糖靶向的GSH响应型埃博霉素B纳米前药及制备方法与应用
文献发布时间:2024-04-18 19:52:40
技术领域
本发明属于生物医药技术领域,涉及糖靶向的谷胱甘肽(GSH)响应型埃博霉素B纳米前药及制备方法与应用。
背景技术
公开该背景技术部分的信息仅仅旨在增加对本发明的总体背景的理解,而不必然被视为承认或以任何形式暗示该信息构成已经成为本领域一般技术人员所公知的现有技术。
化疗仍然是治疗癌症最有效和最常见的治疗方案,但是由于化疗药物较差的选择性和生物相容性,有时会产生较严重的副作用。埃博霉素B是一个拥有强大抗增殖活性的天然产物,对多种肿瘤均有着较好的抑制活性。但埃博霉素B对肿瘤细胞的选择性差,治疗窗不理想,存在着严重的副作用,这限制了其进一步的临床应用。
发明内容
为了解决现有技术的不足,本发明的目的是提供糖靶向的GSH响应型埃博霉素B纳米前药及制备方法与应用,本发明提供的糖靶向的GSH响应型埃博霉素B纳米前药不仅可以在肿瘤细胞中高浓度的GSH环境中断裂并释放出活性药物,而且能够实现药物在肿瘤部位的富集。
为了实现上述目的,本发明的技术方案为:
一方面,一种糖靶向的GSH响应型埃博霉素B纳米前药,其化学结构由鼠李糖片段和埃博霉素B通过连接基共价键合形成;所述鼠李糖片段为含有鼠李糖化学结构的基团,所述连接基为含二硫键的连接基团。
本发明中埃博霉素B和鼠李糖片段通过含有GSH响应的二硫键连接基连接在一起,这种连接基可以在肿瘤细胞中高浓度的GSH环境中断裂并释放出活性药物。同时,埃博霉素B为疏水性,鼠李糖片段为亲水性,因而连接后的化学结构具有两亲性,可以在水溶液中因亲疏水作用自组装为均匀的前药纳米粒。并通过鼠李糖的主动靶向和纳米粒的EPR效应,实现药物在肿瘤部位的富集。
进一步地,所述鼠李糖片段为鼠李糖单糖衍生片段或鼠李糖三糖衍生片段,鼠李糖单糖衍生片段的化学结构如式I所示,鼠李糖三糖衍生片段的化学结构如式II所示,
其中,n为1~5。
进一步地,所述连接基的化学结构如式(III)或式(IV)所示,
其中,R
进一步地,化学结构如式(V)或(VI)所示,
其中,n为1~5。更进一步地,n=3。
另一方面,一种上述糖靶向的GSH响应型埃博霉素B纳米前药的制备方法,将糖片段与埃博霉素片段通过Click反应获得;
其中,糖片段的合成为单糖衍生片段的合成或三糖衍生片段的合成;
所述单糖衍生片段的合成为:将全乙酰化保护鼠李糖供体与PEG-OTs发生糖苷化反应后经叠氮基取代和保护基脱除得到关键的单糖衍生片段;
所述三糖衍生片段的合成为:将全苯甲酰化保护的葡萄糖供体与PEG-OTs发生糖苷化反应,叠氮基取代后脱除苯甲酰基保护基,随后进行选择性3,6-苯甲酰化保护,与鼠李糖供体发生糖苷化反应后脱除保护基即生成三糖衍生片段;
埃博霉素片段的合成为埃博霉素B-烷基二硫片段的合成或埃博霉素B-芳基二硫片段的合成;
所述埃博霉素B-烷基二硫片段的合成为:二硫二乙醇与炔丙基溴发生取代反应后在三光气的条件下生成活性酯,随后活性酯与埃博霉素B发生酯化反应得到相应产物;
所述埃博霉素B-芳基二硫片段的合成为:吡啶二硫代丙酸与炔丙胺发生酰胺缩合反应,随后与巯基苯乙酸发生巯基二硫键交换反应得到关键中间体,最后与埃博霉素B通过酯化反应生成相应产物。
