上孵育2min。
单链环化,配置以下反应液,并按照以下程序进行:
反应条件如下:
酶切消化,配置以下反应液,并按照以下程序进行:
反应条件如下:
环化产物纯化,利用DNA Clean Beads进行。
环化产物质检:利用Qubit2.0荧光定量仪进行。
(7.2)DNB制作
每张 MGISEQ-2000RS 测序载片含有 4 条 lane,一张载片上的每条 lane 都必须是同一份 DNB,每条 lane 需要 50 μL 的DNB。DNB的制作步骤具体操作如下:
计算每个DNB制备体系所需要的ssDNA文库的投入量,按照V=60fmol/C计算。
在冰上配置如下体系的反应液:
将反应混合液用漩涡振荡器震荡混匀,离心机瞬离,然后置于PCR仪中进行如下程序:
取出 DNB 聚合酶混合液 II(LC)置于冰上,离心机瞬离,置于冰上备用,使用前吹打混匀。
在上述反应液中加入如下组分:
反应混合液用漩涡振荡器震荡混匀,离心机瞬离,然后置于PCR仪中进行如下程序:
当 PCR 仪温度达到4℃后立即加入20μL DNB 终止缓冲液,用阔口吸头缓慢地吹打混匀。
DNB 制备完成后,取用2μL DNB,使用Qubit ssDNA Assay Kit和Qubit 2.0荧光定量仪进行浓度检测,浓度≥8ng/μL即为合格。
(7.3)加载DNB
从-20℃冰箱中取出测序载片彩盒,且在室温环境下放置60分钟到24小时。
在0.2ml EP管中配置DNB加载体系,如下表:
DNB加载体系用阔口吸头缓慢混匀,然后加载到测序载片里室温放置30min,即可转移至测序仪上使用。
(7.4)准备测序试剂槽
取出测序试剂槽,于常温水浴解冻,置于4℃冰箱备用。
使用无尘纸擦净盖板及孔位处的冷凝水。
将2.4mL的dNTPs混合液加入新的15mL灭菌管内,然后将2.4mL的DNA 聚合酶混合液加入管内的 dNTPs 混合液中,轻轻颠倒混匀,将混合液全部加到 1 号孔中。
将2.1mL的dNTPs混合液II加入新的15mL灭菌管内,然后将2.1mL的DNA 聚合酶混合液加入管内的 dNTPs 混合液II中,轻轻颠倒混匀,再将混合液全部加到2号孔中。
使用配套的透明封口膜将1号和2号加样孔封住。
用1 mL移液器移取500μLMDA聚合酶混合液加入到MDA试剂的试剂管中,颠倒混匀,然后将混匀液加入15号孔中,加入时确保管底部无气泡。
取出cPAS条形码引物3试剂盒中的cPAS AD153条形码引物3工作液,取 2.90mL加入到 4号孔位中,确保孔位底部无气泡。
将所有试剂加完后,混匀,此时测序试剂槽的上机前的准备工作完成。
8.文库上机检测
质检成功的文库使用MGISEQ-2000采用PE 100程序进行测序。测序结果完成后,使用生物信息学手段进行分析。
9.结果分析
去除核糖体RNA后的纯化RNA使用Qsep100仪器检测,由于经过rRNA去除过程,代表18S rRNA和28S rRNA的峰消失(图2)。对测序结果分析后发现,经过本发明探针并进行建库后,核糖体RNA的残留量仅为2%左右,而经过商业化产品中探针处理后样本的核糖体RNA残留率为70%左右,说明本发明中的探针去除鸡核糖体RNA的效果极佳(图3)。
除上述实施外,本发明还可以有其他实施方式。凡采用等同替换或等效变换形成的技术方案,均落在本发明要求的保护范围。
- 一种环状RNA hsa_circTGFBI_001及其特异性扩增引物和应用
- 在RNA建库过程中封闭核糖体RNA或球蛋白RNA的探针及其应用
- 一组用于鸡源性实时荧光PCR检测的特异性引物和探针