一种靶向测序数据的mTag类别选取的方法、设备和介质
文献发布时间:2024-04-18 19:44:28
相关专利
本申请是申请号为202310069596X,申请日为2023年02月07日,发明名称为“一种基于mTag的靶向测序数据预处理的方法、设备和介质”的中国发明专利申请的分案申请。
技术领域
本发明属于生物数据处理技术领域,具体地,涉及一种靶向测序数据的mTag类别选取的方法、设备和介质。
背景技术
下一代测序(Next-generation sequencing,NGS)又称为高通量测序(High-throughput sequencing),是基于PCR和基因芯片发展而来的边合成边测序技术。高通量测序技术的特点主要有:测序读长短,通量高,准确度高。高通量测序相比一代测序大幅降低了成本,同时保持了较高准确性,并且大幅降低了测序时间,目前高通量测序已经在全组学得到广泛应用。
高通量测序得到的原始图像数据经碱基识别(BaseCalling)转化为原始测序序列(Sequenced Reads),我们称之为Raw Data或Raw Reads,结果以FASTQ格式存储,其中包含测序序列(reads)信息以及其对应的测序质量信息。
FASTQ格式文件中每个read由四行描述,如下所示:
@HWI-ST507:248:D29JDACXX:8:1101:1715:1919:1:1:0:ACTTGA/1
NTAATATTGGGCTAGAAAGTATCTTTGGGATTGCATGTTTTGATGCAGAATCATTGTGCCGTAGAATGC
+
BPYccaceceggghhfhhhhhhhhhhffhfhhgfahhchhhhhhfhbfghh_gfhhhhgghefffhhhh
其中第一行以“@”开头后跟Illumina测序标识符,包括机器型号、上机次数、试剂型号、第几个lane、在flowcell上的坐标、barcode等;第二行是碱基序列;第三行以“+”开头后跟Illumina测序标识符(为节省存储空间,部分fq文件会省略“+”后的信息);第四行是对应第二行碱基序列的质量值,是用来衡量测序准确度的,字符范围[B,h],对应质量范围[2,40]。第四行每个字符对应的ASCII值减去64,即为该碱基的测序质量值,例如h对应ASCII值为104,104-64=40。质量值越高,测序错误率越低。
在NGS过程中可能会出现PCR重复,虽然重复似乎是单独的reads,但它们实际上是由于PCR和测序过程中的错误导致的技术噪音。分子标签(mTag)技术是在文库制备过程中与DNA片段连接的随机短核苷酸序列。这些mTag序列充当唯一识别码,将每个reads标记为来自单个片段的扩增,为确定PCR重复提供了更准确的机制。但目前如何对针对PCR重复引入的分子标签技术进行去噪尚缺乏统一的标准和方法。
发明内容
为了解决上述技术问题中的至少一个,本发明采用的技术方案如下:
本发明第一方面提供一种基于mTag的靶向测序数据预处理的方法,包括以下步骤:
S1,mTag分类与修正:将测序reads基于mTag序列进行分类,包含reads数量小于第一阈值P1的mTag类别为第一类别,其余为第二类别,对于各第一类别中各read,基于其mTag序列中与各第二类别的mTag序列的差异碱基的质量值对其mTag序列进行修正并重新分类;
S2,mTag类别选择:选择包含reads数量大于或等于第二阈值P2的mTag类别;
S3,代表序列选择:从S2选择的各mTag类别中选择一条代表性序列,
其中,P1=3~5;P2=1~10。
在本发明中,靶向测序的测序reads包括接头序列、mTag序列及目标序列。在修正前,首先基于目标序列与参考基因组进行比对,过滤掉无法比对至目标区域的测序reads。
在本发明中,mTag即分子标签(molecule tag),也叫特异性分子标签(UniqueMolecular Indentifier,UMI),是一段随机或固定或随机固定混合的核苷酸短序列,通常设计为完全随机的核苷酸序列(如NNNNNN),也可以设计为包含固定碱基和随机碱基的核苷酸序列(如NNNCNNNNGNNNN)。在靶向测序的文库构建过程中,mTag通过连接的方式导入,如同分子条形码一样特异性地标记每个模板。mTag通过给每一个原始DNA片段加上一段特有的核苷酸标签序列,经过文库构建及PCR扩增过程之后,一起进行测序。根据不同的mTag序列区分不同来源的DNA模板,可以分辨哪些是PCR扩增及测序过程中产生的随机错误造成的假阳性突变,哪些是患者真正携带的突变,从而提高检测的灵敏度和特异性。根据上述分子标签的原理可知,具有相同mTag的测序序列(read)来源于同一DNA模板,因为进行分类时归为一个mTag类别。理论上,不同DNA模板产生的reads数量不会相差太大,如果某个mTag类别包含的reads数量过低,例如低于上述第一阈值P1,有可能是由于PCR过程中产生的错误,导致mTag序列出错,因此需要修正。