第三方面,一种糖靶向的GSH响应型埃博霉素B纳米前药颗粒,由上述糖靶向的GSH响应型埃博霉素B纳米前药形成纳米颗粒,粒径为50~400nm。
第四方面,一种上述糖靶向的GSH响应型埃博霉素B纳米前药颗粒的制备方法,将糖靶向的GSH响应型埃博霉素B纳米前药溶解至有机溶剂中,然后滴加水进行自组装,即得。
进一步地,所述有机溶剂为二甲基亚砜或乙醇。
进一步地,自助装后进行透析去除有机溶剂。
第五方面,一种药物制剂,包括活性成分和药用辅料,所述活性成分为上述糖靶向的GSH响应型埃博霉素B纳米前药或上述糖靶向的GSH响应型埃博霉素B纳米前药颗粒。
所述药用辅料可以为氧化铝、硬脂酸铝、离子交换剂等载体,可以为糖浆、阿拉伯胶、山梨醇等粘合剂,可以为硬脂酸镁、聚乙二醇、滑石等润滑剂,可以为马铃薯淀粉等崩解剂,等等。
所述药物制剂的剂型可以为片剂、胶囊、口服液等。
第六方面,一种上述糖靶向的GSH响应型埃博霉素B纳米前药、糖靶向的GSH响应型埃博霉素B纳米前药颗粒或药物制剂在制备抗肿瘤药物中的应用。
具体地,所述肿瘤为肝癌、肺癌或乳腺癌。
本发明的有益效果为:
本发明提供的糖靶向的GSH响应型埃博霉素B纳米前药在不同还原条件下能够释放不同量的埃博霉素B,即对还原条件有响应性。经过MTT法检测表明,本发明提供的糖靶向的GSH响应型埃博霉素B纳米前药的抗肿瘤活性与埃博霉素相当。经过动物体内抗肿瘤活性研究表明,本发明提供的糖靶向的GSH响应型埃博霉素B纳米前药不仅具有良好的抗肿瘤活性,而且具有更好的安全性。
附图说明
构成本发明的一部分的说明书附图用来提供对本发明的进一步理解,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。
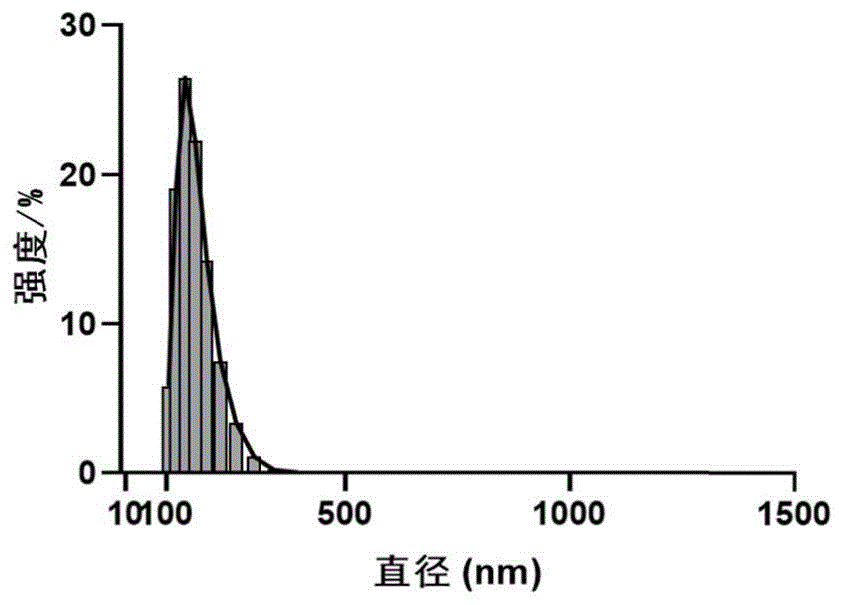
图1为本发明实施例中纳米前药V-a纳米颗粒的动态光散射表征图;
图2为本发明实施例中纳米前药V-a纳米颗粒的透射电镜照片;
图3为本发明实施例中纳米前药V-a在不同DTT浓度下的埃博霉素B药物释放曲线;
图4为本发明实施例中纳米前药V-a对HepG2、NCI-H460、NCI-H460/Taxol和MCF-7的细胞毒性曲线;
图5为本发明实施例中纳米前药V-a的体内抗肿瘤活性表征图,A为肿瘤体积变化曲线,B为体重曲线,C为生存曲线,D为肿瘤重量统计图,E为肿瘤照片。
具体实施方式
应该指出,以下详细说明都是示例性的,旨在对本发明提供进一步的说明。除非另有指明,本文使用的所有技术和科学术语具有与本发明所属技术领域的普通技术人员通常理解的相同含义。
为了使得本领域技术人员能够更加清楚地了解本发明的技术方案,以下将结合具体的实施例详细说明本发明的技术方案。
实施例
中间体A-3的合成
在氮气保护下,将糖基供体A-1(200mg,0.46mmol)、A-2(160mg,0.46mmol)以及适量
中间体A-4的合成
将化合物A-3(320mg,0.52mmol)和四丁基碘化铵(20mg,0.05mmol)溶于DMF,加入叠氮化钠后置于60℃下反应。待反应完全后,向反应溶液中加入大量水,乙酸乙酯萃取。合并有机相后,经浓缩、柱层析得透明油状物(200mg,79%)。
中间体A-5的合成
