尼替西农的制备方法
文献发布时间:2024-01-17 01:24:51
技术领域
本发明涉及药物合成技术领域,具体而言,涉及一种尼替西农的制备方法。
背景技术
尼替西农,是HPPD抑制剂,通过抑制HT-1患者酪氨酸的正常代谢,可防止毒性极大的马来酰乙酰乙酸、延胡索酰乙酰乙酸及其旁路代谢产物琥珀酰丙酮蓄积,适用于1型遗传性酪氨酸血症(HT-1)。
目前,关于尼替西农制备的普遍策略是先将2-硝基-4-三氟甲基苯甲酸制备成羧酸衍生物(酰氯或甲酯),然后与1,3-环己二酮缩合得到尼替西农。然而,该缩合反应却受到酯化副反应的竞争,进而会降低产品的纯度和分离得率,其反应方程式为:
仅使用无机碱促进该缩合反应(US9783485),单步的得率不足50%,经研究发现,该反应在前3h会有10-20%的烯醇酯异构体产生,随着反应的继续,烯醇酯异构体水解为2-硝基-4-三氟甲基苯甲酸和1,3-环己二酮,这大大降低了原料的转化率。
为了将酯异构体转化为尼替西农以提高底物的转化率和分离得率,US5550165、WO2015101794使用有机氰化物三甲基氰基硅烷(TMSCN)作为催化剂,获得了单步反应产率85%、纯度95%的理想结果。该反应还可以使用有机氰化物丙酮氰醇(Organic&Biomolecular Chemistry,2021,19(47),10432-10443)为转化酯异构体的催化剂。然而,有机氰化物却很容易水解产生极毒的氢氰酸,它们的使用不符合现代制药对环境友好以及安全生产的发展要求。
CN201210492081.2则开发了三氯化铝、三乙胺体系促进该异构体的转化方法,获得了高纯度的尼替西农,但并没有报道该方法的分离得率。经研究发现,该反应在经过无机碱参与的缩合反应及提纯后,再与三氯化铝、三乙胺反应转化异构体,两步反应的摩尔得率不足50%(自2-硝基-4-三氟甲基苯甲酸计)。
鉴于上述问题的存在,有必要提供一种新的尼替西农的制备方法。
发明内容
本发明的目的是为了克服上述现有技术存在的缺陷而提出的一种尼替西农的制备方法。
本发明是这样实现的:
本发明提供一种尼替西农的制备方法,其包括:在无水氟化钾的参与下,2-硝基-4-三氟甲基苯甲酰氯和1,3-环己二酮于有机溶剂中发生缩合反应得到尼替西农。
本发明具有以下有益效果:
本发明提供一种尼替西农的制备方法,其包括:在无水氟化钾的参与下,2-硝基-4-三氟甲基苯甲酰氯和1,3-环己二酮于有机溶剂中发生缩合反应得到尼替西农,本发明提供的尼替西农的制备方法,避免使用含氰基的剧毒、高污染试剂。无水氟化钾的参与,提高了缩合反应的选择性,并可将2-硝基-4-三氟甲基苯甲酰氯和1,3-环己二酮发生缩合反应过程中产生的烯醇酯异构体转化为尼替西农,从而提高尼替西农的得率。
附图说明
为了更清楚地说明本发明实施例的技术方案,下面将对实施例中所需要使用的附图作简单地介绍,应当理解,以下附图仅示出了本发明的某些实施例,因此不应被看作是对范围的限定,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他相关的附图。