在本发明中,第一阈值P1的选择与测序深度也有一定的关系,一般情况下,由于目的区域靶向测序更加经济高效,可以实现500~1000×,甚至更高的测序深度,本发明设定第一阈值P1=3~10,即包含reads数目低于所述第一阈值时,预设该mTag是由于PCR扩增引入的错误(噪声)或者由于测序错误引入。但是如果经过与第二类别的mTag对比,在综合考虑mTag序列差异碱基的质量值后无法进行修正,则相应的mTag类别可能真实来自某个模板,包含的reads数目少是由于PCR的偏好性导致的,该mTag类别需要被保留用于后续分析。
在本发明中,质量值表示测序质量值,用于衡量测序准确度,即质量值越高,表明测序的错误率越低。测序错误率用E表示,碱基质量值用Q表示,则有下列关系:
Q=-10log10(E)
若碱基的质量值为10,则错误率为10%;若碱基质量值为20,则错误率为1%;若碱基质量值为30,则错误率为0.1%。
在本发明的一些实施方案中,步骤S1中基于各第一分类中各read的mTag序列中与各第二类别的mTag序列的差异碱基的质量值对其mTag序列进行修正并重新分类的步骤具体包括:
S11,将待修正的第一类别的mTag序列分别与各第二类别的mTag序列进行比对,找相似性最高的第二类别,针对所述待修正的第一类别中的各read分别进行修正:
①如果该read的mTag序列与相似性最高的第二类别的mTag序列的差异碱基位于该第二类别的mTag序列的固定碱基位点,则无论该read的分子标签该差异碱基质量值如何,均修正为该第二类别的mTag序列相应位置上的碱基类型;对于一些捕获测序,引入的mTag序列可能全部是随机的序列,则无需根据本步骤进行修正;
②如果该read的mTag序列与相似性最高的第二类别的mTag序列的差异碱基位于该第二类别的mTag序列的随机碱基位点,并且该差异碱基的质量值小于第三阈值P3,则将该差异碱基修正为该第二类别的mTag序列相应位置上的碱基类型,否则不进行修正;
③若经步骤①~②进行修正后,该read的mTag序列与相似性最高的第二类别的mTag序列的差异碱基占mTag序列长度比例小于第四阈值P4,则无论差异碱基质量值如何,直接将差异碱基修正为该第二类别的mTag序列相应位置的碱基类型,否则该read的mTag序列所有碱基均不进行修正;
④对于经步骤①~③无法完成修正的reads,利用相似性第二高的第二类别重复步骤①~③,直至与所有第二类别进行上述修正步骤后均无法完成修正,
S12,基于修正后的mTag序列对reads重新进行分类,
其中,P3=10~13;P4=10%~30%。
在本发明的一些实施方案中,位于随机碱基位点的差异碱基,若其质量值小于第三阈值P3,则认为该差异碱基的质量值过低,可能是测序错误,依据所述相似性最高的第二类别的mTag序列相应位置的碱基进行修正。在本发明的一些具体实施方案中,P3设置为10,即错误率不高于10%,否则需要进行修正,但也不能苛求质量值过高,否则会导致分类错误,当质量值为13时,错误率仅为5%,质量值更高时,错误率更低,此时,再进行修正可能会导致分类错误。因此,设置P3=10~13。
在本发明的一些实施方案中,P4值设置过低,会导致一些错误的reads的mTag序列不能被正确修正,P4值设置过高,会导致一些reads的mTag序列被错误修正。发明人经过反复探索,发现P4设置10%~30%之间时,修正的准确率最高。
在本发明的一些具体实施方案中,所述mTag序列的长度为16,P4设置为10%。即若经步骤①~②进行修正后,该read的mTag序列与相似性最高的第二类别的mTag序列的差异碱基占mTag序列长度比例小于10%(即仅1个差异碱基),则无论差异碱基质量值如何,直接将差异碱基修正为该第二类别的mTag序列相应位置的碱基类型。
