分步合成硅杂烃的方法
文献发布时间:2024-04-18 19:53:33
相关申请的交叉引用
本申请要求于2020年5月29日提交的欧洲专利申请号EP20177581.4的优先权,其全部内容通过引用并入本文。
技术领域
本发明涉及一种分步生产硅杂烃的方法,特别地涉及分步生产带有至多四个不同烃基的硅杂烃。更具体地,本发明涉及一种分步合成硅杂烃的方法,该方法能够选择性和有效地生产带有一个、两个、三个或四个不同烃基的硅杂烃,其中通过单独的氢化硅烷化步骤引入硅杂烃的至少两个取代基。
背景技术
硅杂烃构成一类通常表现出优异的粘度指数和热稳定性特性的功能流体。发现它们是在-54℃至315℃温度范围内可用的液压流体的出色候选物。它们具有优异的粘度-温度特性和热稳定性,并且预期它们与常规体系相比,在物理和化学性质上足够像烃,以允许设计具有最小再设计的液压系统。
硅杂烃被定义为被四个烃基SiR
因此,硅杂烃的性质,诸如蒸气压、沸点、粘度和热稳定性通过选择合适的取代基R来控制。例如,取代基R中碳链的长度影响沸点和粘度以及热稳定性质,该热稳定性性质也是硅杂烃分子的分子形状的函数。硅杂烃及其碳类似物的熔点比较的表明,硅杂烃具有较低的熔点。另外,硅杂烃的比质量和沸点通常高于相应的碳类似物的那些。
例如,四烷基硅烷(其中烷基中的两个或更多个具有八至三十个碳原子)已被证明是有用和有效的液压流体和润滑剂,尤其是在航空航天和太空交通工具中。
通常,涉及有机金属中间体诸如格氏试剂或有机锂和/或铝化合物的合成方法提供了最简单的合成程序:在US 4973724中,公开了通过使三烷基铝化合物与氯硅烷或烷基氯硅烷反应来制备化合物RSiR’
US 5177235公开了使由熔融金属和三烃铝获得的四烃基铝酸钠在T=100-130℃下与有机基三卤硅烷反应合成四烷基硅烷。
US 4650891公开了一种用于生产硅杂烃的催化方法,其中使卤素取代的硅烷与有机镁化合物在催化有效量的氰化物存在下反应。
在C.E.Snyder,L.J.Geschwender,C.Tamborski,G.J.Chen and D.R.Anderson(1982):Synthesis and Characterization of Silahydrocarbons–AClass of ThermallyStable Wide-Liquid-Range Functional Fluids,ASLE Transactions,9,299-308中提供了不同有机基取代的硅杂烃的一般合成方法以及它们的化学和物理性质的综述。
最近,氢化硅烷化化学反应有利于硅杂烃的构建。这些反应涉及甲硅烷基氢化物和不饱和有机基团之间的反应。这是合成商业硅基产品的一条基本路线,其中氢化硅烷化反应通常通过贵金属催化剂催化。
US 4578497公开了使用氧化的含Pt催化剂由单硅烷RSiH
在J.Am.Chem.Soc.119,(1997)906-917中,LaPointe等人报道了由叔硅烷和烯烃进行的钯催化的四烷基硅烷的氢化硅烷化合成。
在US 2014/0330036 A1中,公开了使用含铁或含钴的催化剂合成饱和和不饱和硅杂烃的方法。从伯硅烷RSiH
Tamborski等人在Ind.Eng.Chem.Prod.Res.Dev.1983,22,172-178中公开了带有不同取代基的硅杂烃的分步生产程序,其中烷基三氯硅烷随后与不同的烷基锂或烷基镁卤化物反应。
Onopchenko等人在J.Chem.Eng.Data,1988,33,64-66中公开了另一种生产带有一个短烷基和三个长烷基的四烷基硅烷的分步合成方法,该方法涉及两步氢化硅烷化程序。其中,将烷基二氯氢硅烷用α-烯烃氢化硅烷化,然后将二烷基二氯硅烷用LiAlH
然而,用于制备在一个硅中心处具有至多四个不同有机取代基的硅杂烃(R
对于各种各样不同烷基取代的硅杂烃的具体设计和一般合成途径而言,强烈需要一种新的合成方案,该方案涉及通过氢化硅烷化反应在硅中心处分步引入至多四个不同的基团。
迄今为止,所有文献报道都缺乏通过氢化硅烷化反应序列分别从伯硅烷RSiH
要解决的问题
本发明要解决的问题是提供一种经由双官能氢氯硅烷作为中间体分步生产硅杂烃、特别是带有至多四个不同有机基团R的硅杂烃的方法。特别地,本发明的一个目的是提供一种新的方法,该方法在反应的总产率、所获得的产物的纯度、转化的选择性、反应程序的便利性、后处理程序的便利性、试剂的易处理性、原子效率和方法的成本效率方面具有优于传统方法的改善性能。
根据本发明,该问题如下解决。
发明内容
本发明涉及生产通式(I)的硅杂烃的方法
SiR
其中
R
R
并且其中R
所述方法包括
a)至少一个生产通式(II)的双官能单硅烷中间体的步骤
SiR
其中R
并且R
其是通过
-在再分配催化剂的存在下和任选地在一种或多种溶剂的存在下使通式(III)的有机基全氯单硅烷与通式(IV)的有机基全氢单硅烷进行再分配反应(重分布反应,redistribution reaction)
SiR
其中R
并且R
SiR
其中R
并且R
或通过
-在再分配催化剂的存在下和任选地在一种或多种溶剂的存在下使通式(III)的有机基全氯单硅烷与原位形成的氢化产物进行再分配反应,所述原位形成的氢化产物是通过使通式(III)的单硅烷与有机金属氢化物供体或通式MH
在通式(III)中,R
并且R
在通式MH
所述有机金属氢化物供体选自二异丁基氢化铝、Me
或通过
-氯化反应,所述氯化反应包括在至少一种催化剂的存在下、任选地在一种或多种溶剂的存在下使通式(IV)的有机基全氢单硅烷与四氯硅烷(SiCl
SiR
其中R
并且R
或通过
-通式(IV)的有机基全氢单硅烷的选择性部分氯化反应
SiR
其中R
并且R
其是通过使所述化合物与HCl/醚试剂任选地在一种或多种其它溶剂的存在下反应,
和
b)至少一个以下步骤:使由步骤(a)获得的通式(II)的双官能单硅烷中间体或HSiCl
SiR
其中R
R
或以获得式R
c)通过步骤b)中获得的通式(V)的化合物的氢化反应生产通式(VI)的中间体的步骤
SiR
其中在通式(V)和(VI)中
R
R
并且R
或通过式R
和
d)使从步骤c)获得的通式(VI)或R
SiR
其中所述中间体优选为其中R
具体实施方式
在下文中,将详细描述本发明。
除非另有限制,否则根据本发明,残基R
R
R
它们可以在表示如以上所定义的基团的任何通式结构中使用。例如,这意味着在式(II)中,R
根据本发明,有机基团是在其碳原子处具有一个自由价的任何有机取代基团,无论其官能类型如何。
根据本发明,由所述方法获得的产物是通式(I)的硅杂烃:SiR
其中式(I)的R
当通式(II)、(III)、(IV)、(V)和(VI)的基团R
通式(I)中的基团R
通式(I)中的基团R
独立地构成R
根据本发明,通常,术语“未取代”意味着各自的烃基残基不包含除H和C之外的任何杂原子,既不作为取代基诸如卤素取代基、氨基或羟基,也不作为烃基的碳支架中包含的官能团(诸如醚基、酯基或酰胺基)的一部分。根据本发明的术语“取代的”通常定义了烃基可以含有除C和H之外的杂原子,和含除C和H之外的杂原子的官能团,诸如卤素取代基、羟基、氨基、酯基、酰胺基、醚基和杂环基。