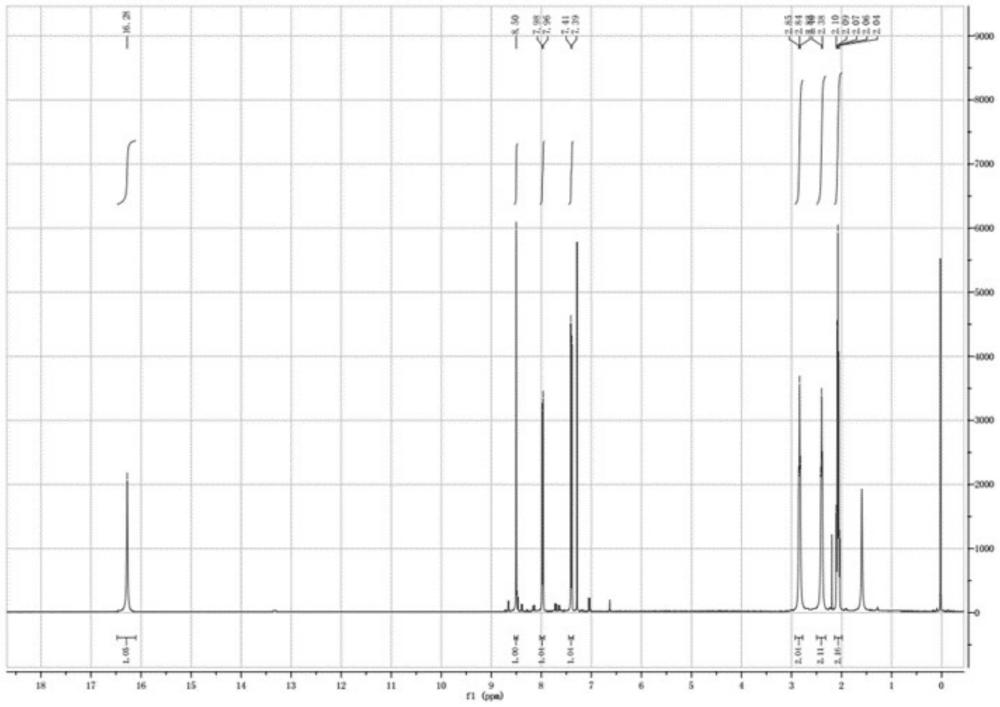
图1为实施例1中尼替西农的核磁氢谱图;
图2为实施例1中尼替西农的核磁碳谱图。
具体实施方式
为使本发明实施例的目的、技术方案和优点更加清楚,下面将对本发明实施例中的技术方案进行清楚、完整地描述。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
本发明的目的在于提供一种尼替西农的制备方法。
为实现本发明的上述目的,特采用以下的技术方案。
本发明实施例提供一种尼替西农的制备方法,其包括:在无水氟化钾的参与下,2-硝基-4-三氟甲基苯甲酰氯和1,3-环己二酮于有机溶剂中发生缩合反应得到尼替西农。
日前,国内外也有关于尼替西农合成方面的文献报道,但公开的文献仅简单介绍尼替西农的制备方法,并无详细说明。且尼替西农的合成过程中,或者为了得到较高的收率和纯度使用有毒有害的三甲基氰基硅烷(TMSCN)作为催化剂,或者为了避免使用有毒有害的催化剂而使用其他催化剂,但是尼替西农的得率又较低,如何在避免使用有毒有害的催化剂的同时,又获得较高的纯度和得率的尼替西农是本领域亟待解决的问题。发明人经长期实践,提出一种新的尼替西农合成方法,该合成方法是在无水氟化钾和有机溶剂存在下,使2-硝基-4-三氟甲基苯甲酰氯和1,3-环己二酮发生缩合反应得到尼替西农。无水氟化钾的参与,一方面,足够弱的碱性保证动力学控制产物烯醇酯被抑制到较低的水平,另一方面,氟离子参与交换,可将烯醇酯异构体转化为产物结构提高尼替西农的得率。可见,本发明实施例提供的尼替西农的合成方法,缩短了合成路线,成功避免了氰化物的使用,解决了目前因使用有毒有害的三甲基氰基硅烷(TMSCN)作为催化剂导致的产品毒性残留量较大、服用时副作用较大的问题,不仅提高了实验操作的安全性和稳定性,更重要的是,提高尼替西农产品服用时的安全性和稳定性,同时采用无机弱碱无水氟化钾制备尼替西农有着较好的反应选择性和转化率,由此可见本发明实施例提供的尼替西农的制备方法是一个绿色环保、易于进行产业化的合成工艺。
同时,尼替西农的合成过程中,需使用无水氟化钾,这是由于氟化钾本身易潮解,在空气中极易吸水导致用量发生变化,同时,反应原料2-硝基-4-三氟甲基苯甲酰氯极易水解,因此,反应需使用无水氟化钾。
在可选的实施方式中,尼替西农的制备包括以下步骤:将2-硝基-4-三氟甲基苯甲酰氯用有机溶剂溶解得到的溶液,滴加到1,3-环己二酮、无水氟化钾和有机溶剂混合形成的混悬液中,以使2-硝基-4-三氟甲基苯甲酰氯和1,3-环己二酮发生缩合反应得到尼替西农。
在可选的实施方式中,2-硝基-4-三氟甲基苯甲酰氯与无水氟化钾的投料量摩尔比为1.0:1.0-6.0,优选为1.0:2.0-5.0,进一步优选为1.0:3.5-5.0。
在可选的实施方式中,2-硝基-4-三氟甲基苯甲酰氯与1,3-环己二酮的投料量摩尔比为1.0:1.0-1.5,优选为1.0:1.0-1.2。需要说明的是,缩小或放大上述优选的物料比例,会引起原料转化率不足或者副产物增加的不良结果。