在本发明的一些实施方案中,所述第二阈值P2利用以下方法确定:
S21,选择具有至少N个reads的mTag类别进行分析时,得到在n个样本中位点的均方根误差,均方根误差的计算公式如下:
其中,N为1~5的正整数;RMSE为均方根误差,
S22,N取不同值时,得到不同的RMSE值,将RMSE值最小时对应的N值设置为第二阈值P2。
在本发明的一些具体实施方案中,若使得RMSE值最小时对应的N值为2,则设置第二阈值P2=2,即选择包含reads数量大于或等于2的mTag类别进行后续分析。
在本发明的另一些实施方案中,所述第二阈值P2利用以下方法确定:
S21’,基于每种mTag类别对应的reads数和累计reads数绘制散点图;
S22’,从散点图中第一个点开始,计算任意两个相邻点连线的斜率,当首次出现某条连线的斜率低于前一条连线的斜率时,该连线的起点对应的横坐标值设置为所述第二阈值P2。
在本发明的一些具体实施方案中,当首次出现某条连线的斜率低于前一条连线的斜率时,表明该连线的终点对应的横坐标值(每种mTag类别对应的reads数)的reads数目增加开始变缓甚至不变(斜率0),则该连线的起点对应的横坐标值的mTag也需要被考虑纳入后续分析中。
在本发明的又一些实施方案中,所述第二阈值P2利用以下方法确定:S21’’,基于每种mTag类别对应的reads数和累计reads数绘制散点图;
S22’’,对散点图进行拟合得到曲线方程f(x),对曲线方程f(x)进行求导得到f'(x),当f’(x)的斜率从小到大首次开始下降时,与对应的横坐标最接近的正整数设置为所述第二阈值P2。
在该实施方案中,与上述实施方案核心思想一致,只不过是本实施方案对散点图进行拟合,使得更加精准。在本发明的一些具体实施方案中,采用4阶函数进行曲线拟合
在本发明的一些实施方案中,步骤S3中,利用以下步骤选取代表性序列:
S31,对mTag类别中的序列根据目标序列进行计数,计算数目最多的reads在所述mTag类别中的占比,若占比低于第五阈值P5,则舍弃该mTag类别所有reads,否则选择数目最多的reads中的任意一条作为代表性序列,
其中,P5=75%~90%。
在本发明的一些具体实施方案中,P5是基于四分位法的一种优化方法。由四分位法可知,排在后1/4(即75%)位置上的数叫做第三四分位数,为了进一步提高数据分析质量,对临床基因诊断提供精准辅助,将P5设置为75%~90%,优选地,P5=80%。在本发明的一些实施方案中,若数目最多的reads在所述mTag类别中的占比超过第五阈值,表明其序列确实能够代表该mTag类别,其他reads(如有)可能是由于PCR扩增错误或测序错误引入的。也可能是mTag序列存在错误导致被错误分类至该mTag类别。
然而,由于第五阈值P5设置相对较高,可能会导致一些mTag类别由于一些目标序列在PCR过程中引入错误或测序引入错误导致整个mTag类别被舍弃,造成后续分析结果不准确。因此,在本发明的另一些实施方案中,步骤S3中,利用以下步骤选取代表性序列:
S31’,从目标序列第一个碱基位置开始,统计所述mTag类别中所有reads该位置的碱基类型及数目:
1)若该位置只有一种碱基类型,则无需修正,继续下一碱基的修正;
2)若该位置不止一种碱基类型,则对数目非第一多的碱基类型进行修正:
①若该碱基质量值不小于第六阈值P6,则无需修正;
②若该碱基质量值小于第六阈值P6,则将其修正为数目第一多的碱基类型;
③统计该mTag中所有reads该位置修正后的碱基类型及数目,若数目第一多的碱基类型占比不低于第七阈值P7,则将所有非第一多的碱基类型均修正为数目第一多的碱基类型,继续下一碱基的修正;若数目第一多的碱基类型占比低于第八阈值P8,则直接舍弃该mTag类别所有reads,
S32’,若按照步骤S31’完成所有碱基的修正,则选择其中任意一条reads作为代表性序列,
其中,P6=15~20;P7=75%~90%。
在本发明的一些具体实施方案中,由于靶向测序可能是为了寻找变异位点,因此,对碱基的质量值要求更高,不能接受错误率超过3%,则质量值不能低于15。当然,同样不能设置P6过高,否则会导致错误修正。当设置P6=15~20时,修正的正确率最为理想。
在本发明的一些具体实施方案中,设置P7的核心思想与设置P5的核心思想相同。