在根据上述定义的取代残基的情况下,根据本发明,在测定残基的碳数时,考虑了含杂原子的官能团中所含的所有碳原子。例如,如果残基R
同样根据本发明,术语“脂族”是指所有非芳族的烃基取代基。
优选的是,当R
以相同的方式优选的是,当R
根据本发明,术语“烷基”通常包括直链、支链和环状烷基。未取代的烷基的优选实例是甲基、乙基、丙基、己基、辛基、异丁基和叔丁基。
残基R
残基R
被一个或多个甲氧基、乙氧基、丙氧基、具有2-10个、优选2-6个(CH
被一个或多个乙酰氧基或未取代的直链C3、C4、C16、C18、C19或C20乙酰氧基取代的直链烷基,
被一个或多个NH
被一个或多个氟基团、氯基团或溴基团取代的直链烷基,
被一个或多个SiMe
很明显,根据本发明,通常,取代的基团可以带有选自不同类型的官能团或杂原子取代基的几个取代基,因此取代的烷基也可以带有选自不同类型的官能团和杂原子取代基中的几个取代基。
根据本发明,术语“脂环族”通常包括所有类型的环状有机基取代基,不包括环状芳族取代基和环状杂环取代基。
根据本发明,术语“烷芳基”通常描述其中一个或多个氢原子被相同数量的烷基取代的芳基,所述烷基可以彼此相同或不同。烷芳基的优选实例是甲苯基、二甲苯基和均三甲苯基,特别是对甲苯基。
根据本发明,术语“芳烷基”通常描述其中一个或多个氢原子被相同数量的芳基取代的烷基,所述芳基可以彼此相同或不同。芳烷基的优选实例是苄基和苯乙基。
根据本发明,术语“芳基”通常被定义为任何已除去一个氢原子的芳香烃。芳基可以具有通过单键或其它基团连接的一个或多个可以稠合的芳环。芳基的优选实例是苯基、联苯基、萘基,苯基是最优选的。
根据本发明,术语“烯基”通常定义为含有一个或多个碳-碳双键的任何直链、支链或环状烯基,其中取代点可以在碳-碳键处或基团中的其它位置。具体地,关于根据本发明的式(I)至(VI)的残基R
烯基的优选实例是乙烯基、丙烯基、烯丙基、甲代烯丙基和亚乙基降莰烷基(ethylidenyl norbornane),其中乙烯基是最优选的。
还优选的是,残基R
在根据本发明的方法的步骤a)中,提供了通式SiR
在根据本发明的方法的步骤a)中,通式SiR
根据本发明,术语“再分配反应”描述了这种反应的反应混合物中所含的硅烷化合物的键合到硅原子的氢和氯取代基通过这些取代基的交换而再分配。交换可以特别通过
在实验室规模上,再分配反应在惰性条件(N
因此,术语“再分配催化剂”适用于增加上述再分配反应速率而自身不发生永久化学变化的任何化合物或化合物混合物。
优选地,也可以是两种或更多种单独催化剂的混合物的再分配催化剂选自由以下化合物组成的组:
-R
-膦类R
-胺类R
-N-杂环胺,优选甲基咪唑,诸如2-甲基咪唑、4-甲基咪唑和1-甲基咪唑,以及
-铵化合物,诸如R
该再分配反应可以净地(即,在不存在另外的溶剂的情况下)进行或在溶剂的存在下进行,该溶剂优选地是在反应条件下实际上是惰性的有机溶剂。
根据本发明,术语“有机溶剂”是指在室温下为液态并且适合作为在其中进行步骤a)的再分配反应的介质的任何有机化合物或其混合物。因此,有机溶剂优选在反应条件下对有机基氢硅烷、有机基氯硅烷、有机基氢氯硅烷和根据本发明的再分配催化剂是惰性的。此外,通式(III)、(IV)的起始材料和通式(II)的产物优选分别溶于有机溶剂或与有机溶剂完全混溶。
优选地,有机溶剂选自任选取代的、优选未取代的直链或环状脂族烃、芳香烃或醚化合物,但不限于此。
在本文中,术语“醚化合物”应指含有醚基-O-(一个或多个醚基是可能的)的任何有机化合物,特别是式R
通常,有机基团R可以选自例如任选取代的、优选未取代的烷基、芳基、烯基、炔基、烷芳基、芳烷基、芳烯基、芳炔基、环烷基、环烯基、环炔基、环芳烷基、环芳烯基和环芳炔基,优选选自烷基、烯基和芳基。
优选地,R
醚化合物可以相对于醚基-O-处的取代基对称或不对称。
根据本发明的术语“醚化合物”还包括直链醚化合物,其中可以包括一个以上的醚基,从而形成二醚化合物、三醚化合物、低聚醚化合物或聚醚化合物,其中R
此类化合物的优选实例是二甲氧基乙烷、乙二醇二醚(甘醇二甲醚(glyme)),特别是二甘醇二甲醚或四甘醇二甲醚,但不限于此。
根据本发明,术语“高沸点醚化合物”被定义为根据上述定义在1.013巴(标准大气压)下具有优选至少65℃,更优选至少85℃,甚至更优选至少100℃,最优选至少120℃的沸点的醚化合物。
高沸点醚在本发明中的应用是有利的,因为它有助于从含有溶剂和残留起始材料的反应混合物中分离所需的通式(I)的产物。通式(I)的产物通常具有比本文所定义的高沸点醚更低的沸点。
例如,所选的通式(I)的代表性产物的沸点在大气压下为35℃(Me
在根据本发明的方法的步骤a)中,通式SiR
虽然使用与上述通式(III)和(IV)化合物的再分配反应相同的再分配催化剂和溶剂,但是通式(III)化合物的再分配配对物(伙伴,partner)是原位形成的。在本文中,根据本发明的术语“原位形成”是指氢化类似物,即其中通式(III)化合物在硅原子处的一个至所有氯取代基被氢取代基代替的化合物,其是由通式(III)的化合物通过在其中进行这种反应步骤a)的反应容器中使这些化合物和如上所述的通式MH
根据本发明,术语金属氢化物试剂是指含有至少一个金属原子或金属离子的任何氢化物供体,其中规定根据本发明的金属氢化物试剂具有通式MH
根据本发明的术语“络合金属氢化物”是指其中阴离子包含金属原子或阳离子和氢化物阴离子二者的金属盐。典型地,络合金属氢化物含有多于一种类型的金属或准金属元素原子。由于既没有准金属的标准定义,也没有对这样适当分类的元素的完全赞同,在本发明的意义上,术语“准金属”包括元素硼、硅、锗、锡、砷、锑、碲、碳、铝、硒、钋和砹。络合金属氢化物的最优选实例是LiAlH
根据本发明的术语“有机金属氢化物供体”是指含有至少一个与至少一个有机残基键合的金属原子的任何化合物,其中进一步地至少一个金属原子或离子与氢原子共价地或以有机金属阳离子-氢化物离子对的形式键合。
或者,在根据本发明的方法的步骤a)中,通式SiR
SiR
其中R
并且R
根据本发明,该反应中的至少一种催化剂选自由以下组成的组:
-一种或多种式R
-一种或多种式R
-一种或多种式R
-一种或多种N-杂环胺,优选非N-取代的甲基咪唑,诸如2-甲基咪唑和4-甲基咪唑,
-一种或多种碱金属卤化物,诸如LiCl,和
-一种或多种碱土金属卤化物。
关于式R
由式R
根据本发明,上述氯化反应在至少一种溶剂的存在或不存在下进行,该溶剂优选为在上述反应条件下实际上是惰性的有机溶剂。
氯化反应优选在约-40℃至约250℃范围内,更优选约0℃至200℃范围内,最优选在约40℃至160℃范围内的温度下进行。
同样,根据本发明,氯化反应优选在约0.1至约10巴的压力下,更优选在1至10巴的压力下进行。
根据本发明,作为根据反应的方法的所有步骤,氯化反应优选在惰性条件下进行。
本文中的术语“惰性条件”是指排除湿气和氧气,特别是来自环境空气的湿气和氧气的存在的条件。优选地,惰性条件是通过在惰性气体气氛诸如氮气气氛或氩气气氛中进行根据本发明的反应来建立。
进一步地,在根据本发明的方法的步骤a)中,通式SiR
