用于分析基因数据的计算机实现方法和装置
文献发布时间:2024-01-17 01:26:37
本发明涉及分析关于生物体的基因和表型数据以获得关于该生物体的信息,特别是在能够获得对于感兴趣表型的改进的多基因风险评分(polygenic risk score,PRS)的背景下。
PRS是生物体的遗传DNA对其可能表现出的表型的贡献的定量总结。PRS在其计算中可能包括与感兴趣表型相关(直接地或间接地)的所有DNA变体,或者如果它们与生物体生物学的特定方面(包括细胞、组织或其他生物单位、机制或过程)更相关,则可以使用其组成部分。PRS可以直接使用,或作为关于该生物体的多个测量结果或记录的一部分,以推断其过去、当前和未来生物学的方面。
PRS作为一种疾病预防、分层和诊断的工具,越来越受到重视。在改善人类健康和医疗保健的背景下,PRS具有一系列实际用途,包括但不限于:预测疾病或表型发展的风险、预测表型发生年龄、预测疾病严重程度、预测疾病亚型、预测对治疗的反应、为个体选择适当的筛查策略、选择适当的药物干预和为其他预测算法设置先验概率。
PRS可以直接用作人工智能和机器学习方法的应用中的输入源,以根据其他高维输入数据(例如成像)进行预测或分类。它们可用于帮助训练这些算法,例如识别基于非基因数据的预测测量。除了在对个体做出预测性说明方面具有实用性外,它们还可用于通过计算大量个体的PRS,然后基于PRS对个体进行分组来识别个体群组(包括但不限于上述应用)。
PRS可以直接用作人工智能和机器学习方法的应用中的输入源,以根据其他高维输入数据(例如成像)进行预测或分类。它们可用于帮助训练这些算法,例如识别基于非基因数据的预测性测量。除了在对个体做出预测性说明方面具有实用性外,它们还可用于通过计算大量个体的PRS,然后基于PRS对个体进行分组来识别个体群组(包括但不限于上述应用)。
PRS还可以帮助选择个体进行临床试验,例如通过招募更有可能发展相关疾病或表型的个体来优化试验设计,从而增强对新治疗的功效的评估。PRS携带有关他们计算的个体的信息,也包括他们的亲属(其共享他们的遗传DNA的一部分)的信息。有关个体DNA对其表型的影响的信息可以源自对携带任何特定DNA变体组合的潜在影响的任何相关评估。
在下文中,我们专注于对源自遗传关联研究(genetic association studies,GAS)的近期大量信息的分析。这些研究系统地评估了DNA变体对表型的遗传基础的潜在贡献。
自2000年代中期(mid-2000s)以来,已经在数以百万计的个体中对成千上万(主要是人)的表型进行了GAS(通常是全基因组关联研究:GWAS,或靶向单个变体或基因组区域中的变体的关联研究,或限于基因组特定区域的GWAS),从而在基因型和表型之间产生数十亿的潜在联系。然后通常将得到的原始数据简化以产生汇总统计数据。对于每个基因变体(不论是插补的还是观察到的),GAS汇总统计数据由基因变体对GAS表型的推断效应量和推断效应量的标准误差组成。在其它情况下,由研究中个体的完整基因概况和关于其表型的信息组成的个体水平数据可直接利用。然而,由于对个体数据的隐私的要求,个体水平数据通常不太被广泛地利用。
PRS由大量基因变体的效应的聚集体组成,通常每个基因变体具有小的个体效应,以构建感兴趣特征的综合预测因子。PRS可以使用根据GWAS确定的变体的效应量来计算。包括在这种评分中的变体可以是“因果变体(causal variant)”,意思是变体直接影响特征(弱的,但直接的),或“标记变体”,这意味着它们与其它未知的因果变体强烈相关,但标记变体本身对表型没有直接效应。
PRS构建策略正在扩展,但构建精确PRS的公认通用方法包括通过研究最佳捕获潜在生物关联的变体的组合对所有关联区域中的信号进行去卷积。关联的数目将变化,其中许多基因组区域含有单个潜在关联,而一些基因组区域将含有多个独立的关联(已报道多达10个,但这是罕见的)。
识别负责区域中所有关联的变体的正确组合的技术挑战在于这些变体可以相互关联。相关性越大,分解这些相关性所需的样本数量就越高。
一些构建PRS的工具设计成利用汇总统计数据。LDpred软件(Vilhjálmsson等人2015,https://github.com/bvilhjal/ldpred)推广的一种方法是基于单个GWAS在全基因组范围内对合理变体的多个随机选择进行迭代,并在选择或删除变体时估计残余信号。
基于汇总统计数据的战略的优势是在共享个体水平数据时没有限制,这意味着科学界可以获得更大的样本量。这就是为什么大多当前的PRS设计基于这些大的汇总统计数据集。
然而,对于所有基于汇总统计数据的方法,相关变体是通过参考描述变体之间预期是何种相关性的外部数据源来处理的。基因变体之间的相关性模式被称为连锁不平衡(linkage disequilibrium,LD)。依赖外部数据集来描述LD模式的局限性在于不同的亚群具有不同的LD模式。例如,欧洲祖先的个体可能与东南亚祖先的个体具有不同的LD模式。考虑到真正的因果变体的同一性通常永远无法绝对确定,LD的这些差异可能导致不同祖先中PRS预测准确性的差异。此外,特定变体对表型的影响可能在亚群之间变化。例如,在男性中给定的因果基因变体对给定表型的影响可能比在女性中大,或者在较老年个体中的影响比较年轻个体中小。因此,对一个亚群做出的推断,或基于来自混合亚群的个体的数据做出的推断不太可能对不同的亚群同样精确。例如,支持PRS构建的数据集通常基于欧洲祖先的大型同类者(cohort)。因此,这些分数在非欧洲祖先中往往表现不佳。