在本发明的一些实施方案中,在步骤S31’中,若数目第一多的碱基类型不止一种,则先选择一种碱基类型作为数目第一多的碱基类型,其余作为数目非第一多的碱基类型进行修正,若无法完成修正,则再选择另外一种碱基类型作为数目第一多的碱基类型进行修正,直至完成修正或者舍弃该mTag类别所有reads。
在本发明的一些具体实施方案中,若对于一个碱基,G=40%,C=40%,A=20%。此时,先将G作为数目第一多的碱基类型,将C和A均作为数目非第一多的碱基类型,按照上述方法进行修正;若无法完成修正(即修正后数目第一多的碱基类型占比仍低于第八阈值P8),则再针C作为数目第一多的碱基类型,将G和A作为数目非第一多的碱基类型,按照上述方法进行修正,若仍然无法完成修正(占比低于第八阈值P8),则直接舍弃该mTag类别所有reads。若能完成修正,则继续下一碱基的修正。
本发明第二方面提供一种计算机设备,包括:
存储器,用于存储计算机程序;
处理器,用于执行所述计算机程序时实现如本发明第一方面任一所述的一种基于mTag的靶向测序数据预处理的方法的步骤。
本发明第三方面提供一种计算机可读存储介质,所述计算机可读存储介质上存储有计算机程序,所述计算机程序被处理器执行时实现如本发明第一方面任一所述的一种基于mTag的靶向测序数据预处理的方法的步骤。
在本发明中,步骤S1、步骤S2和步骤S3可以作为单独的技术方案分别解决相应的技术问题。
本发明的有益效果
相对于现有技术,本发明取得了以下有益效果:
利用本发明的方法,可以对mTag进行修正,避免PCR过程中或测序过程中引入的错误,降低mTag被错误分类的可能性,可以分辨哪些是PCR扩增及测序过程中产生的随机错误造成的假阳性突变,哪些是患者真正携带的突变,从而提高检测的灵敏度和特异性。
利用本发明的方法,可以精准地选择合适的mTag类别进行后续分析,进一步提升分析的准确度。
利用本发明的方法,可以精准选择mTag类别中的代表性序列,数据利用率更高,分析准确性也得到大大提升。
附图说明
图1示出了靶向测序下机的reads序列结构示意图。
图2示出了本发明实施例2中针对4种位点当mTagCutOff=2时,RMSE随测序数据量变化的情况。
图3示出了本发明实施例2中针对4种位点在不同测序数据量不同mTagCutOff时的RMSE变化情况。
图4示出了本发明实施例2中针对p.G719S在不同测序数据量不同mTagCutOff时的RMSE变化情况。
图5示出了本发明实施例2中针对p.E746_A750delELREA在不同测序数据量不同mTagCutOff时的RMSE变化情况。
图6示出了本发明实施例2中针对p.T790M在不同测序数据量不同mTagCutOff时的RMSE变化情况。
图7示出了本发明实施例2中针对p.L858R在不同测序数据量不同mTagCutOff时的RMSE变化情况。
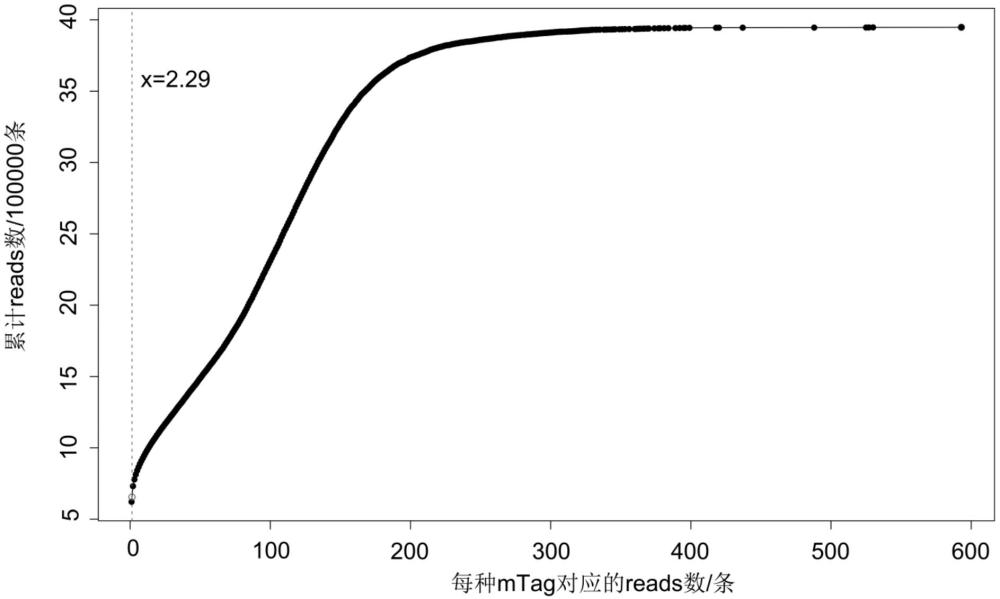
图8示出了本发明实施例3中readsPerTag数和累计reads数的散点图。
具体实施方式
除非另有说明、从上下文暗示或属于现有技术的惯例,否则本申请中所有的份数和百分比都基于重量,且所用的测试和表征方法都是与本申请的提交日期同步的。在适用的情况下,本申请中涉及的任何专利、专利申请或公开的内容全部结合于此作为参考,且其等价的同族专利也引入作为参考,特别这些文献所披露的关于本领域中的相关术语的定义。如果现有技术中披露的具体术语的定义与本申请中提供的任何定义不一致,则以本申请中提供的术语定义为准。
为了使本发明所解决的技术问题、技术方案及有益效果更加清楚明白,以下结合实施例,对本发明进行进一步详细说明。
实施例
以下例子在此用于示范本发明的优选实施方案。本领域内的技术人员会明白,下述例子中披露的技术代表发明人发现的可以用于实施本发明的技术,因此可以视为实施本发明的优选方案。但是本领域内的技术人员根据本说明书应该明白,这里所公开的特定实施例可以做很多修改,仍然能得到相同的或者类似的结果,而非背离本发明的精神或范围。
除非另有定义,所有在此使用的技术和科学的术语,和本发明所属领域内的技术人员所通常理解的意思相同,在此公开引用及他们引用的材料都将以引用的方式被并入。
那些本领域内的技术人员将意识到或者通过常规试验就能了解许多这里所描述的发明的特定实施方案的许多等同技术。这些等同将被包含在权利要求书中。