将化合物A-4(85mg,0.17mmol)置于反应瓶中,加入甲醇将其溶解后,滴加甲醇钠的甲醇溶液调pH为10,室温反应。待反应完全后,加入酸性阳离子树脂调pH为中性,减压抽滤,滤液浓缩后经柱层析纯化后得黄色油状物(55mg,88%)。
中间体B-2的合成
将化合物B-1(1g,1.3mmol)、A-2(546mg,1.6mmol)和适量
3.77(m,1H),3.70–3.63(m,4H),3.60–3.56(m,2H),3.52(dd,J=5.6,3.4Hz,2H),3.47–3.43(m,4H),3.36(t,J=4.7Hz,2H),2.42(s,3H).
中间体B-3的合成
将化合物B-2(1.1g,1.2mmol)和四丁基碘化铵(37mg,0.1mmol)溶于DMF,加入叠氮化钠(115mg,1.78mmol)后置于60℃下反应。待反应完全后,向反应溶液中加入大量水,用乙酸乙酯萃取水相。合并有机相后,经减压浓缩得透明油状物。
中间体B-4的合成
将化合物B-3溶于适量甲醇,滴加适量的甲醇钠的甲醇溶液调节pH为10,室温反应1h。加入酸性阳离子树脂调pH为中性,滤液浓缩后经柱层析纯化得透明油状物(260mg,57%)。
中间体B-5的合成
将化合物B-4(260mg,0.68mmol)、适量分子筛置于反应瓶中,氮气保护后加入适量二氯甲烷。将反应瓶置于-65℃,并在该温度下加入三乙胺(190μl,1.36mmol)和BzCN(178mg,1,36mmol),滴加完毕后保持该温度继续反应。TLC监测反应完全后,向反应溶液中加入适量甲醇淬灭反应,随后将反应液减压抽滤,滤液浓缩后经柱层析纯化得透明油状物(340mg,85%)。
中间体B-6的合成
取一两颈瓶,将化合物A-1(217mg,0.5mmol)、B-5(60mg,0.1mmol)和适量
中间体B-7的合成
将化合物B-6(55mg,0.048mmol)置于反应瓶中,加入适量甲醇将其溶解,缓慢滴加甲醇钠的甲醇溶液调节pH为10,室温反应1h。反应完全后,向反应液中加入适量酸性阳离子树脂将pH调至中性。将反应液减压抽滤,滤液浓缩后经柱层析纯化得透明油状物(24mg,74%)。
中间体C-2的合成
取一两颈瓶,将化合物C-1(3.85g,24.96mmol)和3-溴丙炔(1.48g,12.48mmol)置于瓶中,加入适量四氢呋喃将其溶解。0℃下,将氢化钠分批加入到反应液中,缓慢升至室温过夜反应。加水淬灭反应,将溶液浓缩后经柱层析得C-2(1g,43%)。
中间体C-3的合成
将适量的化合物C-2(100mg,0.52mmol)、三乙胺(72μl,0.52mmol)溶于二氯甲烷,在冰浴下缓慢滴加三光气(54mg.0.18mmol)的二氯甲烷溶液,室温下反应4h。随后向反应液中加入Hobt(70mg,0.52mmol)和三乙胺(72μl,0.52mmol),室温反应过夜,待反应完全后加水淬灭反应。用二氯甲烷萃取水相,合并有机相经柱层析得黄色油状物(70mg,38%)。
中间体C-5的合成
将化合物C-4(300mg,0.59mmol)和DMAP(87mg,0.71mmol)溶于二氯甲烷,在冰浴下加入适量的C-3(208mg,0.59mmol),缓慢升至室温反应。反应完全后,加入蒸馏水淬灭反应,用二氯甲烷萃取水相,将有机相浓缩后经柱层析纯化得黄色油状物(200mg,46%)。