处理这个问题的现有方法是基于使用来自合适亚群的训练数据集来创建PRS。然而,可用于特定亚群的数据量可能差异很大。因此,这些方法的样本量要低得多,这反过来限制了它们的预测能力。由于较小研究的统计能力降低,与简单地使用从具有更多可用数据的不同亚群获得的结果相比,尝试计算具有较少可用数据的特定亚群的PRS可能产生不太可靠的结果。例如,在许多情况下,来自欧洲祖先的同类者的较大样本量可以克服与使用不匹配训练集相关的偏差,并且在欧洲祖先上训练的PRS实际上可以在非欧洲同类者中提供最佳的PRS选项,即使这在原则上是次优的。
本发明的目的是改进关于生物体的基因数据的分析和/或允许为属于特定亚群的个体获得更稳健和/或准确的PRS。
根据本发明的方面,提供了一种分析关于生物体的基因数据的计算机实现方法。所述方法包括接收多个输入单元,其中每个输入单元包括关于生物体基因组的感兴趣区域中的多个基因变体与生物体的目标表型之间关联的信息;进行一次或多次迭代,包括:对于所述多个基因变体中的每一个,基于所述多个输入单元确定所述基因变体对所述目标表型是否具有因果关系;并且,如果基因变体确定为因果关系的,则基于所述多个输入单元和关于所述感兴趣区域中的多个基因变体之间的相关性的信息,确定所述基因变体对于每个输入单元的目标表型的采样效应量,所述基因变体对目标表型的采样效应量对于所有输入单元都是非零的;并且对于每个基因变体,基于输入单元的基因变体的采样效应量的迭代的至少一个子集的平均值或使用所述采样效应量计算的所述输入单元的基因变体的后验效应量的平均值,确定所述基因变体对每个输入单元的目标表型的预测效应量。
通过使用来自多个输入单元的数据确定哪些变体是因果关系,可以以更大的置信度识别因果变体。然而,为每个输入单元单独地确定预测效应量仍然允许该方法解释不同亚群的不同效应量的可能性。因此,使用大型数据集的统计能力可以与生成亚群特异性结论的能力相结合。通过获得更准确的预测效应量,随后可以计算出更准确的PRS。
在一些实施例中,确定基因变体是否具有因果关系包括:计算来自多个输入单元的假设基因变体是因果关系的信息的概率和来自所述多个输入单元的假设基因变体不是因果关系的信息的概率,以及随机地确定所述基因变体是因果关系的,其概率取决于根据假设所述基因变体是因果关系的输入数据的概率与假设所述基因变体不是因果关系的输入数据的概率的比率。使用随机的采样允许该方法考虑因果变体的许多不同组合,以确定最能解释观测数据的总体效果。
在一些实施例中,来自所述多个输入单元的假设所述基因变体是因果关系的信息的概率,取决于预期是因果关系的多个基因变体的比例(proportion)、所述多个输入单元以及所述基因变体对每个输入单元的目标表型的效应量之间的相关性。在一些实施例中,来自所述多个输入单元的假设所述基因变体不是因果关系的信息的概率,取决于预期具有因果关系的多个基因变体的比例和所述多个输入单元。这些术语(term)允许预先存在的关于因果变体比例的信息纳入分析,并允许输入单元之间的预测效应量变化。在非因果关系的情况下,效应量为零,因此效应之间没有相关性是合适的。
在一些实施例中,预期为因果关系的多个基因变体的比例是预先确定的。在一些实施例中,基因变体对每个输入单元的目标表型的效应量之间的相关性是预先确定的。使使用参数的预定值允许以计算上高效的方式将预先存在的知识并入该方法中。
在一些实施例中,在每次迭代时更新预期具有因果关系的多个基因变体的比例。在一些实施例中,其中在每次迭代时更新所述基因变体对每个输入单元的目标表型的效应量之间的相关性。在每次迭代时学习和更新参数允许该方法收敛(converge)于真实的参数值,其可以提供更准确的结果,但可能在计算上更昂贵。
在一些实施例中,所述输入单元从各个个体群组确定,并且来自多个输入单元的假设基因变体是因果关系的消息的概率取决于量化各个输入单元对之间的个体群组中的重叠的一个或多个参数。根据所使用的数据,一些个体可能出现在多个输入单元中,这可能会扭曲得出的结论。添加参数来说明这点提高得到的效应量的准确性。
在一些实施例中,确定所述基因变体的采样效应量包括计算所述基因变体对输入单元的目标表型的效应量的概率分布,以及从该概率分布中采样输入单元的效应量的值。使用概率分布允许该方法对不同的效应量进行采样,同时仍然鼓励在被认为最有可能正确的范围内选择值。
在一些实施例中,概率分布是多元正态分布。使用多元正态分布提供了一种方便的方式来允许不同输入单元的不同采样效应量。
在一些实施例中,每次迭代中效应量的值的采样取决于来自一个或多个先前迭代的采样效应量。这种类型的依赖性可以允许采样有效地探索可能值的空间。在一些实施例中,使用蒙特卡罗吉布斯采样器(Monte-Carlo Gibbs sampler)来执行效应量的值的采样。这种类型的采样算法特别适合于本申请。
在一些实施例中,概率分布取决于基因变体对每个输入单元的目标表型的效应量之间的相关性。这允许待控制输入单元之间的效应量的可能差异范围,以提高准确性和计算效率。
在一些实施例中,基因变体对每个输入单元的目标表型的效应量之间的相关性是预先确定的。使用参数的预定值允许以计算上高效的方式将预先存在的知识并入该方法中。
在一些实施例中,在每次迭代时更新基因变体对每个输入单元的目标表型的效应量之间的相关性。在每次迭代中学习和更新参数允许该方法收敛于真实的参数值,这可以提供更准确的结果,但可能在计算上更昂贵。