下述实施例中的实验方法,如无特殊说明,均为常规方法。下述实施例中所用的仪器设备,如无特殊说明,均为实验室常规仪器设备;下述实施例中所用的试验材料,如无特殊说明,均为自常规生化试剂商店购买得到的。
实施例 1 分子标签的分类和修正
VariantPro
对测序下机数据进行处理的第一步是将拥有相同分子标签(mTag)的reads分类到一起。VariantPro
在本实施例中,已知其中一条分子标签的模板序列为:NNTNNNTNNGNCTNNN,其中N为随机碱基,其他位置为固定碱基,其测序后可能得到的序列如下:
上述9条分子标签序列中1-5号序列完全一致,6号序列中只有1个碱基和1-5号序列不一致,7号序列有2个碱基和1-5号序列不一致,8号序列和9号序列总共有3个碱基不一致。发明人利用分子标签序列和测序质量值对这些分子标签进行修正和分类,步骤如下:
(1)首先将测序reads按照分子标签进行分类,即将完全相同的分子标签归为一个类别,其中,包含reads数量较小的类别(<5条)为第一类别,属于第一类别组,包含reads数量较大的类别(≥5条)为第二类别,属于第二类别组。如上述1~9号序列,1~5号归为一个类别,为第二类别,属于第二类别组;8~9号序列归为一个类别,6号序列和7号序列分别归为另外不同的类别,均为第一类别,属于第一类别组;
(2)针对第一类别组中的各第一类别(如上述6号序列组成的类别、7号序列组成的类别、8号序列和9号序列组成的类别),分别进行如下处理:
1)将该第一类别的分子标签分别与第二类别组中的各第二类别的分子标签进行比对,找出相似性最高的第二类别,如与上述6号序列组成的类别、7号序列组成的类别、8号序列和9号序列组成的类别相似性最高的类别均为上述1~5号序列组成的第二类别,针对该第一类别中每条read分别进行处理:
①如果该read的分子标签与相似性最高的第二类别的分子标签的差异碱基位于该第二类别的固定碱基位点,即与该第二类别的分子标签在固定碱基位点的碱基类型不同,则无论该read的分子标签该差异碱基质量值如何,都修正为该第二类别的分子标签的固定碱基。如第6、7、8号序列,与该第二类别的分子标签不同的第7位碱基A位于该第二类别的分子标签的固定碱基T所在位点,则不管该read此位点的质量值如何,都修正为T,由此,6号序列经修正后,与1~5号序列归为同一类别。
②如果该read的分子标签与相似性最高的第二类别的分子标签的差异碱基位于该第二类别的随机碱基位点,即与该第二类别的分子标签在随机碱基位点的碱基类型不同,并且该差异碱基的质量值小于10,则将该差异碱基修正为该第二类别的分子标签相应位置上的碱基类型,否则不进行修正。例如,7号序列,第11位碱基的质量值小于10,则将该差异碱基C修正为A。由此,7号序列经修正后,与1~5号序列也归为同一类别;再如,8号序列,第11位碱基和第15位碱基均为差异碱基,且第11位碱基质量小于10,则将第11位碱基由C修正为A,但是第15位碱基质量值大于10,则第15位碱基不进行修正;又如,9号序列,同8号序列,第11位碱基和第15位碱基均为差异碱基,但质量值均不小于10,则第11位碱基和第15位碱基均不进行修正。
③若经步骤①~②进行修正后,该reads的分子标签只有1个碱基与相似性最高的第二类别的分子标签不同,则同样无论该碱基质量值如何,直接将该碱基修正为该第二类别的分子标签相应位置的碱基类型。如第8号序列,此时只有第15位碱基与第二类别分子标签不同,则直接将第15位碱基G修正为第二类别分子标签相应位置的C,由此,8号序列经修正后,与1~5号序列也归为同一类别。若经步骤①~②进行修正后,该reads的分子标签仍然存在2个或2个以上碱基与相似性最高的第二类别的分子标签不同,则该reads分子标签所有碱基均不进行修正(经步骤①和②修正的碱基也恢复至原来的碱基)。如9号序列,经上述修正后,仍有第11位碱基和第15位碱基2个差异碱基,则该reads分子标签所有碱基均不进行修正(在第②步修正的第7位碱基也恢复至原来的碱基)。
④对于无法完成修正的reads,利用相似性第二高的第二类别重复步骤①~③,直到与第二类别组中所有第二类别进行上述修正步骤后均无法完成修正,将该第一类别中剩余的reads作为独立的类别,上述序列中,9号序列作为独立的类别。
经过上述修正后,1~8号序列重新分类为一个类别,9号序列单独成为另外一个类别。
发明人根据如上方法将测序下机数据分成若干个类别,分在一个类别里的序列代表这些序列是由于PCR扩增时基于偏好性导致的重复序列。