6.98(s,1H),6.61(s,1H),5.48(t,J=4.9Hz,1H),5.15–5.08(m,1H),4.40(td,J=6.6,4.1Hz,2H),4.27(d,J=7.2Hz,1H),4.18(d,J=2.4Hz,2H),4.10(dd,J=9.3,5.0Hz,1H),3.78(t,J=6.4Hz,2H),3.49(dd,J=8.8,6.7Hz,1H),2.97(t,J=6.6Hz,2H),2.92(t,J=6.4Hz,2H),2.85(t,J=6.4Hz,1H),2.70(s,3H),2.57(dd,J=13.9,9.9Hz,1H),2.51–2.43(m,2H),2.10(s,3H),1.99(q,J=6.2Hz,2H),1.82–1.76(m,2H),1.66(dd,J=13.1,7.0Hz,2H),1.57(q,J=8.3,6.2Hz,2H),1.46(dd,J=13.5,6.0Hz,1H),1.36(s,3H),1.27(s,3H),1.12(d,J=6.8Hz,3H),1.07(s,3H),0.97(d,J=6.7Hz,3H).
中间体D-3的合成
将化合物D-1(250mg,1mmol)和D-2(168mg,1mmol)溶于THF中,室温反应过夜。反应完全后,将溶液浓缩并经柱层析纯化得到白色固体(100mg,30%)。
将化合物D-3(58mg,0.19mmol)、C-4(80mg,0.157mmol)、DCC(48mg,0.23mmol)和DMAP(38mg,0.31mmol)溶于DCM中,室温反应过夜,将反应液减压抽滤,滤液浓缩后经柱层析纯化得目标产物(50mg,40%)。
V-a的合成
将化合物C-5(10mg,0.014mmol)、A-5(5mg,0.014mmol)、TBTA(cat.)溶于叔丁醇中,加入适量的硫酸铜(2.5mg,0.01mmol)水溶液并搅拌10min。随后滴加维C钠(11mg,0.056mmol)的水溶液,室温反应过夜,将反应液浓缩后经柱层析得V-a(10mg,62%)。
V-b由中间体B7与C5参照V-a合成,白色固体(19mg,38%)。
2.10(m,2H),2.08(d,J=1.4Hz,3H),2.06–1.95(m,2H),1.29–1.22(m,12H),
1.13(d,J=6.7Hz,3H),1.05–0.98(m,6H).
216.24,170.70,155.55,151.74,138.20,129.46,119.26,116.52,101.62,100.89,82.26,78.52,77.86,76.88,76.49,75.29,73.40,72.72,72.66,72.33,71.33,71.04,70.97,70.81,70.17,70.11,70.05,69.86,69.28,68.97,68.40,68.27,68.18,65.38,63.38,62.16,61.70,60.56,53.54,53.43,50.17,42.90,38.88,38.33,36.95,35.08,32.60,31.76,29.83,29.34,22.57,21.29,21.24,18.62,17.37,16.74,16.63,16.50,14.17,14.07,7.84.