在可选的实施方式中,有机溶剂为非质子性有机溶剂,优选地,非质子性有机溶剂包括甲苯、乙腈、二氯甲烷、乙酸乙酯、丙酮、二氧六环和四氢呋喃中的至少一种,进一步优选为甲苯、乙腈和乙酸乙酯中的至少一种。
在可选的实施方式中,缩合反应的反应温度-20-50℃、反应时间不低于3h,选优反应温度为-10-40℃、反应时间为3-72h,进一步优选反应温度为0-30℃、反应时间为8-32h。需要说明的是,缩短或延长上述优选的反应时间,会引起原料转化率不足或者产物降解的不良结果。
在可选的实施方式中,2-硝基-4-三氟甲基苯甲酰氯通过以下步骤制备得到:
在非质子性有机溶剂存在下,以N,N-二甲基甲酰胺为催化剂,使2-硝基-4-三氟甲基苯甲酸与氯化亚砜发生氯化反应,得到2-硝基-4-三氟甲基苯甲酰氯;
优选地,2-硝基-4-三氟甲基苯甲酸和氯化亚砜的投料量摩尔比为1.0:1.1-3.0,优选为1.0:1.1-2.0,进一步优选为1.0:1.3-1.8;2-硝基-4-三氟甲基苯甲酸和N,N-二甲基甲酰胺的投料量摩尔比为1.0:0.01-0.1;
优选地,控制氯化反应的反应温度为40-120℃,反应时间为3-8h;
优选地,非质子性有机溶剂包括酯类、芳香族类、醚类和烷烃类有机溶剂中的至少一种,优选为乙酸丙酯、乙酸乙酯、甲苯、苯、二甲苯、二氧六环、四氢呋喃、甲基叔丁基醚、二氯甲烷和氯仿中的至少一种,进一步优选为乙酸乙酯、甲苯和二氯甲烷中的至少一种。
在可选的实施方式中,还包括:缩合反应结束后,反应混合物经洗涤、萃取、干燥、过滤、浓缩后得到尼替西农粗品,再将尼替西农粗品进行重结晶精制,得到药用级尼替西农。
在可选的实施方式中,重结晶精制使用的溶剂为丙酮和水的混合溶剂,并且丙酮和水的体积比为1.0:0.5-3.0,优选为1.0:0.6-2.4,进一步优选为1.0:0.8-1.5。
在可选的实施方式中,重结晶精制的温度为30-100℃,优选为35-80℃,进一步优选为40-65℃。
本发明实施例中将获得的尼替西农粗品以丙酮和水混合作为溶剂进行重结晶精制,其中大量的有机杂质留在了丙酮中、无机杂质则留在水中,这一纯化过程相比于乙酸乙酯做溶剂的重结晶效果更好,HPLC测试的结果显示产品的纯度大幅提高,可以得到满足现行质量标准要求的尼替西农原料药。
以上,本发明实施例提供一种新的尼替西农的制备方法,包括:在无水氟化钾和有机溶剂存在下,使2-硝基-4-三氟甲基苯甲酰氯和1,3-环己二酮发生缩合反应得到尼替西农。该方法避免了使用极毒氰化试剂,采用无机弱碱氟化钾参与反应制备尼替西农的方法有着较好的反应选择性、转化率以及良好纯度和分离得率,具有原料易得、环境友好、原子和过程经济等优点。
以下结合实施例对本发明的特征和性能作进一步的详细描述。
本发明提供了一种尼替西农的制备新方法,首先以2-硝基-4-三氟甲基苯甲酸为原料与氯化亚砜发生氯代反应制备2-硝基-4-三氟甲基苯甲酰氯,不经分离,将制得的2-硝基-4-三氟甲基苯甲酰氯与1,3-环己二酮在无水氟化钾的参与下发生缩合反应,得到尼替西农粗品后再经重结晶提纯得到满足现行质量标准要求的尼替西农原料药。反应方程式如下:
实施例1
氮气保护下,35ml干燥甲苯、催化量的N,N-二甲基甲酰胺(3滴)室温溶解4.70g 2-硝基-4-三氟甲基苯甲酸,冰浴下,缓慢滴加20ml干燥甲苯溶解的3.60g氯化亚砜溶液,滴加结束升温至110℃回流,反应4h后降至室温,减压蒸尽溶剂,蒸尽后,再每次以20ml甲苯带尽残留的二氯亚砜3次。蒸干后,即得无色油状的2-硝基-4-三氟甲基苯甲酰氯,加入50ml甲苯溶解待用。
氮气保护下,2.52g 1,3-环己二酮、5.2g经烘干至恒重的氟化钾粉末,100ml干燥甲苯于25℃搅拌混合,再于冰浴下,向此悬浮液中缓慢滴加上述2-硝基-4-三氟甲基苯甲酰氯的甲苯溶液,滴加结束后,再升至25℃反应30h。反应结束后,向反应混合物中加入100ml10%氯化钠溶液,分液,弃水相后,依次以2mol/L盐酸150ml(2次)、饱和氯化钠溶液150ml以及纯化水50ml洗涤有机相,无水硫酸钠干燥,过滤、浓缩,得到6.35g淡黄色粉末状尼替西农粗品,HPLC纯度96.2%。
于50℃,以200ml丙酮/水(5:3,v/v)重结晶上述粗品,降温至5℃析出固体,过滤,以20ml冰水泼洗滤饼,所得滤饼在40℃下真空干燥(0.01MPa)至恒重,得到白色针状固体4.4g,摩尔总收率66%(自2-硝基-4-三氟甲基苯甲酸计),HPLC纯度:99.9%,最大单个杂质:0.04%。熔点:140.1-141.5℃;
核磁氢谱参见图1,
碳谱参见图2,
实施例2
氮气保护下,40ml干燥乙酸乙酯、催化量的N,N-二甲基甲酰胺(3滴)室温溶解4.70g 2-硝基-4-三氟甲基苯甲酸,冰浴下,缓慢滴加4.30g氯化亚砜,滴加结束升温至70℃,反应8h后降至室温,减压蒸尽溶剂,蒸尽后,再每次以20ml正庚烷带尽残留的二氯亚砜3次。蒸干后,即得无色油状的2-硝基-4-三氟甲基苯甲酰氯,加入50ml乙腈溶解待用。
氮气保护下,2.77g 1,3-环己二酮、4.50g经烘干至恒重的氟化钾粉末,100ml干燥乙腈于25℃搅拌混合,10℃下向此悬浮液中缓慢滴加上述2-硝基-4-三氟甲基苯甲酰氯的乙腈溶液,滴加结束后,再升至35℃反应12h。反应结束后,向反应混合物中加入100ml 10%氯化钠溶液,分液,弃水相后,依次以2mol/L盐酸150ml(2次)、饱和氯化钠溶液150ml以及纯化水50ml洗涤有机相,无水硫酸钠干燥,过滤、浓缩,得到6.14g淡黄色粉末状尼替西农粗品,HPLC纯度95.5%。
于55℃,以350ml丙酮/水(1:1,v/v)重结晶上述粗品,降温至5℃析出固体,过滤,以20ml冰水泼洗滤饼,所得滤饼在40℃下真空干燥(0.01MPa)至恒重,得到白色针状固体4.8g,摩尔总收率74%(自2-硝基-4-三氟甲基苯甲酸计),HPLC纯度:99.9%,最大单个杂质:0.02%。目标化合物的核磁氢谱、碳谱、高分辨质谱等表征数据参见实施例1。
实施例3