在一些实施例中,一次或多次迭代中的每一次还包括,对于被确定为因果关系的每个基因变体,从关于每个其他基因变体与每个输入单元的目标表型之间的关联的信息中减去加权效应量;所述加权效应量是由所述基因变体和每个其它基因变体之间的各个相关因子进行加权的所述基因变体对所述输入单元的目标表型的采样效应量;并且基于关于感兴趣区域中的多个基因变体之间的相关性的信息来确定相关因子。从关联的变体中减去被确定为因果关系的变体的效应,确保不会基于单个因果关系错误地识别多个因果变体。使用输入单元特异的相关因子允许该方法解释亚群(subpopulations)之间的基因相关性的变化。
在一些实施例中,输入单元是从各个个体群组中确定的,并且基因变体和每个其他基因变体之间的相关因子取决于输入单元的个体群组的祖先。在一些实施例中,至少一个输入单元的个体群组包括具有共同祖先的个体,对于具有共同祖先的个体,相关因子是基于感兴趣区域中的基因变体之间的相关性来确定的。使用基于祖先的相关因子是特别有用的,因为具有不同祖先的个体通常具有不同的在基因变体之间的相关性模式。
在一些实施例中,至少一个输入单元的个体群组包括具有不同祖先的个体,对于具有每个不同祖先的个体,相关因子是基于感兴趣区域中的基因变体之间的相关性的平均值来确定的。一些输入单元可能来自未进行祖先分层的研究。使用一组混合的相关因子仍然允许该数据纳入该方法中并改善结果。
在一些实施例中,至少一个输入单元的个体群组包括具有相同特征值的个体。在一些实施例中,至少一个输入单元的个体群组包括具有不同特征值的个体。在一些实施例中,特征是性别、年龄、体重、分子生物标志物或行为特征中的一个。亚群也可以基于特征来定义,并且基于来自具有这些特征的个体的数据的输入单元允许得出关于不同亚群之间的效应量的差异的结论。
在一些实施例中,执行一次或多次迭代包括进行预定次数的迭代。执行预定次数的迭代可以为已知类型的问题提供足够的结果,同时保持计算上的高效性。
在一些实施例中,一次或多次迭代中的每一次还包括评估收敛参数的步骤,并且执行一次或多次迭代包括执行迭代直到收敛参数的预定条件得到满足。在合适的迭代次数不确定的情况下,计算收敛参数可能是有利的。
在一些实施例中,关于多个基因变体和所述目标表型之间的关联的信息包括,对于多个基因变体中的每一个,所述基因变体和目标表型之间关联强度的估算以及所述关联强度的估算中的误差。如上所述,使用这种类型的汇总统计数据在获得大量数据的可用性方面具有优势。
根据另一方面,提供了一种确定目标个体的目标表型的多基因风险评分的方法,包括:接收关于目标个体的基因组的感兴趣区域的基因信息;接收使用所述分析基因数据的方法确定的在所述感兴趣区域中多个基因变体对目标表型的预测效应量;以及基于目标个体的基因信息和预测效应量来确定多基因风险评分。如上所述,计算多基因风险评分是对于基因变体确定的预测效应量的特别理想的用途,并且可以用于各种临床应用。在一些实施例中,在分析基因数据的方法中接收的输入单元是从各个个体群组中确定的,并且使用从与目标个体最相似的个体群组中所确定的输入单元的预测效应量来确定个体的多基因风险评分。使用最适合个体的输入单元的预测效应量可以相对于使用针对未分层数据确定的一般效应量确定的多基因风险评分来提高多基因风险得分的准确性。
根据本发明的另一方面,提供了一种用于分析关于生物体的基因数据的装置。所述装置包括接收单元,配置为接收多个输入单元,其中每个输入单元包括关于生物体基因组的感兴趣区域中的多个基因变体和生物体的目标表型之间的关联的信息;以及数据处理单元,配置为:执行一次或多次迭代,包括:对于所述多个基因变体中的每一个,基于所述多个输入单元确定所述基因变体对于所述目标表型是否是因果关系的;并且如果所述基因变体确定为是因果关系的,则基于所述多个输入单元和关于所述感兴趣区域中的多个基因变体之间的相关性的信息,确定所述基因变体对每个所述输入单元中的所述目标表型的采样效应量,对于所有输入单元所述基因变体对所述目标表型的采样效应量都是非零的;并且对于每个基因变体,基于输入单元的基因变体的采样效应量的迭代的至少一子集的平均值或使用所述采样效应量计算的所述输入单元的基因变体的后验效应量的平均值,确定对于每个所述输入单元的所述基因变体对所述目标表型的预测效应量。
本发明还可以体现在包括使计算机执行该方法的指令的计算机程序中,或者体现在包括当由计算机执行时使计算机执行该方法的指令的计算机可读介质中。
将仅参考附图,通过示例的方式进一步描述本发明的实施例,其中:
图1是根据本发明的分析关于生物体的基因数据的方法的流程图;
图2是示出在图1的方法中执行迭代的步骤中的每次迭代的步骤的流程图;
图3是根据本发明的确定多基因风险评分的方法的流程图;
图4是示出使用分析基因数据的现有技术方法为两个不同亚群估算的效应量的图;和
图5是示出使用根据本发明的方法为两个不同亚群估算的效应量的图。
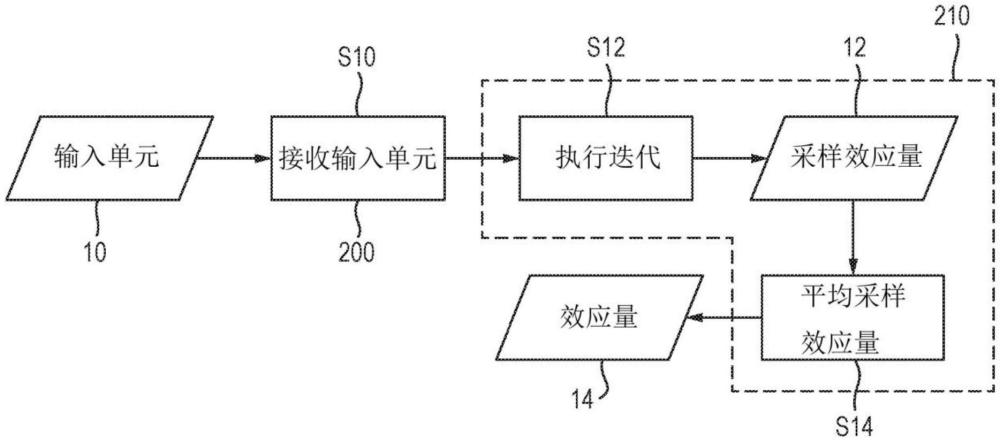
图1示出了分析关于生物体的基因数据的计算机实现方法。通常,生物体是人,尽管该方法也可以应用于其他生物体。尽管该方法指的是“生物体”,但它可能不是指特定个体的生物体,而是一般地指生物体或一群生物体。
该方法包括接收多个输入单元10的步骤S10。输入单元10包括关于生物体基因组的感兴趣区域中的多个基因变体与生物体的目标表型之间的关联的信息。目标表型可以包括可能感兴趣的任何生理(physical)、行为或其他表型。基因变体通常是单核苷酸多态性,但也可以包括其他类型的基因变体,例如生物体基因组部分的插入或缺失。