在该反应步骤中的任选其它溶剂优选是有机溶剂,根据本发明,该有机溶剂被定义为在反应条件下为液态并且适合作为在其中进行部分氯化步骤的介质的任何有机化合物。因此,有机溶剂优选对根据本发明在反应条件下应用的有机基氢硅烷和HCl/醚试剂以及所得的有机基氢氯硅烷是惰性的。通常,溶剂可以与针对上述再分配步骤定义的相同,其中醚化合物或含有至少一种醚化合物的溶剂的混合物是优选的。
实现部分氯化反应的HCl/醚试剂是通过醚化合物吸收或溶解HCl获得的,这可以在将HCl/醚试剂引入步骤a)的反应容器中之前进行,或者通过使气态HCl或HCl溶液与醚化合物或含有至少一种醚化合物的混合物在其中进行步骤a)的反应容器中原位接触而原位进行。
在本发明中,术语“醚化合物”应指含有醚基-O-的任何有机化合物,特别是式R
醚化合物可以相对于醚基-O-处的取代基对称或不对称,并且醚化合物选自直链醚化合物和环醚化合物。在本文中,直链醚化合物是含有如以上所定义的醚基R
根据本发明的环醚化合物是其中一个或多个醚基包含在由一系列原子形成的环中的化合物,例如四氢呋喃、四氢吡喃或1,4-二噁烷,其可以例如被烷基取代。
优选地,选自直链醚化合物和环醚化合物的醚化合物是脂族化合物。
还优选地,R
更优选地,R
具体HCl/醚试剂的使用主要取决于所形成产物的沸点。为了简化产物分离,对于高沸点产物,使用氯化氢/乙醚试剂是有利的,而对于通式(II)的低沸点有机基氯硅烷,高沸点醚,例如二甘醇二甲醚是优选的。
优选地,在至少一种醚化合物存在下用氯化氢的部分氯化反应在不存在含金属催化剂的情况下进行,更优选在不存在含第13族金属原子的路易斯酸化合物的情况下进行。
在根据本发明的至少一个步骤b)中,将如以上所定义的式(II)的有机基氢氯硅烷或HSiCl
根据本发明,金属氢化硅烷化催化剂可以是含有通过通式(II)的有机氯氢硅烷或通过HSiCl
在本发明中,金属铂、铱、钯、锇、铑、钌、铜、银、金和汞被认为是贵金属,因此根据本发明的步骤b)的氢化硅烷化催化剂可以基于金属,特别是基于上述贵金属。
优选地,根据本发明的方法的步骤b)的氢化硅烷化催化剂选自Mn、Fe、Co、Ni、Ir、Rh、Ru、Os、Pd和Pt化合物,如US 3,159,601;US 3,159,662;US 3,419,593;US 3,715,334;US 3,775,452;US 3,814,730;US 20130158281A1;WO 2013090548 A1;WO 2011006049 A1;US 20110009573 A1;WO 2011006044A2;US 20110009565 A1;US 9,387,468;US20180015449;US 20180201634;US 9,890,182和US 9,371,339中所教导的,所有均通过引用并入本发明。最优选的是铂化合物。
步骤b)的氢化硅烷化催化剂是有助于通过通式(II)的有机氯氢硅烷或通过HSiCl
在本发明的步骤b)中应用的典型的含铂催化剂组分是能够形成络合物的铂(0)、(II)或(IV)化合物的任何形式。优选的络合物是Pt-
铂络合物的特别有用的形式是Pt
如通过引用并入本文的例如US 3,419,593所公开的,特别优选环己烯-Pt、环辛二烯-Pt和四乙烯基四甲基-四环硅氧烷(Vinyl-D4)-Pt,例如Ashby催化剂,经验式为Pt[(C
还优选的是所谓的Lamoreaux催化剂,其是铂(II)络合物,由氯铂酸六水合物和辛基醇获得(如例如US 3,197,432或US 3,220,972中所述的)。进一步优选的是Pt(0)或Pt(II)催化剂,优选Ashby和Lamoreaux铂催化剂。
在本发明的组合物中使用的含铂催化剂组分的量不受严格限制,只要存在足够的量以在步骤b)的所需时间(B)内在期望温度下加速C-C-不饱和化合物与通式(II)的有机氯氢硅烷之间的氢化硅烷化,或通过HSiCl
氢化硅烷化步骤b)可以在热和光的辅助下进行。然后通过用光,特别是波长最大值在300和550nm之间的UV光照射来引发光固化。照射引发的氢化硅烷化优选在室温(25℃)下进行。
因此,氢化硅烷化催化剂也可以选自能够被光活化的催化剂组。这些光活化性催化剂优选含有至少一种选自Pt、Pd、Rh、Co、Ni、Ir或Ru的金属。能够被光活化的催化剂优选包含铂化合物。能够光活化的催化剂优选选自有机金属化合物,即包含含碳配体或其盐。优选地,光活化氢化硅烷化催化剂(C)具有金属碳键,包括σ和π键。还优选地,能够被光活化的催化剂(C)是具有至少一个金属碳σ键的有机金属络合物化合物,还更优选具有优选一个或多个σ键合的烷基和/或芳基、优选烷基的铂络合物化合物。σ键合的配体特别包括σ键合的有机基团,优选σ键合的C
能够被光活化的其它催化剂包括(η-二烯烃)-(σ-芳基)-铂络合物(参见例如US4,530,879)。
能够被光活化的催化剂可以这样使用或负载在载体上。
能够被光活化的催化剂的实例包括η-二烯烃-σ-芳基铂络合物,如US 4,530,879、EP 122008、EP 14630(对应于US 4510094和其中引用的现有技术文件)或US 2003/0199603中公开的;以及铂化合物,其反应性可以通过例如使用如US 4,640,939中公开的偶氮二羧酸酯,或二酮酸酯来控制。
此外,可以使用的能够被光活化的铂化合物是选自具有选自二酮的配体的组的那些(例如苯甲酰基丙酮或乙炔二羧酸酯)和嵌入可光降解有机树脂中的铂催化剂。在US 3,715,334或US 3,419,593、EP 1 672 031 A1和Lewis,Colborn,Grade,Bryant,Sumpter,andScott in Organometallics,1995,14,2202-2213(所有均通过引用在这里并入)中通过示例方式提及了其它Pt催化剂。
能够被光活化的催化剂也可以通过使用Pt
然而,这里可以使用的能够被光活化的催化剂不限于这些上述实例。
要在本发明的方法中使用的能够被光活化的最优选催化剂是(η
能够光活化的催化剂的量优选为1至500ppm,并且优选在与针对上述热活化性氢化硅烷化催化剂所定义的相同的较低范围内。
进行步骤b)的氢化硅烷化反应的化合物可以是在氢化硅烷化反应中被转化为针对R
通常,含有至少一个C-C双键或C-C三键的化合物可以选自各自具有2至30个碳原子的单不饱烯烃和或多不饱和烯烃、单不饱和炔烃或多不不饱和炔烃以及含有C-C双键和C-C三键两者的化合物。
在步骤b)的氢化硅烷化反应中应用的含有至少一个C-C双键或C-C三键的优选化合物是
-1-烯烃,优选直链C2-C16 1-烯烃,更优选1-丁烯、1-戊烯、1-己烯、1-庚烯、1-辛烯、1-壬烯、1-癸烯、1-十一烯和1-十二烯,
-1-炔烃,优选直链C2-C12 1-炔烃,更优选1-丁炔、1-戊炔、1-己炔、1-庚炔、1-辛炔、1-壬炔、1-癸炔、1-十一炔和1-十二炔,以及
-直链酯基取代的烯烃,优选C8-C24单不饱和烯基酯。
在根据本发明的至少一个步骤c)中,将如以上所定义的通式(V)或通式R
根据本发明,步骤c)的氢化反应是其中起始材料的硅原子处的所有氯取代基被氢取代基取代的任何类型的反应。
优选地,有机基氯硅烷的氢化是通过使所述化合物与金属氢化物、混合金属氢化物、有机金属氢化物供体反应或在氢化催化剂存在下与氢气反应来实现。
优选的金属氢化物是LiH、NaH、KH、CaH