氮气保护下,40ml干燥乙酸乙酯、0.10g N,N-二甲基甲酰胺室温溶解4.70g 2-硝基-4-三氟甲基苯甲酸,冰浴下,缓慢滴加4.85g氯化亚砜,滴加结束升温至80℃,反应6h后降至室温,减压蒸尽溶剂,蒸尽后,再每次以20ml正庚烷带尽残留的二氯亚砜3次。蒸干后,即得无色油状的2-硝基-4-三氟甲基苯甲酰氯,加入50ml乙酸乙酯溶解待用。
氮气保护下,2.33g 1,3-环己二酮、5.00g经烘干至恒重的氟化钾粉末,80ml干燥乙酸乙酯于25℃搅拌混合,-5℃下向此悬浮液中缓慢滴加上述2-硝基-4-三氟甲基苯甲酰氯的乙腈溶液,滴加结束后,再升至40℃反应15h。反应结束后,向反应混合物中加入100ml10%氯化钠溶液,分液,弃水相后,依次以2mol/L盐酸150ml(2次)、饱和氯化钠溶液150ml以及纯化水50ml洗涤有机相,无水硫酸钠干燥,过滤、浓缩,得到6.50g淡黄色粉末状尼替西农粗品,HPLC纯度93.4%。
于65℃,以250ml丙酮/水(4:3,v/v)重结晶上述粗品,降温至5℃析出固体,过滤,以20ml冰水泼洗滤饼,所得滤饼在40℃下真空干燥(0.01MPa)至恒重,得到白色针状固体4.5g,摩尔总收率68%(自2-硝基-4-三氟甲基苯甲酸计),HPLC纯度:99.8%,最大单个杂质:0.06%。目标化合物的核磁氢谱、碳谱、高分辨质谱等表征数据参见实施例1。
实施例4
氮气保护下,30ml干燥二氯甲烷、0.05g N,N-二甲基甲酰胺室温溶解4.70g 2-硝基-4-三氟甲基苯甲酸,冰浴下,缓慢滴加5.0g氯化亚砜,滴加结束升温至40℃,反应12h后降至室温,减压蒸尽溶剂,蒸尽后,再每次以20ml正庚烷带尽残留的二氯亚砜3次。蒸干后,即得无色油状的2-硝基-4-三氟甲基苯甲酰氯,加入50ml二氯甲烷溶解待用。
氮气保护下,2.46g 1,3-环己二酮、4.80g经烘干至恒重的氟化钾粉末,120ml干燥二氯甲烷于室温搅拌,3℃下向此悬浮液中缓慢滴加上述2-硝基-4-三氟甲基苯甲酰氯的二氯甲烷溶液,滴加结束后,再于室温下反应40h。反应结束后,向反应混合物中加入100ml10%氯化钠溶液,分液,弃水相后,依次以2mol/L盐酸150ml(2次)、饱和氯化钠溶液150ml以及纯化水50ml洗涤有机相,无水硫酸钠干燥,过滤、浓缩,得到6.08g淡黄色粉末状尼替西农粗品,HPLC纯度95.4%。
于50℃,以150ml丙酮/水(2:1,v/v)重结晶上述粗品,降温至5℃析出固体,过滤,以20ml冰水泼洗滤饼,所得滤饼在40℃下真空干燥(0.01MPa)至恒重,得到白色针状固体3.9g,摩尔总收率60%(自2-硝基-4-三氟甲基苯甲酸计),HPLC纯度:99.9%,最大单个杂质:0.03%。目标化合物的核磁氢谱、碳谱、高分辨质谱等表征数据参见实施例1。
实施例5
氮气保护下,40ml干燥甲苯、0.05g的N,N-二甲基甲酰胺室温溶解4.70g2-硝基-4-三氟甲基苯甲酸,冰浴下,缓慢滴加4.5g氯化亚砜,滴加结束升温至90℃,反应16h后降至室温,减压蒸尽溶剂,蒸尽后,再每次以20ml正庚烷带尽残留的二氯亚砜3次。蒸干后,即得无色油状的2-硝基-4-三氟甲基苯甲酰氯,加入50ml乙腈溶解待用。