每个输入单元10可以来自一个或多个全基因组关联研究(genome-wideassociation studies,GWAS),因此也可能被称为研究(study)或GWAS。每个输入单元10将包括关于个体群组(例如参与相应GWAS的个体)的多个基因变体与目标表型之间关联的信息。
输入单元10的至少一个子集是从特定亚群的个体群组中确定的。例如,至少一个输入单元10的个体群组可以包括具有共同祖先的个体。可替选地或额外地,至少一个输入单元10的个体群组可以包括具有相同特征值的个体。该特征可以是例如性别、年龄、体重、分子生物标志物或行为特征(例如个体是否吸烟)中的一个。在例如年龄或体重的连续特征的情况下,特征值可以被划分为任意的箱(bins)以产生离散的类别数量,并且将可获得数据的个体划分为相应的用于定义输入单元10的离散组。
由于箱的定义不是由生物学固定的,而是任意的,因此所述方法的一些实施例可以包括用不同的箱定义(以及相应地修改的输入单元10)执行所述方法的步骤多次,并且比较用不同箱定义生成的效应量的预测能力。具有最大预测能力的效应量然后可以作为该方法的输出返回。
并非所有的输入单元10都可以从特定亚群的个体群组中确定。例如,至少一个输入单元10的个体群组可以包括具有不同祖先的个体。可替选地或额外地,至少一个输入单元10的个体群组可以包括具有不同特征值的个体。包括来自不是分层亚群的研究的一个或多个额外输入单元10可以允许该方法利用来自亚群之间不可能分离的个体群组的额外信息。例如,这可能是因为基础数据不包括研究中个体的特定特征的信息,因此不可能对其进行分层。
在本文所述的实施例中,关于多个基因变体和目标表型之间的关联的信息包括,对于多个基因变体中的每一个,基因变体与目标表型之间的关联强度的估算值,以及关联强度的估算值的误差(error)。因此,每个输入单元10包括,对于编号为1至n的每个变体i,变体i与目标表型之间关联强度的估算值
每个输入单元10中的关联强度的估算值
希望确定每个给定变体i的未知真实效应量β
在本方法中,通过在执行一次或多次迭代的步骤S12中探索可能的空间(X
如上所述,目前可用的分析基因数据的方法(如LDpred)一次考虑一个GWAS,并对哪些变体是因果关系的进行随机采样,例如通过蒙特卡罗采样。LDpred依赖于能够解决用于一项研究和一种基因变体的贝叶斯(Bayesian)计算。然后,它使用吉布斯(Gibbs)采样技术将方法从一个相关变体扩展到多个相关变体。准确地说,对于给定的基因变体,LDpred使用了先验假设:
-基因变体对表型的效应为0的概率(1-p)(即变体不具有因果关系)。
-对结果的效应为具有均值0和方差σ
利用这些假设和用于相关表型的训练GWAS中的汇总统计
然而,这种方法有局限性,特别是对于可能导致某些亚群的较差结果的较小研究。例如,对非欧洲祖先的个体的研究不太常见且与欧洲祖先的研究相比通常较小,这导致非欧洲祖先个体的预测结果较差。
在为相同的目标表型考虑多项研究时,目前可用的方法包括将多项研究合并为单个元分析,并对该元分析进行进一步处理,例如确定PRS。基于多项研究,解释变体和目标表型之间的关联证据的工具的示例是GWAS的多特征分析(MTAG,Turley等2018)。MTAG结合了一组GWAS,并为每个输入GWAS生成一种类型的元分析,从而为每个输入的GWAS生成更新的汇总统计数据。这些更新的汇总统计数据可以输入任何标准的PRS构建方法,包括LDPred(Craig等人,Nature Genetics 2020)。然而,MTAG使用边际效应量和标准误差,而没有同时考虑LD信息,这意味着该方法没有充分利用可用输入数据集的丰富性。结合多项研究的另一种现有方法是在另一种背景下开发的单变量贝叶斯计算(Trochet等,GeneticEpidemiology 2019)。在这种方法中,目的不是预测效应量,而是将研究相结合,以提高检测基因关联的能力。因此,基因变体是单独考虑的,没有动机控制它们之间的相关性模式。
现有方法的局限性也可以通过一些示例使用案例来证明。
在第一种情况下,由于历史原因,较强的GWAS存在于第一祖先中,通常是欧洲祖先的个体。第二不太强的研究存在于另一针对相同的目标表型的祖先中。较强的研究不能容易地与使用现有方法的第二项研究相结合。首先,变体之间的相关性模式因祖先变化,因此两项研究的结合导致了难以分析的未定义研究。其次,研究之间的基因和环境差异可能导致群体特异性变体或这些人群的效应量差异。现有方法无法对此进行解释。
在第二种情况下,将生成捕获对于群体的子集特定的风险因子的预测算法。目前的方法可能无法充分利用潜在的基因数据。可能是使用对于个人的年龄、性别、种族或任何其他健康的社会决定因素特定的效应量计算的“背景特异性的”PRS可能更准确。例如,心血管疾病(CVD)的决定因素因性别、BMI存在差异、血压、饮酒量和运动模式而不同。
现有的方法通过获取已经分层为亚群特异性研究的样本来解决这个问题,然后从这些样本中单独推导PRS。例如,在上述CVD示例中,当前的方法将分别分析两个性别特异性同类者(男性和女性)的GWAS,并使用这些同类者中的每一个生成PRS。然而,许多基因决定因素是跨性别共享的。因此,对男性和女性同类者进行联合分析(其将解释性别差异并产生性性别特异性的PRS)将更适合最大限度地提高预测能力。例如,如果某人对非吸烟者的肺癌的PRS感兴趣,那么在现有方法中也有类似的选择:1)具有多个样本(包括吸烟者),或2)使用仅包括非吸烟者的较小研究。