在根据本发明的方法的步骤d)中,将通式SiR
其中,优选在步骤d)中应用如针对步骤b)所定义的金属催化剂,以提高通式(VI)的化合物和含有一个或多个C-C双键或C-C三键的化合物的氢化硅烷化反应的反应速率,并且含有一个或多个C-C双键和C-C三键化合物同样如针对步骤b)所定义的。
根据本发明,优选的是进行步骤d)的中间体是如以上所定义的三有机基氢硅烷SiR
步骤d)必然是生产硅杂烃的方法的最后步骤。
应注意,根据本发明,通常,残基R
同样应注意的是,根据本发明,残基R
在根据本发明的优选实施方案中,通式(I)的硅杂烃产物的硅中心处的四个有机基取代基R
通过本发明的方法,可以选择性方式制备带有至多四个不同取代基的硅杂烃。其中,该方法可以通过对通过使用SiHCl
在根据本发明的另一优选实施方案中,通式(I)的硅杂烃产物的硅中心处的四个有机基取代基R
根据本发明,当通过HSiCl
优选的是,在带有四个不同基团作为取代基的硅杂烃中,碳原子的总数在8至80的范围内,更优选在10至50的范围内、甚至更优选在12至40的范围内。
在根据本发明的另一优选实施方案中,通式(I)的硅杂烃产物的残基R
如以上所定义的通式(III)SiR
优选的是当通式(I)的硅杂烃产物中的R
还优选的是,当R
在根据本发明的又一优选实施方案中,通式(I)的硅杂烃产物的残基R
其中,优选的是R
在根据本发明的优选实施方案中,通式(I)的硅杂烃产物的取代基R
通过使含有两个或更多个C-C双键的化合物进行步骤b)和d)的氢化硅烷化反应,或通过使炔基化合物进行所述方法步骤来引入烯基取代基。特别地,当使1-炔基化合物进行氢化硅烷化步骤时获得1-烯基取代基。烯基取代基的存在可用于控制化合物物理性质,并允许硅杂烃的进一步官能化。
优选地,烯基取代基选自丁烯基、2-甲基丁烯基、-2-氯丁烯基、环己烯基、乙烯基、烯丙基、丙烯基、戊烯基、己烯基、辛烯基、壬烯基、癸烯基、十一烯基和十二烯基。乙烯基、烯丙基、1-丙烯基、2-丁烯基、1-戊烯基、1-己烯基、1-辛烯基、1-壬烯基和1-十二烯基取代基是特别优选的。
在根据本发明的另一优选实施方案中,通式(I)的硅杂烃产物的取代基R
通过卤素取代基官能化可用于更改和调整硅杂烃化合物的物理性质,例如通过应用完全或部分全氟化的烷基链。引入与根据本发明的方法的氢化硅烷化条件相容的氯和溴取代基也可用于更改化合物的物理性质,并且另外,氯或溴取代基的存在可用作硅杂烃进一步官能化的起点。
优选的是,残基R
还优选的是,残基R
在根据本发明的又一优选实施方案中,通式(I)的硅杂烃产物的取代基R
根据本发明的该实施方案,R
在根据本发明的甚至进一个优选实施方案中,通式(I)的硅杂烃产物的取代基R
其中,优选的是,R
在根据本发明的另一优选实施方案中,通式(I)的硅杂烃产物的硅中心处的所有四个有机基取代基R
当所有四个取代基R
优选的是取代基R
在根据本发明的又一优选实施方案中,通式(I)的硅杂烃产物选自由Me
在根据本发明的优选实施方案中,步骤a)中通式(II)的双官能单硅烷中间体是式SiR
其中R
通过使这样的中间体进行方法步骤b),获得式SiR
在根据本发明的还优选的实施方案中,步骤a)中通式(II)的双官能单硅烷中间体是式SiR
其中R
通过使这样的中间体进行下一步骤b),获得式SiR
在根据本发明的另一优选实施方案中,步骤a)中通式(II)的双官能单硅烷中间体是式SiR
优选地R
通过使这样的中间体进行下一步骤b),获得式SiR
在根据本发明的又一优选的实施方案中,步骤a)中通式(II)的双官能单硅烷中间体选自由MeSiHCl
上面列出的一组化合物是容易获得的,并且构成了合成带有至多四个不同取代基的硅杂烃的起点,因此允许设计具有用于各种应用的物理和化学性质的化合物。以上所列组的化合物的取代基R
在根据本发明的另一优选实施方案中,通式(III)的步骤a)的起始材料是通式R
优选地,通式(III)的起始材料通过HSiCl
容易获得的化合物HSiCl
在根据本发明的优选实施方案中,通式(IV)的步骤a)的起始材料是通式R
当根据该实施方案的通式(IV)的化合物与通式(III)的化合物进行再分配反应时是优选的,通式(III)的化合物是通式(IV)的化合物的全氯化类似物,即,R
在根据本发明的另一优选实施方案中,在步骤a)的反应中应用的通式(III)和(IV)的起始材料中的一种或两种是从HSiCl
在用于生产高纯多晶硅的西门子法中,将98%–99%的纯硅粉碎,并在反应器中与气态氯化氢在300–350℃下反应,以获得三氯硅烷。然后,将三氯硅烷在T>1000℃和氢气(H
HSiCl
在根据本发明的还优选的实施方案中,根据通式(III)的步骤a)的起始材料是MeSiCl
Me
因此,MeSiCl
优选的是,使起始材料MeSiCl
在根据本发明的另一优选实施方案中,根据通式(IV)的步骤a)的起始材料是MeSiH
特别优选的是,化合物MeSiH
在根据本发明的另一优选实施方案中,至少一种通式(II)的中间体通过如以上所定义的通式(III)的化合物和通式(IV)的化合物的再分配反应获得,其中再分配催化剂选自一种或多种选自由以下组成的组的化合物:
-鏻卤化物,优选鏻氯化物R
-膦类R
-胺类R
-N-杂环胺,优选甲基咪唑,诸如2-甲基咪唑、4-甲基咪唑和1-甲基咪唑,以及
-铵卤化物,优选式R
优选地,在再分配反应中应用的通式(III)的化合物是所应用的通式(IV)的化合物的氯化类似物。
在根据本发明的优选实施方案中,至少一个步骤a)在溶剂的存在下进行,其中溶剂选自由醚、烷烃或芳族溶剂组成的组,更优选地选自由THF、1,4-二噁烷、二甘醇二甲醚、四甘醇二甲醚、己烷和苯组成的组,最优选地该溶剂是THF。
特别地,对于步骤a)是再分配反应,THF、二甘醇二甲醚、1,4-二噁烷和四甘醇二甲醚是优选的溶剂;
对于步骤a)是具有氯硅烷的原位还原的再分配反应,THF、二甘醇二甲醚和四甘醇二甲醚是优选的溶剂;并且
对于步骤a)是使用醚/HCl的部分氯化,1,4-二噁烷、n-Bu
如果SiCl
在根据本发明的另一优选实施方案中,至少一个步骤a)中的反应温度在0℃至180℃,优选20℃至160℃,最优选60℃至120℃的范围内。
特别地,对于步骤a)是再分配反应,温度优选在50至160℃,更优选60至120℃的范围内;
对于步骤a)是具有氯硅烷的原位还原的再分配反应,温度优选在70至100℃的范围内;
对于步骤a)是使用醚/HCl的部分氯化,温度优选在0至80℃的范围内,更优选在20至60℃的范围内,并且对于步骤a)是使用SiCl
根据本发明,步骤a)中的反应温度是反应混合物的温度,即在进行反应的反应容器内测量的温度。
在根据本发明的另一个优选实施方案中,至少一个步骤a)中的再分配配对物选自MeSiCl
以上列出的配对表明,使这些特定的化合物对中的一个进行步骤a),而没有在反应之前或期间向反应混合物中加入通式(III)或(IV)的任何其它化合物。
在根据本发明的另一优选实施方案中,步骤a)中的至少一种通式(II)的中间体通过在再分配催化剂的存在下通式(III)的化合物和原位形成的氢化产物的再分配反应获得,所述原位形成的氢化产物通过使一种或多种通式(III)的单硅烷与通式MH
-R
-膦类R
-胺类R