本实施例对针对一个9基因25个突变位点的靶向测序数据进行处理,处理前,reads小于5条的类别有1412785个,共有reads数目为1681888条;处理后,reads小于5条的类别有1200656个,共有reads数目为1412537条。
实施例2 基于均方根误差设置分子标签阈值
经过实施例1分子标签的修正和分类,发明人需要判断当一个类别中reads数量有多少时可以利用其进行后续的数据分析。这是由于对于一些类别,如果只有一条或者若干条reads,那么其与拥有数十条甚至数百条reads的类别在可信度上会存在巨大差异。尤其是需要针对不同类别挑选代表性序列进行分析时,选择具有多少条reads的类别将会变得更加重要。
为此,发明人对样本测序数据量(SampleDataSize)和分子标签阈值(mTag阈值,mTagCutOff)这两个因素设置梯度,其中,mTagCutOff是指选择作为分析基础的类别(mTag)的最少reads数目。评估测序数据量的变化和mTagCutOff值的变化对检测精度的影响。发明人通过计算位点的观察频率(observed frequency)与期望频率(expected frequency,实际频率)的均方根误差(RMSE,root-mean-square error)来描述检测精度,RMSE越小,检测精度越高。RMSE的计算公式如下:
其中,
具体的测试方案如下:
(1)测试样品数据量统计:
Q20与Q30表示质量值大于等于20或30的碱基所占百分比。
(2)阈值设置
SampleDataSize取值范围为15M~270M(由于L8样品的数据总量只有270M,为保证每个样品都能同等取值,所以上限设定为270M),步长15M。mTagCutOff取值范围为1~10,步长为1。
(3)位点期望频率
(4)RMSE变化趋势
发明人对EGFR基因的所有4个位点及单独每个位点均进行了统计。结果发现,当mTagCutOff固定时,RMSE随着测序量的增加而减小并逐渐趋于稳定;在测序数据量一定的情况下,mTagCutOff为1或2时RMSE达到最低点,即检测精度最高点,之后随着mTagCutOff的增加而增加。详细如下:
①针对所有4个位点
当数据量到达165M后,RMSE波动开始变得很小趋于稳定(当mTagCutOff=2时,RMSE的变化如图2所示),并且在数据量相同的前提下,mTagCutOff=2时RMSE最小,如图3所示。
②针对p.G719S位点
同样地,当数据量到达165M后,RMSE波动开始变得很小趋于稳定,并且在数据量相同的前提下,mTagCutOff=2时RMSE最小,如图4所示。
③针对p.E746_A750delELREA位点
当数据量到达225M后,RMSE波动开始变得很小趋于稳定,并且在数据量相同的前提下,mTagCutOff=1时RMSE最小,如图5所示。
④针对p.T790M位点
同样地,当数据量到达225M后,RMSE波动开始变得很小趋于稳定,并且在数据量相同的前提下,mTagCutOff=1时RMSE最小,如图6所示。
⑤针对p.L858R位点
同样地,当数据量到达225M后,RMSE波动开始变得很小趋于稳定,并且在数据量相同的前提下,mTagCutOff=1时RMSE最小,如图7所示。
然而,当mTagCutOff设置为1时,会包含过多的假阳性,影响突变检测的特异性。
综合考虑,发明人将20ng左右文库起始量约200M测序量的mTagCutOff设置为2,即一个类别中(拥有同一个mTag)的reads条数需不小于2才能用于下游的数据分析,共计360196个类别(mTag)进入下游分析。
实施例3 基于readsPerTag曲线设置分子标签阈值
关于如何设置分子标签阈值,发明人基于每种mTag对应的reads数(readsPerTag)提供另外一种设置方法。
具体地,发明人统计readsPerTag并进行绘图,如图8所示。其中标题为样本名,横坐标为readsPerTag,纵坐标是累计reads数(Cumulative reads)。部分数据如下:
首先,发明人采用4阶函数进行曲线拟合,