然而,PRS的预测能力也是基础研究的大小的函数。因此,将研究样本限制在数据的子集内通常是有害的。在吸烟的示例中,第一种选择使用了有偏见的研究(从参与者中吸烟者的比例来看,PRS将表明与成瘾相关的变体的较大的效应量),但第二种选择可能较弱(underpowered)(因为80%的肺癌患者是吸烟者)。这产生了反对亚群特异性PRS的对立论点。
这些使用示例并不是相互排斥的。例如,某人可能希望确定PRS来预测给定种族群体的性别或社会定义的子集的临床结果。
为了克服这些限制,本方法允许在确定因果变体及其效应量时组合多个研究的信息,但显著地,允许每个基因变体的确定的效应量在输入单元10之间不同。这允许将更大的研究的更大的统计能力与来自较小研究的数据一起使用,以在较小研究中改进哪些变体是因果关系的估算,但是仍然可以确定对于不同的亚群的不同的效应量。
这涉及到将来自LDPred(Vilhjálmsson等人2015)的贝叶斯计算从一项研究扩展到针对相同表型但在不同亚群中的任意数量的研究。在这样做时,Trochet等人的单变体多研究工作与Vilhjálmsson等人的多变体单研究工作之间建立了联系。通过理解两种方法论方法之间的关系,可以以灵活的方式整合多个研究,并创建基于多个GWAS,而不是单个研究的预测算法。
如图2所示,本方法步骤S12中的每次迭代包括,对于多个基因变体中的每一个,基于多个输入单元10确定基因变体对目标表型是否是因果关系的。对于现有的方法,基因变体逐一进行考虑,例如按物理顺序或随机采样,尽管其他选择也是可能的。然而,在每个变体中,本方法结合了多个研究而不是单个研究,并且评估该变体对每个输入单元10的因果关系和效应量的模型的概率(例如,通过贝叶斯分析,如下文进一步讨论的)。因此,本方法通过将所有输入单元10一起分析,而不是一次只考虑一个输入单元10,或者像现有方法那样将输入单元10组合成单个元分析,来确定每个基因变体是否具有因果关系。
如果确定基因变体是因果关系的,执行基于多个输入单元10和关于感兴趣区域中的多个基因变体之间的相关性的信息来确定基因变体对每个输入单元10的目标表型的采样效应量12的步骤。因此,在探索因果变体和联合效应量的空间时,当变体被选择为具有因果关系时,对每个研究采样不同的效应量。
在图1的实施例中,确定基因变体是否是因果关系的包括计算来自多个输入单元的假设基因变体是因果关系的信息的概率和来自多个输入单元的假设基因变体不是因果关系的信息的概率的步骤S120,以及随机地确定基因变体是因果关系的步骤S122,其概率取决于来自多个输入单元的假设基因变体是因果关系的信息的概率与来自多个输入单元的假设基因变体不是因果关系的信息的概率的比率。
在步骤S120中,来自多个输入单元的假设基因变体是因果关系的信息的概率可以取决于预期是因果关系的多个基因变体的比例、多个输入单元10以及对于每个输入单元10基因变体对目标表型的效应量之间的相关性。来自多个输入单元的假设基因变体不是因果关系的信息的概率可以取决于预期是因果关系的多个基因变体的比例和多个输入单元10。可以使用先验值来计算概率。
例如,在一实施例中,对于任何给定的变体考虑两个较早的模型:
●无效假设(null hypothesis),其中概率(1-p),该变体对于所有输入单元10具有0的效应量;
●替代方案,其中概率p,基因变体对输入单元10的效应量遵循多元高斯分布。
参数p是预期是因果关系的多个基因变体的比例。在一些实施例中,预期为因果关系的多个基因变体的比例是预先确定的。如果估算是可用的,则这可能在计算上是更高效的。在一些实施例中,在每次迭代时更新预期为因果关系的多个基因变体的比例。这允许该方法收敛于p的真实值,这潜在地提高了准确性。
在无效假设下,对于所有输入单元10,采样效应量12的值等于0。因此,变体i的采样效应量β
其中SE
在替代方案下,变体i的采样效应量β
其中
ρ
在其他实施例中,在每次迭代时更新基因变体对每个输入单元10的目标表型的效应量之间的相关性。这允许该方法收敛于真实的参数值,潜在地导致更准确的结果。可替代地,可以考虑相关性值的网格,并且通过具有结果的个体水平数据的数据集中的最大化预测可以来选择这些相关性的最佳参数值。在本文给出的示例中,效应量之间的相关性是单个参数,这对于输入单元10的所有组合也是相同的。
相关性也可以是相关性矩阵,其允许相关性在输入单元10的不同组合之间不同。例如,对于年龄等连续特征,可以使用相关性来平滑变量的各个箱(bins)。对于如年龄这样的连续特征,可以从相邻的年龄箱借用信息,以便改善任何给定箱的效应量和相应的PRS。由于存在先验预期,即相邻或附近的箱应该比更远的箱具有更高的基因相关性,因此可以使用连续变量的不同箱之间的相关性的不同值来解释这一点。
一旦这两个先验模型被定义,就可以计算来自多个输入单元的假设基因变体是因果关系的信息的概率和来自多个输入单元的假设基因变体不是因果关系的信息的概率,并将其与这些先验相结合。
在步骤S122的实施例中,对于每个变体i,使用在步骤S120中确定的概率可以计算贝叶斯因子:
然后基于贝叶斯因子执行变体是否是因果关系的随机采样。在这些等式中的β
假设因果基因变体在输入单元10(及其相应的亚群)之间是共享的,并且这些变体的效应量在跨输入单元10相关的同时是变化的。换句话说,变体对于所有输入单元10具有因果关系,或者对所有输入单元10都不具有因果关系。因此,如果基因变体被确定为因果关系的,则对于所有输入单元10,基因变体对目标表型的采样效应量12被确定为非零。