-N-杂环胺,优选甲基咪唑,诸如2-甲基咪唑、4-甲基咪唑和1-甲基咪唑,以及
-铵化合物,诸如R
特别优选的是,再分配催化剂选自n-Bu
在根据本发明的另一优选实施方案中,涉及全氯化起始材料的原位还原的再分配反应中的溶剂选自醚溶剂,更优选THF、二甘醇二甲醚、1,4-二噁烷、三甘醇二甲醚、四甘醇二甲醚、DME(二甲氧基乙烷),最优选THF、1,4-二噁烷或二甘醇二甲醚。
在根据本发明的还优选的实施方案中,涉及全氯化起始材料的原位还原的再分配反应中的反应温度在0℃至180℃,优选20℃至160℃,最优选60℃至140℃的范围内。
在根据本发明的另一优选实施方案中,通式(III)的化合物选自由MeSiCl
在根据本发明的优选实施方案中,在通式(IV)的化合物的选择性部分氯化反应中通过在步骤a)中使所述化合物与HCl/醚试剂反应来获得至少一种通式(II)的中间体,其中HCl/醚试剂优选选自THF/HCl、二乙醚/HCl、二甘醇二甲醚/HCl、1,4-二噁烷/HCl、二丁醚/HCl,更优选选自二甘醇二甲醚/HCl、二乙醚/HCl、1,4-二噁烷/HCl、二丁醚-HCl,最优选选自二乙醚/HCl或二甘醇二甲醚/HCl。
优选地,根据本发明实施方案进行部分氯化的通式(IV)化合物选自SiR
在根据本发明的另一优选实施方案中,在至少一种催化剂存在下,在通式(IV)SiR
用SiCl
在根据本发明的另一优选实施方案中,在至少一个步骤a)中,在至少一种催化剂存在下与HCl/醚试剂或与SiCl
在根据本发明的又一优选实施方案中,在至少一种催化剂存在下与HCl/醚试剂或与SiCl
在根据本发明的另一优选实施方案中,使用Rh或Pt基催化剂,更优选使用固定在载体上的Pt催化剂,甚至更优选使用固定在二氧化硅上的Pt催化剂,最优选使用包含共价键合到二氧化硅载体的含金属的硅氧烷聚合物基质的固定在二氧化硅上的Pt催化剂,特别是包封在共价键合到二氧化硅载体的硅氧烷聚合物基质中的Pt纳米颗粒,进行至少一个金属催化的氢化硅烷化步骤(b)。
根据本发明优选的铂基催化剂公开于专利申请US 2015/0051357 A1中,其通过引用整体并入本文。特别地,其中在实施例2中公开的催化剂是根据本发明特别优选的。
通常,当在步骤b)中应用固定在载体上的Pt基催化剂时,优选的是当金属负载为载体材料的约0.1至约5重量%时。
当应用包含共价键合到二氧化硅载体的含金属的硅氧烷聚合物基质的固定在二氧化硅上的Pt催化剂时,优选的是当金属负载为载体材料的约0.1至约1重量%时。在这种情况下,还优选的是当含金属聚合物基质经由选自烷基二硅氮烷、含乙烯基硅氮烷或其组合的疏水官能团共价键合到载体材料时。
在根据本发明的又一优选实施方案中,进行步骤b)的通式(II)的双官能单硅烷中间体选自R
根据本发明的该实施方案,进一步优选的是通过氢化硅烷化反应步骤引入的残基R
根据本发明的该实施方案,还进一步优选的是通过氢化硅烷化步骤引入的残基R
在根据本发明的优选实施方案中,步骤b)的氢化硅烷化反应中的含有至少一个C-C双键或C-C三键的化合物选自由烯烃、环烯烃、多烯烃、炔烃、环炔烃、多炔烃组成的组,优选烯烃、环烯烃、炔烃、环炔烃,更优选烯烃、环烷烃、炔烃,甚至更优选烯烃,最优选单不饱和末端烯烃。
在根据本发明的另一优选实施方案中,至少一个步骤b)在0℃至180℃、优选20℃至140℃、最优选60℃至100℃范围内的温度下进行。
其中,优选的是当不使用另外的溶剂或溶剂选自THF、二甘醇二甲醚、1,4-二噁烷、苯或甲苯,优选选自THF、二甘醇二甲醚或1,4-二噁烷,更优选选自THF或1,4-二噁烷,最优选地该溶剂是THF。
在根据本发明的另一优选实施方案中,在步骤c)中,通式(V)的中间体通过与通式MH
根据本发明,LiH是用于还原步骤c)的最优选的金属氢化物试剂,因为它相对容易处理,在便利的反应条件下,即在低温下还原氯硅烷,并且所得氯化锂可以进行再循环过程以回收LiH。NaH也是优选的,因为其在氯硅烷的还原方面的低成本和令人满意的性能。
在根据本发明的又一优选实施方案中,步骤d)的氢化硅烷化反应的催化剂选自Rh或Pt基催化剂,更优选选自固定在载体上的Pt催化剂,甚至更优选选自固定在二氧化硅上的Pt催化剂,最优选选自包含共价键合到二氧化硅载体的含金属的硅氧烷聚合物基质的固定在二氧化硅上的Pt催化剂,特别是包封在共价键合到二氧化硅载体的硅氧烷聚合物基质中的Pt纳米颗粒。
根据本发明优选的铂基催化剂公开于专利申请US 2015/0051357 A1中,其通过引用整体并入本文。特别地,其中在实施例2中公开的催化剂是根据本发明特别优选的。
通常,当在步骤b)中应用固定在载体上的Pt基催化剂时,优选的是当金属负载为载体材料的约0.1至约5重量%时。
当应用包含共价键合到二氧化硅载体的含金属的硅氧烷聚合物基质的固定在二氧化硅上的Pt催化剂时,优选的是当金属负载为载体材料的约0.1至约1重量%时。在这种情况下,还优选的是当含金属聚合物基质经由选自烷基二硅氮烷、含乙烯基硅氮烷或其组合的疏水官能团共价键合到载体材料时。
在根据本发明的另一优选实施方案中,进行步骤d)的氢化硅烷化反应的含有一个或多个C-C双键或C-C三键的化合物选自由烯烃、环烯烃、多烯烃、炔烃、环炔烃、多炔烃组成的组,优选烯烃、环烯烃、炔烃、环炔烃,更优选烯烃、环烷烃、炔烃,甚至更优选烯烃,最优选单不饱和末端烯烃。
在根据本发明的另一优选实施方案中,步骤c)的氢化反应和步骤d)的氢化硅烷化反应在一步程序中进行。
实施例
通过以下实施例进一步说明本发明,但不限于此。
概述
在反应之前,根据从文献中已知的程序仔细干燥所用溶剂。通过标准程序,尤其是通过NMR光谱和GC/MS分析来分析和表征产物。
本文中称为固定的Pt催化剂“Y1EX2”的氢化硅烷化催化剂是根据专利US 9,993,812B2(对应于申请US 2015/0051357 A1)的实施例2中公开的程序制备的多相铂基催化剂。
本文中称为“B770011”的氢化硅烷化催化剂是名为3.6R210的商品,其含有在210型二氧化硅上的3.6%铂金属(500nm),如购自Johnson Matthey(JM)。
虽然双官能起始材料HSiCl
通过NMR光谱测量的摩尔比和加入的起始材料的量来估计所形成的产物的量。
通过NMR管实验确定了最佳反应条件,并将其转移到封闭玻璃安瓿或开放系统中的制备规模的反应中。示例性地描述了以制备规模由Me
双官能单硅烷与1-烯烃和1-炔烃的氢化硅烷化反应通常如下进行。将0.1mL的烯烃或炔烃(基于双官能单硅烷的量为1.1-3.5当量)与0.05-0.15mL的单硅烷(1.0当量)和10重量%(基于硅烷底物的量)的氢化硅烷化催化剂(Y1EX2,或Karstedt催化剂,或B770011)在NMR管中的0.2-0.3mL的作为溶剂的THF中混合。用液氮(约-196℃)冷却样品后,将管抽真空(约0.1毫巴)并密封。温热至室温后,记录起始混合物的NMR光谱,随后加热样品。反应过程由NMR光谱控制,测量的频率取决于反应时间和温度。通过对指定给该混合物中的特定产物的相关NMR信号进行积分来确定所形成的产物的摩尔比。在氢硅烷完全加在碳-碳双键上的情况下,转化率定义为100%;在仅消耗一半氢硅烷的情况下,氢硅烷的转化率相应地为50%。在一些情况下,当烯烃反应物被部分异构化和/或由于所施加的高反应温度而氢化(来自脱氢甲硅烷化的H