VI-a中间体A5及D4参照V-a合成,透明油状物(20mg,57%)。
151.76,138.09,136.58,134.26,131.15,129.73,129.47,128.03,127.56,119.28,
116.52,100.40,78.37,76.89,72.61,71.28,70.99,70.81,70.28,70.17,70.09,70.02,69.01,68.42,66.37,62.18,61.72,53.52,50.09,42.89,38.90,35.07,34.76,34.37,33.67,32.62,31.87,29.86,29.43,29.34,28.93,26.72,22.52,22.34,21.38,21.30,18.57,17.35,16.83,16.70,14.33,14.07.
VI-b由中间体B7及D4参照V-a合成。白色固体(18mg,40%),mp:101–
103℃。
3.51(m,20H),3.43–3.38(m,3H),3.00(t,J=7.1Hz,2H),2.89(dd,J=8.7,3.8Hz,1H),2.70(s,3H),2.64(t,J=7.1Hz,2H),2.56(d,J=3.5Hz,1H),2.48(dd,J=15.2,10.3Hz,1H),2.23–2.10(m,2H),2.08(d,J=1.3Hz,3H),2.05–1.91(m,2H),1.61(d,J=11.0Hz,2H),1.45(d,J=11.1Hz,2H),1.28–1.23(m,12H),1.05(d,J=6.8Hz,3H),1.01(s,3H),0.89(d,J=6.7Hz,3H).
将上述制得的两亲性前药V-a为实施实例进行了纳米自组装性质、药物释放及抗肿瘤活性的评价。
将前药V-a溶于适量DMSO,在超声下逐滴滴加到适量水中,随后经透析去除有机溶剂DMSO即得到相应纳米前药颗粒。
本实施实例制备的纳米前药V-a纳米颗粒的动态光散射数据如图1所示,平均粒径为224nm。纳米前药V-a纳米颗粒的透射电镜照片如图2所示,纳米颗粒粒径与动态光散射数据基本一致。
纳米前药V-a的体外释放实验
以含有5%吐温的pH为7.4的磷酸盐缓冲溶于为释放介质,向释放介质中加入一定浓度的二硫苏糖醇(DTT),将上述纳米前药V-a加入到适量的上述介质后在37℃下设定的时间点取样,并通过高效液相(HPLC)色谱监测埃博霉素B的含量,以考察在不同还原条件下的释放情况。
结果如图3所示,前药V-a变现出了较好的还原条件响应性,在不含DTT的介质里,前药性质稳定,几乎不降解。当DTT的浓度为2mM,在6小时时便释放了75%的埃博霉素B。当DTT的浓度为10mM时,前药的释放速度明显加快,1小时内便释放了超过80%的埃博霉素B。
纳米前药V-a的细胞毒性
采用MTT法测定了纳米前药V-a对人肝癌细胞(HepG2)、人大细胞肺癌细胞(NCI-H460)、人大细胞肺癌细胞紫杉醇耐药株(NCI-H460/Taxol)和人乳腺癌细胞(MCF-7)的细胞毒性。将上述制得的两亲性纳米前药与埃博霉素B配置成不同浓度的细胞培养液,分别对上述细胞培养72小时后,用MTT法测定细胞活力。结果如图4所示,纳米前药表现出与埃博霉素相当的抗肿瘤活性。
纳米前药V-a的体内抗肿瘤活性
将MCF-7细胞悬液接种于裸鼠腋下。待肿瘤体积生长至50mm
结果如图5所示,与生理盐水组相比,埃博霉素B与纳米前药均表现出很好的抗肿瘤活性。体重曲线(图5B)和生存曲线(图5C)均表明前药组具有更好的安全性。肿瘤重量(图5D)和肿瘤照片(图5E)均表明前药很好的体内有效性。
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 一种酶响应型结肠靶向载药凝胶的制备方法
- 一种具有靶向光热治疗和可控释药的多重作用纳米材料的制备方法及应用
- 一种GSH响应型纳米钻石靶向药物及其制备方法和应用
- 一种GSH响应型纳米钻石靶向药物及其制备方法和应用