在输入单元10是从各个个体群组中确定的情况下,并且取决于用于确定输入单元10的研究,一个潜在的问题是研究之间的样本重叠。例如,“组合性别”研究可用于推导一个输入单元10,并随后与从其他“仅男性”和“仅女性”研究中得出的输入单元10联合进行分析。男性和女性特异性的研究可能是更大的综合性别研究集的子集,而综合性别研究可能包括没有提供性别信息的额外样本,或者仅仅是两项性别特异性研究的结合。为了说明这一点,在一些实施例中,来自多个输入单元的假设基因变体是因果关系的信息的概率取决于一个或多个量化各个输入单元10对之间的个体群组中的重叠的参数。
例如,说明这种可能性的一种方法是将上面所示的协方差矩阵V
其中r
如果基因变体被确定是因果关系的,则可以计算所有输入单元10的联合效应量的后验均值和方差。确定基因变体的采样效应量12的步骤包括计算基因变体对输入单元10的目标表型的效应量的概率分布的步骤S124,以及根据概率分布采样效应量的值的步骤S126。
使用采样效应量12是因为在实践中不可能在合理的时间内完全探索所有可能的因果变体和所有可能的对应效应量的空间。因此,采样技术,例如蒙特卡罗模拟(MonteCarlo simulations),用来探索因果变体及其相应的效应量的空间。在一些实施例中,每次迭代中效应量的值的采样取决于来自一个或多个先前迭代的采样效应量12。这可以用来指导采样技术以充分探索可能值的空间。在一些实施例中,使用蒙特卡罗吉布斯采样器来执行效应量的值的采样。
在优选实施例中,概率分布是多元正态分布。概率分布可以取决于基因变体对每个输入单元10的目标表型的效应量之间的相关性。如对上述概率所讨论的,基因变体对每个输入单元10的目标表型的效应量之间的相关性可以是预先确定的。可替代地,基因变体对每个输入单元10的目标表型的效应量之间的相关性可以在每次迭代时更新,从而允许该方法学习相关性的合适值。
在具体示例中,概率分布是效应量的后验均值,并且分布为多元正态分布:
在以计算PRS为目的的用于分析基因数据的方法的一些实施例中,重要的步骤是控制基因变体之间的相关性的能力。如上所述,变体之间的相关性可能导致一些变体具有大的边际效应量,即使它们对目标表型没有因果关系时。
为了说明这一点,在一些实施例中,一个或多个迭代中的每一个还包括,对于确定为因果关系的每个基因变体,从关于每个其他基因变体和每个输入单元10的目标表型之间的关联的信息中减去加权效应量的步骤S128。因此,当确定基因变体i是因果关系时,为基因变体i确定采样效应量β
在特定的实施例中,这导致以下校正被应用于每个其他基因变体j的边际效应量:
在上述公式中,β
这种校正的效果是,当确定变体是否是因果关系的时,其边际效应量将根据迄今为止确定为该迭代中具有因果关系的所有变体的采样效应量并使用上述公式进行校正。因此,在这样的实施例中,等式(4)和(6)中使用的效应量β
重要的是,通常不可能直接从数据本身计算基因变体之间的相关因子(上述示例中的值r
然而,在本方法中,效应量减法步骤S128正在应对的挑战在于以与变体相关性的祖先特异性模式一致的方式解释跨基因变体的相关性。为了克服该挑战,在适当的情况下,本方法可以并行处理多个参考LD图。一旦变体被确定具有因果关系,则减法步骤S128然后以祖先特异性方式被应用。因此,如果输入单元10是由各自的个体群组确定的,那么基因变体和其他基因变体之间的相关因子取决于输入单元10的个体群组的祖先。一对一映射可以使用在进行每项研究时的祖先和其匹配的LD图(协方差结构)之间。
例如,至少一个输入单元10的个体群组包括具有共同祖先的个体时,基于具有共同祖先的个体感兴趣区域中的基因变体之间的相关性来确定相关因子。
在另一示例中,多个输入单元10来源于包含祖先混合体的个体的研究。至少一个输入单元10的个体群组包括具有不同祖先的个体时,基于具有各个不同祖先的个体在感兴趣区域中基因变体之间的相关性的平均值来确定相关因子。该方法将混合的输入单元10的LD图确定为多个“主要(primary)”LD图的平均值,这些“主要”LD图中的每个根据基因变体之间相关性的明确定义的参考祖先集确定。
在输入单元10的个体群组具有共同祖先,但具有如性别的另一特征的不同值的情况下,可能不需要同时处理多个LD图,因为用于共同祖先的单个LD图是足够的。
根据所使用的输入数据,并非所有多个基因变体都可以以对所有祖先有意义的频率存在。例如,一些基因变体可能只在特定祖先的个体中发现。当这种情况发生时,并且因果效应被分配给这些低频变体中的一个时,可以假设给定祖先中不存在的这个变体与同一祖先的其他变体不相关。因此,低频变体和所有其他变体之间的相关性的相关因子r
一旦完成了一次或多次迭代,对于每个基因变体,该方法包括步骤S14:基于基因变体对输入单元10的采样效应量12的平均值,确定基因变体对每个输入单元10的目标表型的预测效应量14。预测效应量14还可以基于使用采样效应量12计算的输入单元的基因变体的后验效应量的平均值。在任何情况下的平均值都是在迭代的至少一个子集上计算的。可以使用任何合适的平均方法。使用多次迭代并对结果求平均值克服了效应量采样的随机性。一旦已经确定了因果变体的集及其效应量14,基于效应量14来确定PRS变得简单。在一实施例中,采样效应量的平均值可以是加权平均值,其中被确定为因果关系的每个变体的采样效应量通过该变体是因果关系的后验概率进行加权。