作为封闭系统中的氢化硅烷化反应的替代(其构成了使低沸点化合物反应的优选程序),如果具有相对高沸点的反应配对物进行反应,则反应也可以在开放系统(反应烧瓶、磁力搅拌器、回流冷凝器和滴液漏斗,在惰性气氛下,例如Ar或N
在某些情况下,产物没有被进一步纯化或分离(在THF或其它硅烷化合物的杂质中稀释),因此,产率基于所用起始材料的量,或者给出与起始材料相关的相应产物比例。
化合物的鉴定
通过
使用与ITQ 900MS质谱仪联用的Thermo Scientific Trace GC Ultra进行GC-MS分析。固定相(Macherey-Nagel PERMABOND硅烷)长度为50m,内径为0.32mm。注入1μL的分析物溶液,将其1/200转移到由氦气运载的流速为1,7mL/min的柱上。首先将柱的温度保持在50℃持续10分钟。然后以20℃/min的速度将温度升高至250℃,并在该温度下再保持25–160分钟(取决于空间需求或硅中心处的烷基取代基的长度)。离开柱后,用70eV电离物质,并在34–600m/z(每电荷质量)的范围内测量阳离子碎片。起始材料和所形成的反应产物的
表1:起始材料和反应产物的列表
/>
/>
1)
续表1:起始材料和反应产物的列表
/>
1)
表2显示了在随后描述的实施例中合成的所选硅杂烃的分子结构以及它们在合成中的前体和中间体。用星号(*)标记根据本发明的方法的目标化合物。
表2:所选硅杂烃(*)和它们的前体。
/>
/>
/>
步骤a):双官能单硅烷的合成
1)氢硅烷和氯硅烷的再分配反应以产生双官能单硅烷
/>
条目A1-A3/表3:目标反应:1Me2SiH2+1Me2SiCl2→2Me2SiHCl
从表3可以看出,再分配反应以与所反应的氢硅烷相关的优良产率(约85%)并且在温和条件(100℃/24h)下得到了Me
条目A4/表3:目标反应:Me
在80℃下2小时后,将Me
条目A5/表3:目标反应:1Me
条目A5表明,在80℃/16h下,再分配平衡强烈移动,以得到55%的单甲基硅烷MeSiH
条目A6和A7/表3:目标反应:1MeSiH
如条目A6和A7、表3以及下面的表3/A7
表3/A7
表3/A7
*
条目A8/表3:目标反应:HexSiH
在80℃下14小时后,以与使用的起始材料HexSiH
条目A9/表3:目标反应:1MeSiHeptH
在密封的NMR管中,使MeSiHeptH
条目A10
在120℃(17h)下,MeSiBuH
条目A11/表3:目标反应:1BuSiHexH
将124mg的n-Bu
条目A12/表4:目标反应:Ph
将Me
条目A13/表4:目标反应:Me
在Me
条目A14/表4:目标反应:PhMeSiCl
在类似于条目A12的再分配反应中,使二甘醇二甲醚Me
条目A15/表4:目标反应:ViMeSiCl
Me
放大到制备规模:
通过Me
通过在THF中用LiH还原Me
在配有滴液漏斗、回流冷凝器和磁力搅拌器的250ml三颈烧瓶中放置7.22g(0.88mol,97%)的在惰性氮气气氛下悬浮于100ml完全干燥的四氢呋喃(THF)中的氢化锂(LiH)。通过真空脱气并且用气态氮再填充以建立惰性条件而从氧气/空气小心地缩放THF/LiH悬浮液。通过滴液漏斗向剧烈搅拌的悬浮液中缓慢加入56.84g(53.6ml,0.44mol)二甲基二氯硅烷(Me
在Me
Me
Me
用氢化锂进行氯硅烷还原后,形成氯化锂并从溶液中沉淀。通过过滤分离LiCl并真空干燥。获得36.05g(LiH 96%转化为LiCl;100%转化后的理论产率:37.56g)的所形成的LiCl,其与形成的Me
经由在THF中Me
在玻璃安瓿中放置0.41g(1.39mmol)n-Bu
Me
Me
Me
为了分离Me
Me
Me
Me
蒸馏后残留93.95g残留物,其也通过
Me
Me
将产物馏出物和残留物合并,Me
总之,Me
2)一步氯硅烷还原和随后的再分配反应以产生双官能单硅烷
条目A16/表5:MeSiBuCl
将LiH(150mg,18.3mmol,1当量)和n-Bu
条目A17/表5:MeSiCl
将LiH(340mg,43mmol,1.9当量)和n-Bu
条目A18/表5:BuSiCl
将LiH(290mg,0.38当量,36.5mmol)和n-Bu
3)用Et
将MeSiBuH
4)氢单硅烷R
表6:氢硅烷R
a)
条目A20,表6,目标反应:OctSiH
使OctSiH
条目A21,表6,目标反应:HexSiH
使HexSiH
条目A22,表6,目标反应:MeHexSiH
使MeHexSiH
条目A23,表6,目标反应:OctHexSiH
使OctHexSiH
步骤b):双官能单硅烷HSiCl
1)HSiCl
条目B1/表7:目标反应:HSiCl
将192mg的氢化硅烷化催化剂(Y1EX2)置于配备有NMR管的安瓿中。加入10mL无水THF、0.8mL C
条目B2/表7:目标反应:HSiCl
使HSiCl
条目B3/表7:目标反应:HSiCl
将HSiCl
2)R
条目B4/表7:目标反应:MeSiHCl
在附带有NMR管的安瓿中混合MeSiHCl
条目B5/表7:目标反应:MeSiHCl
将Pt催化剂(260mg)置于安瓿中,并用70mL无水THF和63mL(0.5mol,1.1当量)1-己烯悬浮。用液氮冷冻混合物,随后加入MeSiHCl
条目B6/表7:目标反应:MeSiHCl
将120mg催化剂(Y1EX2)置于烧瓶中,并用30mL无水THF、7mL(1.0当量,64mmol)MeSiHCl
条目B7/表7:目标反应:BuSiHCl
在Schlenk烧瓶中,向与THF(表5,条目A18)混合并被少量BuSiCl
条目B8/表7:目标反应:HexSiHCl
将HexSiHCl
条目B9/表7:目标反应:OctSiHCl
将OctSiHCl
3)R
条目B10/表7:目标反应:Me
将Pt催化剂(Y1EX2,230mg)置于安瓿中,并用60mL(0.48mol,1.5当量)1-己烯和36mL(0.32mol,1.0当量)Me
条目B11/表7:目标反应:Me
将催化剂(Y1EX2,12mg)、Me
条目B12/表7:目标反应:MeSiBuHCl+1-丁烯→MeSiBu
将从条目A10
条目B13/表7:目标反应:MeSiBuHCl+1-丁烯→MeSiBu
将从条目A10
条目B14/表7:目标反应:MeHexSiHCl+1-庚烯→MeHexSiHeptCl
将Pt催化剂(100mg)悬浮在40mL(0.28mol,1.2当量)1-庚烯和41g(0.23摩尔,1.0当量)MeHexSiHCl中。将反应混合物加热至100℃持续19小时,使挥发性化合物在真空中冷凝出,并且分离出59.3g的所需产物(0.22mol,97%产率,δ
条目B15/表7:目标反应:MeSiHeptHCl+1-辛烯→MeSiHeptOctCl
将40mg催化剂(Y1EX2)置于烧瓶中,并加入MeSiHeptCl
条目B16/表7:目标反应:OctHexSiHCl+1-戊烯→OctHexSiPentCl