发明人还用一种更简单的方法确定mTagCutOff的阈值:基于mTagCutOff和累计reads数绘制散点图;从散点图中第一个点开始,计算任意两个相邻点连线的斜率,当首次出现某条连线的斜率低于前一条连线的斜率时,该连线的起点对应的横坐标值设置为mTagCutOff的阈值。在上述例子中,第一个点和第二点之间连线的斜率109746,第二个点和第三个点之间的斜率为47034,斜率开始下降,则第二个点对应的横坐标值设置为mTagCutOff的阈值,即mTagCutOff=2。
实施例4 代表序列的选择
根据实施例2和实施例3,发明人选择reads数目不少于2的类别(mTag)进行后续分析。
由图1可知,在mTag之后是目标序列,这些目标序列会直接影响到后续判定位点是否存在突变。发明人接下来会从划分在同一类别里的序列中挑选出一条代表性序列来进行后续的数据比对分析。
在本实施例中,针对其中一个类别的代表性序列的筛选进行详细介绍。该类别(mTag为ACTAGATCCGACTACGACG)共包括10条reads,序列如下所示:
reads-1:ACTAGATCCGACTACGACGAGCTTTTAGACGATTATACCAGCCG
reads-2:ACTAGATCCGACTACGACGAGCTTTTAGACGATTATACCAGCCG
reads-3:ACTAGATCCGACTACGACGAGCTTTTA
reads-4:ACTAGATCCGACTACGACGAGCTTTTA
reads-5:ACTAGATCCGACTACGACGAGCTTTTAGACGATTATACCAGCCG
reads-6:ACTAGATCCGACTACGACGAGCTTTTAGACGATTATA
reads-7:ACTAGATCCGACTACGACGAGCTTTTAGACGATTATACCAGCCG
reads-8:ACTAGATCCGACTACGACGAGCTTTTAGACGATTATACCAGCC
reads-9:ACTAGATCCGACTACGACGAGCTTTTAGACGATTATACCAGCC
reads-10:ACTAGATCCGACTACGACGAGCTTTTAGACGATTATACCAGCC
上述序列中,黑体加粗部分为mTag序列,10条reads的mTag序列相同,在实施例1的步骤中会被归为一类。由上述序列可以看出,拥有相同mTag的reads分类在一起后,中间的目标序列仍然可能会存在一些不完全一致的位点(下划线部分),在选择代表性序列时,需要考虑这些位点。
在本实施例中,发明人采用两种方法来筛选代表性序列。
1. 基于哈希表进行选择
将上述10条reads序列放入到一个哈希列表中,分别对10条序列进行计数(序列完全相同的进行累加),取个数最多的序列作为代表性序列,若个数最多的那一条序列在该mTag中所占的比例不足80%,则舍弃该类别的数据。其中,80%阈值是基于四分位法的一种优化方法:由四分位法可知,排在后1/4(75%)位置上的数叫做第三四分位数,为了进一步提高数据分析质量,对临床基因诊断提供精准辅助,发明人将阈值设置为80%。
该方法的优点为由于采用哈希列表进行键值存储,故选择速度非常快;缺点是该方法不考虑测序质量,如果整条reads的测序质量比较差的话,后续数据分析的准确性会受到较大的影响,另外,也会导致一些序列被舍弃而影响后续分析质量。利用上述类别的10条reads,个数最多的序列(read-1、read-2、read-5、read-7)占比仅40%,不足80%,会导致该类别的数据直接被舍弃。
利用本方法得到的代表序列数目有288156条。
2. 基于位移法对位点修正后进行选择
步骤如下:
(1)从目标序列第一个碱基位置开始,统计该mTag中所有reads该位置的碱基类型及数目:
1)若该位置只有一种碱基类型,则无需修正,继续下一碱基的修正;
2)若该位置不止一种碱基类型,则对数目非第一多的碱基类型进行修正:
①若该碱基质量值不小于20,则无需修正;
②若该碱基质量值小于20,则将其修正为数目第一多的碱基类型;
③统计该mTag中所有reads该位置修正后的碱基类型及数目,若数目第一多的碱基类型占比不低于80%,则将所有非第一多的碱基类型均修正为数目第一多的碱基类型,继续下一碱基的修正;若数目第一多的碱基类型占比低于80%,则直接舍弃该mTag所有reads,不再对其他碱基进行修正。
(2)如能按照步骤(1)的方法完成所有碱基的修正,则选择其中一条reads作为代表性序列(经过修正后,所有reads序列完全相同)。
在本实施例中,针对上述mTag的10条reads,修正方法如下:
针对第20-27位碱基(前19位是mTag序列,第20位开始为目标序列),由于所有reads在这些位置的碱基类型均相同,因此无需进行修正。
针对第28个碱基,该位置的碱基类型及数目为G=8,C=2,由于read3和read4该位置的碱基(C)对应的质量值均大于20,不进行修正;再统计得到该位置的碱基类型和数目分别为:G=8,C=2。其中,G%=80%,C%=20%,满足80%的阈值,则将两个C都修正为G,最终所有reads该位点的碱基类型均为G。
针对第29-37位碱基,同样由于所有reads在这些位置的碱基类型均相同,因此无需进行修正。
针对第38位碱基,该位置的碱基类型及数目为C=9,A=1,由于read-6该位置的碱基(A)对应的测序质量小于20,则直接将A修正为C。修正后,所有reads该位点的碱基类型均为C,占比100%,超过80%。
针对第39-40位碱基,同样由于所有reads在这些位置的碱基类型均相同,因此无需进行修正。
针对第41位碱基,该位置的碱基类型及数目为G=9,C=1,由于read-4该位置的碱基(C)对应的测序质量大于20,不进行修正,不进行修正;再统计得到该位置的碱基类型和数目分别为G=9,C=1,其中G%=90%,C%=10%,G的占比超过80%的阈值,则将read-4该位置的C修正为G,最终所有reads该位点的碱基类型均为G。
针对第42-43位碱基,同样由于所有reads在这些位置的碱基类型均相同,因此无需进行修正。
针对第44位碱基,该位置的碱基类型及数目为G=7,C=3,read-8质量值小于20,则该位置的碱基修正为G,而read-9和read-10的碱基质量值均大于20,不进行修正;再统计得到该位置的碱基类型和数目分别为:G=8,C=2。其中,G%=80%,C%=20%,满足80%的阈值,则将read-9和read-10的该位置的碱基C也都修正为G,最终所有reads该位点的碱基类型均为G。
经过上述修正后,该mTag下所有reads序列均为:ACTAGATCCGACTACGACGAGCTTTTAGACGATTATACCAGCCG,选择任意一条作为代表性序列。可见,利用本发明可以更大限度地利用测序数据,提高分析的准确性。
利用本方法得到的代表序列数目有295071条,比利用基于哈希表进行选择的方法多了2.4%,提高了数据利用率,对于本技术领域而言,这些数据利用率的提高具有非常显著的意义,可以检测出一些低频突变,有助于辅助医生结合其他临床指标进行综合诊断。
实施例5 突变位点检测
发明人以EGFR的p.T790M(c.2369c>t)突变为例,p.T790M的突变属于有非小细胞肺癌的病人在经过针对EGFR基因敏感突变的小分子的酪氨酸激酶抑制剂治疗之后,经常出现的一种耐药性突变。如果在针对EGFR基因治疗之后出现了p.T790M突变的话,往往意味着对原来的小分子酪氨酸激酶抑制剂出现了耐药的现象,这个时候就需要停止原来的小分子酪氨酸激酶抑制剂的治疗,而换用能够对p.T790M突变有效的新的小分子酪氨酸激酶抑制剂,比如可使用第三代的药物进行治疗。该位点具有非常重要的临床用药指导意义。其中一段目标序列的测序结果如下(不含mTag序列):
上述目标序列中,第12位C->T突变的reads覆盖数为8,然而,一般情况下会将10条以上reads覆盖的突变综合判断为真阳性突变,上述位点会被判为阴性位点。发明人发现该位点上有2个C->A的突变,且A碱基的测序质量偏低(均小于10,分别为“)”和“*”,对应的质量值分别是8和9),利用实施例4的方法进行修正,该位点的A突变会被修正成T,那么该位点上的T的覆盖修正成10,满足阈值条件,软件会初步判断为阳性,该结果可被下游的PCR实验进行进一步的验证,以确保减少假阳性的发生,提高检测的灵敏性和特异性。
在本发明提及的所有文献都在本申请中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。
- 一种数据配置方法及装置、一种计算设备及存储介质
- 一种数据存储方法及装置、一种计算设备及存储介质
- 一种数据存储方法及装置、一种计算设备及存储介质
- 一种数据处理方法及装置、一种计算设备及存储介质
- 一种基于大数据的航空数据分析方法、设备及存储介质
- 一种靶向测序数据的mTag类别选取的方法、设备和介质
- 一种基于mTag的靶向测序数据预处理的方法、设备和介质