例如,变体i的平均效应量βi可以计算为:
其中L表示迭代的总次数,可选地在迭代的一些初始磨合(burn)之后。变体是因果关系的后验概率可以用任何合适的方式来确定。例如,它可以使用被确定为因果关系的变体的迭代次数来确定,作为执行的迭代总次数的比例。可替代地,当使用例如等式(4)f(β
以肺癌患者吸烟为例,本方法允许对来自大型的肺癌GWAS(不按吸烟状态分层)的输入单元10与来自非吸烟者的较小的肺癌GWAS的输入单元10进行联合分析。这将有效地为两个亚群(即非吸烟者和普通人群)中的肺癌的表型产生两组预测效应量14。对于大多数基因变体,对于对应于两个亚群的两个输入单元10,预测效应量14将是相同的。然而,对于与上瘾相关的变体,来自较小GWAS的输入单元10的效应量应该清楚地表明这些变体与非吸烟者的肺癌无关。这有效地实现了上述目标,即允许获得与上瘾相关的变体已被减去的肺癌PRS。
通常,如果确定输入单元10的个体群组的大小变化不是太大,则该方法执行得最好。例如,当使用来源于较小和较大个体群组的两个输入单元10时,一旦较小个体群组为较大个体群组大小的约~20%或更大时,则通常观察到显著的性能改进。
在一些实施例中,每个基因变体的一个或多个采样效应量12可以被丢弃,并且不包括在用于获得预测效应量14的平均值中。未包括的数量可以是预先确定的,或者基于采样效应量12的值。丢弃的采样效应量12可以是来自该方法的前面迭代(first iterations)的那些,例如前十次迭代、前二十次迭代或一些其他预定次数。这些通常被称为“老化(burn-in)”迭代,通常被丢弃,因为蒙特卡罗-吉布斯采样器等采样技术采用多次迭代以收敛于有用的采样模式。
考虑到一般确定PRS的可取性,本发明也可用于确定目标个体的目标表型的多基因风险评分的方法,如图3所示。使用上述方法获得的预测效应量的改进的估算值允许确定更准确的PRS。
确定PRS的方法包括接收关于目标个体的基因组的感兴趣区域的基因信息16的步骤S20。这可以包括关于个体在感兴趣区域中表达的基因变体(例如单核苷酸多态性、插入缺失)的信息。
该方法还包括接收使用上述分析基因数据的方法确定的感兴趣区域中的多个基因变体对目标表型的预测效应量14的步骤S22。
该方法还包括基于目标个体的基因信息16和效应量14来确定多基因风险评分20的步骤S24。
在一实施例中,在分析基因数据的方法中接收的输入单位10是从各自的个体群组中确定的,并且使用从与目标个体最相似的一组个体中确定的输入单元10的预测效应量14来确定个体的多基因风险评分20。例如,如果效应量14是为两个输入单位10确定的,这两个输入单元10分别由具有欧洲祖先和东亚祖先的个体群组确定,并且该个体是东亚祖先,那么东亚输入单元10的预测效应量14将用于确定该个体的PRS 20。
在一实施例中,计算PRS 20如下:
其中K是有助于PRS 20的变体的数量,x
分析基因数据的方法可以通过用于分析关于生物体的基因数据的装置来执行,也如图1所示。所述装置包括接收单元200,配置为接收多个输入单元10,其中每个输入单元包括关于生物体基因组的感兴趣区域中的多个基因变体与生物体的目标表型之间的关联的信息。该装置还包括数据处理单元210,配置为执行一次或多次迭代,包括:对于多个基因变体中的每一个,基于多个输入单元确定基因变体对目标表型是否是因果关系的;并且如果基因变体确定为是因果关系的,则基于多个输入单元10和关于感兴趣区域中的多个基因变体之间的相关性的信息,确定基因变体对每个输入单元10的目标表型的采样效应量12。对于所有输入单元10,基因变体对目标表型的采样效应量12都是非零的。数据处理单元210还被配置为对于每个基因变体,基于输入单元10的基因变体的采样效应量12的迭代的至少一个子集的平均值或使用采样效应量12计算的输入单元10的基因变体的后验效应量的平均值,确定基因变体对于每个输入单元10的目标表型的预测效应量14。
本发明还可以体现在包括指令的计算机程序中,当该指令由计算机执行时,该指令使计算机执行分析基因数据的方法。本发明还可以体现在包括指令的计算机可读介质中,当该指令由计算机执行时,该指令使计算机执行分析基因数据的方法。
结果
跨祖先
为了说明本方法在确定不同祖先亚群的效应量方面的有效性,使用现有技术方法确定的效应量的示例如图4所示,并且使用本方法确定的效应量如图5所示。
欧洲祖先的个体存在有力的乳腺癌汇总统计数据,东亚女性存在少得多的同类者,病例数的差异证明了这点(表1)。此外,有两个有力的同类者可用于评估各种表型的效应量:用于欧洲祖先个体的英国生物样本库(UK Biobank)(Bycroft等人)和用于东亚祖先个体的多种族同类者(multi ethnic cohort,MEC)。
图4和图5均示出了从东亚祖先(红色)个体和欧洲祖先(黑色)个体的两项乳腺癌研究中确定的两个输入单元的19号染色体上基因变体的推断效应量。图4示出了使用现有技术方法分别为两个输入单元确定的效应量。图5示出了使用本方法为两个输入单元联合确定的效应量。
当通过分别分析每个输入单元来确定效应量时(图4),已建立的癌症位点(cancerlocus)ELL(图4和图5下部面板(panel)中的放大插入)的基因变体对欧洲人具有较大的权重。然而,在对东亚祖先的个体的研究中,较小的样本量不足以检测到这种信号。当通过联合分析输入单元来确定效应量时(图5),这两项研究的结合为东亚人提供了足够的统计能力,使其在已建立的癌症位点ELL上也具有较大的效应量。