将OctHexSiHCl(85g,0.33mol,1.0当量)、150mL的无水二甘醇二甲醚和143mL(0.75mol,2.3当量)的1-戊烯添加到Schlenk烧瓶中的500mg催化剂(B770011)中。加热至回流(100℃)82小时后,经由真空蒸馏分离二甘醇二甲醚和烯烃。通过真空分馏分离出102.2g的产物(0.31mol,94%产率,沸点:135℃(10
条目B17/表7:目标反应:BuSiHexHCl+1-辛烯→BuSiHexOctCl
将与THF(15mL)混合的BuSiHexHCl(15.7mmol)和硅烷化合物BuSiHexH
4)HSiCl
根据条目B18-B32/表8的一般合成程序
将0.1mL(1当量)氢硅烷、催化剂(8-25mg)、0.3mL THF、0.2mL C
条目B22、B27和B32/表8的附加注释
在这三个实验中,通过GC/MS和
步骤c):通过氯硅烷的还原来合成氢硅烷
表9:通过用氢化锂还原氯硅烷来合成氢硅烷
根据条目C1-C13/表9的一般合成程序
将悬浮在无水THF中的LiH置于配备有磁力搅拌器、回流冷凝器和滴液漏斗的三颈烧瓶中。通过滴液漏斗加入相应的氯硅烷。在几分钟的短引发时间后或通过将反应混合物加热至60-80℃开始,氯硅烷的还原开始于LiCl沉淀。氢硅烷形成完成后(通过
条目C1/表9:目标反应:HexSiCl
将LiH(54g,5.5mol,6.4当量)悬浮于300mL无水THF中。然后加入HexSiCl
条目C2/表9:目标反应:OctSiCl
将LiH(13.8g,1.7mol,3.9当量)悬浮于300mL无水THF中。然后加入OctSiCl
条目C3/表9:目标反应:MeSiHeptCl
将LiH(375mg,3.4当量,47.2mmol)悬浮于10mL无水THF中。随后,在室温下经由滴液漏斗加入3mL(1.0当量,13.8mmol)的MeSiHeptCl
条目C4/表9:目标反应:MeSiBuCl
经由滴液漏斗将MeSiBuCl
条目C5/表9:目标反应:MeHexSiCl
将LiH(13.6g,1.7mol,6当量)悬浮于120mL无水THF中。然后加入MeHexSiCl2(60mL,290mmol),并将反应混合物加热至80℃持续1.5小时。NMR光谱分析证实MeHexSiCl
条目C6/表9:目标反应:OctHexSiCl2→OctHexSiH
将LiH(8.6g,1.1mol,4.2当量)悬浮于150mL无水THF中。加入OctHexSiCl
条目C7/表9:目标反应:BuSiHexCl
将LiH(320mg,40mmol,5.7当量)悬浮于10mL无水THF中。然后加入BuSiHexCl
条目C8/表9:目标反应:MeSiBu
经由滴液漏斗将MeSiBu
条目C9/表9:目标反应:MeSiHeptOctCl→MeSiHeptOctH
将LiH(500mg,62.9mmol,11.8当量)悬浮于10mL无水THF中,随后加入MeSiHeptOctCl(1.55g,5.3mmol,1.0当量)。将反应混合物加热至100℃持续9小时,并在室温下搅拌过夜。GC-MS分析证明MeSiHeptOctCl100%转化为MeSiHeptOctH。通过过滤将该氢硅烷(δ
条目C10/表9:目标反应:BuSiHexOctCl→BuSiHexOctH
将LiH(220mg,28.0mmol,5.8当量)悬浮于5mL无水THF中。随后,加入2.1g的由BuSiHexOctCl(1.5g,4.8mmol)和BuSiHexCl
条目C11/表9:目标反应:MeHexSiHeptCl→MeHexSiHeptH
将LiH(5.1g,0.62mol,3.1当量)悬浮于70mL无水THF中。在70℃下将MeHexSiHeptCl(53g,0.20mol)滴加到剧烈搅拌的悬浮液中。将反应混合物加热至70℃持续15小时,然后通过过滤将液相与LiCl分离。蒸馏出THF,并分离出90%产率的MeHexSiHeptH(41.3g,0.18mol,δ
条目C12/表9:目标反应:Me
将LiH(5.0g,0.63mol,2.0当量)悬浮于60mL无水THF中。然后在70℃下将Me
条目C13/表9:目标反应:OctHexSiPentCl→OctHexSiPentH
将LiH(10.7g,1.3mol,4.6当量)悬浮于200mL无水THF中。向悬浮液中加入OctHexSiPentCl(102g,0.31mmol,1.0当量),加热至90℃持续16小时,并另外在120℃下加热18小时。反应混合物的GC-MS分析证明所有氯取代基对氢化取代基的100%转化率。通过过滤将液相与LiCl分离,并通过分馏分离出76g的OctHexSiPentH(0.26mol,86%产率,沸点:120℃,10
步骤d):通过三烷基氢硅烷的氢化硅烷化反应来合成四有机基硅烷
a)
根据条目D1-D10/表10的一般合成程序
将0.1mL氢硅烷溶液(根据步骤c)条目C8和C9)、催化剂(8-12mg)、0.3mL THF、0.2mL C
条目D1/表10:目标反应:MeSiHeptOctH+1-丁烯→MeSiHeptOctBu
反应时间80℃(22h,转化率:50%);100℃(10h,Si-H完全转化)(δ
条目D2/表10:目标反应:MeSiHeptOctH+1-己烯→MeSiHexHeptOct
反应时间80℃(22h,转化率:50%);100℃(10h,Si-H完全转化)(δ
条目D3/表10:目标反应:MeSiHeptOctH+1-庚烯→MeSiHept
反应时间80℃(22h,转化率:50%);100℃(10h,Si-H完全转化)(δ
条目D4/表10:目标反应:MeSiHeptOctH+1-癸烯→MeSiHeptOctDec
反应时间80℃(22h,转化率:50%);100℃(10h,Si-H完全转化)(δ
条目D5/表10:目标反应:MeSiHeptOctH+1-十六烯→MeSiHeptOctHexdec
反应时间80℃(22h,转化率:60%);100℃(10h,Si-H完全转化)(δ
条目D6/表10:目标反应:BuSiHexOctH+1-丁烯→Bu
反应时间:80℃(16h,转化率:16%);100℃(32h,转化率:31%);120℃(31.5h,转化率:80%);140℃(10h,Si-H完全转化,100%);(δ
条目D7/表10:目标反应:BuSiHexOctH+1-己烯→BuSiHex
反应时间:80℃(16h,转化率:12%);100℃(32h,转化率:29%);120℃(31.5h,转化率:75%);140℃(10h,转化率:80%,没有烯烃残留);(δ
条目D8/表10:目标反应:BuSiHexOctH+1-庚烯→BuSiHexOctHept
反应时间:80℃(16h,转化率:14%);100℃(32h,转化率:54%);120℃(31.5h,转化率:77%);140℃(10h,转化率:80%,没有烯烃残留);(δ
条目D9/表10:目标反应:BuSiHexOctH+1-癸烯→BuSiHeptOctDec
反应时间:80℃(16h,转化率:3%);100℃(32h,转化率:11%);120℃(31.5h,转化率:47%);140℃(10h,转化率:50%,没有烯烃残留);(δ