氮气保护下,2.88g 1,3-环己二酮、3.5g经烘干至恒重的氟化钾粉末,100ml干燥乙腈于25℃搅拌混合,10℃下向此悬浮液中缓慢滴加上述2-硝基-4-三氟甲基苯甲酰氯的乙腈溶液,滴加结束后,再升至40℃反应9h。反应结束后,向反应混合物中加入100ml 10%氯化钠溶液,分液,弃水相后,依次以2mol/L盐酸150ml(2次)、饱和氯化钠溶液150ml以及纯化水50ml洗涤有机相,无水硫酸钠干燥,过滤、浓缩,得到6.27g淡黄色粉末状尼替西农粗品,HPLC纯度96.9%。
于60℃,以250ml丙酮/水(2:1,v/v)重结晶上述粗品,降温至5℃析出固体,过滤,以20ml冰水泼洗滤饼,所得滤饼在40℃下真空干燥(0.01MPa)至恒重,得到白色针状固体4.5g,摩尔总收率70%(自2-硝基-4-三氟甲基苯甲酸计),HPLC纯度:99.9%,最大单个杂质:0.02%。目标化合物的核磁氢谱、碳谱、高分辨质谱等表征数据参见实施例1。
对比例1
与实施例1的步骤相似,主要不同之处仅在于:无水氟化钾替换为无水碳酸钾(2.5摩尔当量),结果如下。
氮气保护下,35ml干燥甲苯、催化量的N,N-二甲基甲酰胺(3滴)室温溶解4.70g 2-硝基-4-三氟甲基苯甲酸,冰浴下,缓慢滴加20ml干燥甲苯溶解的3.60g氯化亚砜溶液,滴加结束升温至110℃回流,反应4h后降至室温,减压蒸尽溶剂,蒸尽后,再每次以20ml甲苯带尽残留的二氯亚砜3次。蒸干后,即得无色油状的2-硝基-4-三氟甲基苯甲酰氯,加入50ml甲苯溶解待用。
氮气保护下,2.52g 1,3-环己二酮、6.50g干燥的碳酸钾粉末,100ml干燥甲苯于25℃搅拌混合,再于冰浴下,向此悬浮液中缓慢滴加上述2-硝基-4-三氟甲基苯甲酰氯的甲苯溶液,滴加结束后,再升至25℃反应30h。反应结束后,向反应混合物中加入100ml 10%氯化钠溶液,分液,弃水相后,依次以2mol/L盐酸150ml(2次)、饱和氯化钠溶液150ml以及纯化水50ml洗涤有机相,无水硫酸钠干燥,过滤、浓缩,得到4.20g黄色粉末状尼替西农粗品,HPLC纯度94.5%。
于50℃,以150ml丙酮/水(5:3,v/v)重结晶上述粗品,降温至5℃析出固体,过滤,以20ml冰水泼洗滤饼,所得滤饼在40℃下真空干燥(0.01MPa)至恒重,得到白色针状固体2.6g,摩尔总收率40%(自2-硝基-4-三氟甲基苯甲酸计),HPLC纯度:99.0%,最大单个杂质:0.11%。
对比例2
与实施例1的步骤相似,主要不同之处在于:无水氟化钾的用量降低至2.5摩尔当量,实验如下。
氮气保护下,35ml干燥甲苯、催化量的N,N-二甲基甲酰胺(3滴)室温溶解4.70g 2-硝基-4-三氟甲基苯甲酸,冰浴下,缓慢滴加20ml干燥甲苯溶解的3.60g氯化亚砜溶液,滴加结束升温至110℃回流,反应4h后降至室温,减压蒸尽溶剂,蒸尽后,再每次以20ml甲苯带尽残留的二氯亚砜3次。蒸干后,即得无色油状的2-硝基-4-三氟甲基苯甲酰氯,加入50ml乙腈溶解待用。