使用本方法的全基因组联合分析提高了两个祖先的预测性能。此外,联合分析显著改变了因果变体定位的准确性。这可以在图4和图5中观察到,在欧洲和东亚祖先中,乳腺癌的大的非零效应量在图5(联合分析)的顶部面板中所跨越的位置距离远小于图4(单独分析)的顶部面板。这反映了对通过结合来自多个祖先的数据获得的因果变体的定位(localisation)的更好理解。
表1示出用于确定欧洲和东亚祖先的女性中乳腺癌PRS的训练群体。
表1
利用这些同类者,评估了确定PRS计算中使用的预测效应量的不同方法的PRS预测能力,即LDPred、MTAG和本方法。结果如表2所示,其中粗体表示每个祖先的最佳性能。由于乳腺癌是二元特征,曲线下面积(under the curve,AUC)被用作预测准确性的度量,以量化乳腺癌病例和对照例之间PRS的分离。最好的执行方法是本方法,其结合了来自多个祖先研究的输入单元,并根据每个输入单元的效应量生成PRS的祖先特异性版本。
表2
背景特异性
如上所述,本方法还可以用于确定对于基于个体的其他特征确定的亚群特异性的预测效应量。群体的不同阶层可以以类似于不同祖先的方式进行处理,也可以计算出这些不同阶层特有的PRS。在下面的示例中,假设用于确定输入单元的研究来源于单个群体。因此,没有必要为每个输入单元考虑不同的相关因子集(即描述基因变体之间的相关结构的LD图)。然而,如上所述,研究之间的个体样本有时候可能是重叠的。
在本示例中,基因变体对BMI的预测效应量是在使用来自GIANT联合体GWAS的训练数据集确定的输入单元上确定的(152893名男性,171977名女性,或332154名组合的)。然后将从效应量获得的PRS应用于评估数据集。由于BMI是定量特征,因此解释的方差(r
-使用现有方法的方法将两个性别组合到单个的元分析,并生成在男性和女性中都进行评估的单个PRS;和
-本方法联合分析男性的BMI研究和另外的女性的BMI研究,并产生不同的效应量和两个不同的PRS(每个性别一个)。
这种比较的结果如表3所示。粗体表示两种性别中每个的表现最好的方法论。
表3
当使用本发明的性别分层方法的男性效应量时,解释的BMI方差对男性更高。同样,当使用本发明的性别分层方法的女性权重效应量时,解释的BMI方差对女性更高。在这两种情况下,使用现有方法对男性和女性进行的元分析效果不佳。此外,与现有的基于元分析的方法相比,使用来自本发明方法的男性效应量和女性效应量中的任何一个解释了在组合的性别评估集中BMI方差的更高百分比。
参考文献
Bayesian meta-analysis across genome-wide association studies ofdiverse phenotypes,Trochet H,Pirinen M,Band G,Jostins L,McVean G,Spencer C,Genetic Epidemiology 2019
Multi-trait analysis of genome-wide association summary statisticsusing MTAG,P Turley et al.Nature Genetics 2018
Vilhjálmsson BJ,Yang J,Finucane HK,et al.Modeling LinkageDisequilibrium Increases Accuracy of Polygenic Risk Scores.Am J Hum Genet2015.
Variable prediction accuracy of polygenic scores within an ancestrygroup,Hakhamanesh Mostafavi,Arbel Harpak Ipsita Agarwal,Dalton Conley,Jonathan K Pritchard,Molly Przeworski,eLife,2020
Bycroft et al,The UK Biobank resource with deep phenotyping andgenomic data,Nature 2018
A correction for sample overlap in genome-wide association studies ina polygenic pleiotropy-informed framework,Marissa LeBlanc,Verena Zuber,WesleyK.Thompson,Ole A.Andreassen,Schizophrenia and Bipolar Disorder Working Groupsof the Psychiatric Genomics Consortium,Arnoldo Frigessi,and Bettina KulleAndreassen,2018
Multitrait analysis of glaucoma identifies new risk loci and enablespolygenic prediction of disease susceptibility and progression,Jamie E.Craiget al,Nature Genetics 2020
- 用于分析基因数据的计算机实现方法和装置
- 分析关于生物体的基因数据的计算机实现的方法