氮气保护下,2.52g 1,3-环己二酮、2.90g经烘干至恒重的氟化钾粉末,100ml干燥乙腈于25℃搅拌混合,再于冰浴下,向此悬浮液中缓慢滴加上述2-硝基-4-三氟甲基苯甲酰氯的乙腈溶液,滴加结束后,再升至25℃反应32h。反应结束后,向反应混合物中加入100ml10%氯化钠溶液,分液,弃水相后,依次以2mol/L盐酸150ml(2次)、饱和氯化钠溶液150ml以及纯化水50ml洗涤有机相,无水硫酸钠干燥,过滤、浓缩,得到3.75g淡黄色粉末状尼替西农粗品,HPLC纯度93.5%。
于50℃,以180ml丙酮/水(4:3,v/v)重结晶上述粗品,降温至5℃析出固体,过滤,以20ml冰水泼洗滤饼,所得滤饼在40℃下真空干燥(0.01MPa)至恒重,得到白色针状固体2.2g,摩尔总收率33%(自2-硝基-4-三氟甲基苯甲酸计),HPLC纯度:99.1%,最大单个杂质:0.15%。
对比例3
与实施例1的步骤相似,主要不同之处仅在于:缩合反应的原料比例提高至1.3摩尔当量的1,3-环己二酮,结果如下。
氮气保护下,35ml干燥甲苯、催化量的N,N-二甲基甲酰胺(3滴)室温溶解4.70g 2-硝基-4-三氟甲基苯甲酸,冰浴下,缓慢滴加20ml干燥甲苯溶解的3.60g氯化亚砜溶液,滴加结束升温至110℃回流,反应4h后降至室温,减压蒸尽溶剂,蒸尽后,再每次以20ml甲苯带尽残留的二氯亚砜3次。蒸干后,即得无色油状的2-硝基-4-三氟甲基苯甲酰氯,加入50ml甲苯溶解待用。
氮气保护下,2.92g 1,3-环己二酮、5.2g经烘干至恒重的氟化钾粉末,100ml干燥甲苯于25℃搅拌混合,再于冰浴下,向此悬浮液中缓慢滴加上述2-硝基-4-三氟甲基苯甲酰氯的甲苯溶液,滴加结束后,再升至25℃反应30h。反应结束后,向反应混合物中加入100ml10%氯化钠溶液,分液,弃水相后,依次以2mol/L盐酸150ml(2次)、饱和氯化钠溶液150ml以及纯化水50ml洗涤有机相,无水硫酸钠干燥,过滤、浓缩,得到6.44g淡黄色粉末状尼替西农粗品,HPLC纯度95.0%。
于50℃,以200ml丙酮/水(5:3,v/v)重结晶上述粗品,降温至5℃析出固体,过滤,以20ml冰水泼洗滤饼,所得滤饼在40℃下真空干燥(0.01MPa)至恒重,得到白色针状固体4.3g,摩尔总收率64%(自2-硝基-4-三氟甲基苯甲酸计),HPLC纯度:99.5%,最大单个杂质:0.10%。
对比例4
与实施例1的步骤相似,主要不同之处在于:缩合反应温度升高至45℃,结果如下。
氮气保护下,35ml干燥甲苯、催化量的N,N-二甲基甲酰胺(3滴)室温溶解4.70g 2-硝基-4-三氟甲基苯甲酸,冰浴下,缓慢滴加20ml干燥甲苯溶解的3.60g氯化亚砜溶液,滴加结束升温至110℃回流,反应4h后降至室温,减压蒸尽溶剂,蒸尽后,再每次以20ml甲苯带尽残留的二氯亚砜3次。蒸干后,即得无色油状的2-硝基-4-三氟甲基苯甲酰氯,加入50ml甲苯溶解待用。
氮气保护下,2.52g 1,3-环己二酮、5.2g经烘干至恒重的氟化钾粉末,100ml干燥甲苯于25℃搅拌混合,再于10℃下,向此悬浮液中缓慢滴加上述2-硝基-4-三氟甲基苯甲酰氯的甲苯溶液,滴加结束后,再升至45℃反应8h。反应结束后,向反应混合物中加入100ml10%氯化钠溶液,分液,弃水相后,依次以2mol/L盐酸150ml(2次)、饱和氯化钠溶液150ml以及纯化水50ml洗涤有机相,无水硫酸钠干燥,过滤、浓缩,得到6.10g淡黄色粉末状尼替西农粗品,HPLC纯度90.8%。
于50℃,以220ml丙酮/水(2:1,v/v)重结晶上述粗品,降温至5℃析出固体,过滤,以20ml冰水泼洗滤饼,所得滤饼在40℃下真空干燥(0.01MPa)至恒重,得到白色针状固体3.3g,摩尔总收率50%(自2-硝基-4-三氟甲基苯甲酸计),HPLC纯度:98.7%,最大单个杂质:0.21%。
对比例5
与实施例1的步骤相似,主要不同之处在于:无水氟化钾的用量升高至6.5摩尔当量,实验如下。
氮气保护下,35ml干燥甲苯、催化量的N,N-二甲基甲酰胺(3滴)室温溶解4.70g 2-硝基-4-三氟甲基苯甲酸,冰浴下,缓慢滴加20ml干燥甲苯溶解的3.60g氯化亚砜溶液,滴加结束升温至110℃回流,反应4h后降至室温,减压蒸尽溶剂,蒸尽后,再每次以20ml甲苯带尽残留的二氯亚砜3次。蒸干后,即得无色油状的2-硝基-4-三氟甲基苯甲酰氯,加入50ml甲苯溶解待用。
氮气保护下,2.52g 1,3-环己二酮、7.60g经烘干至恒重的氟化钾粉末,100ml干燥甲苯于25℃搅拌混合,再于冰浴下,向此悬浮液中缓慢滴加上述2-硝基-4-三氟甲基苯甲酰氯的甲苯溶液,滴加结束后,再升至25℃反应30h。反应结束后,向反应混合物中加入100ml10%氯化钠溶液,分液,弃水相后,依次以2mol/L盐酸150ml(2次)、饱和氯化钠溶液150ml以及纯化水50ml洗涤有机相,无水硫酸钠干燥,过滤、浓缩,得到6.04g淡黄色粉末状尼替西农粗品,HPLC纯度95.0%。
于50℃,以180ml丙酮/水(2:1,v/v)重结晶上述粗品,降温至5℃析出固体,过滤,以20ml冰水泼洗滤饼,所得滤饼在40℃下真空干燥(0.01MPa)至恒重,得到白色针状固体3.31g,摩尔总收率50%(自2-硝基-4-三氟甲基苯甲酸计),HPLC纯度:99.4%,最大单个杂质:0.10%。
综上,本发明实施例提供了一种尼替西农的制备方法,包括:在无水氟化钾的参与下,2-硝基-4-三氟甲基苯甲酰氯和1,3-环己二酮于有机溶剂中发生缩合反应得到尼替西农。本发明实施例提供的尼替西农的制备方法,避免使用含氰基的剧毒、高污染试剂,在氟化钾的参与下,提高了缩合反应的选择性,反应得到的尼替西农粗品再使用丙酮和水的混合溶剂重结晶精制,可以药品级尼替西农,本发明实施例提供的药品级尼替西农的制备方法为其广泛应用提供基础。
以上仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 一种阿尼西坦的制备方法
- 一种球形酒石酸西尼必利结晶的制备方法
- 一种帕博西尼的制备方法及其产品
- 一种含有尼替西农的药物组合物及其制备方法
- 尼替西农的制备方法