条目D10/表10:目标反应:BuSiHexOctH+1-十六烯→BuSiHeptOctHexdec
反应时间:80℃(16h,转化率:2%);100℃(32h,转化率:7%);120℃(31.5h,转化率:40%);140℃(10h,转化率:44%,没有烯烃残留);(δ
条目D11/表10:目标反应:MeHexSiHeptH+1-戊烯→MeHexSiHeptPent
将Pt催化剂(40mg)置于安瓿中,用33mL(0.3mol,3.6当量)的1-戊烯、17.7g(0.08mol,1.0当量)MeHexSiHeptH和5mL无水nBu
条目D12/表10:目标反应:MeHexSiHeptH+1-壬烯→MeHexSiHeptNon
将Pt催化剂(80mg)置于安瓿中,并用3g(0.02mol,1.2当量)的1-壬烯、4.4g(0.02mol,1.0当量)的MeHexSiHeptH和5mL无水nBu
条目D13-D15/表10的一般程序:目标反应:
Me
将Pt催化剂(8-10mg)置于安瓿中,并用Me
条目D16/表10:目标反应:OctHexSiPentH+1-庚烯→OctHexSiPentHept
将OctHexSiPentH(8.2g,0.027mol,1.0当量)、20mL的无水二甘醇二甲醚和7.8mL(0.068mol,2.5当量)的1-庚烯添加到Schlenk烧瓶中的200mg(2.5重量%)催化剂(B770011)中。加热至回流(100℃)60小时后,反应混合物的GC-MS分析证明所需产物以45%形成。为了将氢硅烷完全转化为相应的四烷基硅烷,将反应混合物转移到安瓿中,并与额外当量的1-庚烯(0.027mol)和0.5mL Karstedt催化剂混合。将反应混合物冷却至-196℃,在真空下密封安瓿,并将其置于150℃下的干燥箱中60小时。然后打开安瓿,蒸馏出所有挥发物,并通过真空分馏分离出4.0g的OctHexSiPentHept(0.01mol,37%产率,沸点:140℃(10
条目D17/表10:目标反应:OctHexSiPentH+1-辛炔→OctHexSiPentOctenyl
将OctHexSiPentH(8.2g,0.027mol,1.0当量)、20mL的无水二甘醇二甲醚和6.1mL(0.041mol,1.5当量)的1-辛炔添加到Schlenk烧瓶中的200mg(2.5重量%)催化剂(B770011)中。加热至回流(100℃)60小时后,反应混合物的GC-MS分析证明氢硅烷被定量消耗。冷凝出挥发性组分,并在真空下蒸馏残留物。将OctHexSiPentOctenyl(10.8g)分离为由1-烯烃取代的硅烷(9.2g,89%)和2-烯烃取代的硅烷(1.6g,11%)组成的混合物;通过样品的相应GC和
OctHexSiPent(1-辛烯基):δ
OctHexSiPent(2-辛烯基):δ
条目D18/表10:目标反应:OctHexSiPentH+1-癸烯→OctHexSiPentDec
将OctHexSiPentH(8.0g,0.027mol,1.0当量)、20mL的无水二甘醇二甲醚和12.7mL(0.068mol,2.5当量)的1-癸烯添加到Schlenk烧瓶中的200mg(2.5重量%)催化剂(B770011)中。加热至回流(100℃)60小时后,反应混合物的
条目D19/表10:目标反应:OctHexSiPentH+1-十六烯→OctHexSiPentHexdec
将OctHexSiPentH(8.0g,0.027mol,1.0当量)、20mL的无水二甘醇二甲醚和9.4mL(0.068mol,2.5当量)的1-十六烯添加到Schlenk烧瓶中的200mg(2.5重量%)催化剂(B770011)中。加热至回流(100℃)60小时后,反应混合物的
条目D20/表8:目标反应:MeSiBu
将MeSiBu
条目D21/表10:目标反应:MeSiBu
将50mg催化剂(Y1EX2)置于chlenk烧瓶中,并用3mL无水THF、0.5mL(1.0当量,2.4mmol)的MeSiBu
所选实施例:分步合成具有四个不同有机取代基(R
1)从MeSiHCl
a)MeSiHCl
将Pt催化剂(Y1EX2,260mg)置于安瓿中,并用70mL无水THF和63mL(0.5mol,1.1当量)1-己烯悬浮。用液氮冷冻混合物,随后加入MeSiHCl
1
29
13
b)MeHexSiCl
将LiH(13.6g,1.7mol,6当量)悬浮于120mL无水THF中。然后加入MeHexSiCl
1
29
13
c)MeHexSiH
使MeHexSiH
1
29
13
d)
将Pt催化剂(Y1EX2,100mg)悬浮在40mL(0.28mol,1.2当量)1-庚烯和41g(0.23摩尔,1.0当量)MeHexSiHCl中。将反应混合物加热至100℃持续19小时,使挥发性化合物在真空中冷凝出,并且获得59.3g的所需产物(0.22mol,97%产率,R
1
29
13
e)
将LiH(5.1g,0.62mol,3.1当量)悬浮于70mL无水THF中。在70℃下将MeHexSiHeptCl(53g,0.20mol)滴加到剧烈搅拌的悬浮液中。将反应混合物加热至70℃持续15小时,然后通过过滤将液相与LiCl分离。蒸馏出THF,并分离出90%产率的MeHexSiHeptH(41.3g,0.18mol,R
1
29
13
f)MeHexSiHeptH+1-戊烯→MeHexSiHeptPent
将Pt催化剂(Y1EX2,40mg)置于安瓿中,用33mL(0.3mol,3.6当量)的1-戊烯、17.7g(0.08mol,1.0当量)和5mL无水nBu
1
29
13
g)MeHexSiHeptH+1-壬烯→MeHexSiHeptNon
将Pt催化剂(Y1EX2,80mg)置于安瓿中,并用3g(0.02mol,1.2当量)的1-壬烯、4.4g(0.02mol,1.0当量)的MeHexSiHeptH和5mL无水nBu
1
29
13
2)从HSiCl
a)HSiCl
将HSiCl
1
29
13
b)OctSiCl
将LiH(13.8g,1.7mol,3.9当量)悬浮于300mL无水THF中。然后加入OctSiCl
1
29
13
c)OctSiH
使OctSiH
1
29
13
d)OctSiHCl
将OctSiHCl
1
29
13
e)OctHexSiCl
将LiH(8.6g,1.1mol,4.2当量)悬浮于150mL无水THF中。加入OctHexSiCl
1
29
13
f)OctHexSiH
使OctHexSiH
1
29
13
g)OctHexSiHCl+1-戊烯→OctHexSiPentCl
将OctHexSiHCl(85g,0.33mol,1.0当量)、150mL的无水二甘醇二甲醚和143mL(0.75mol,2.3当量)的1-戊烯添加到Schlenk烧瓶中的500mg催化剂(B770011)中。加热至回流(100℃)82小时后,经由真空蒸馏分离二甘醇二甲醚和烯烃。通过真空分馏分离出102.2g的产物(0.31mol,94%产率,沸点:135℃(10
1
29
13
h)OctHexSiPentCl+LiH→OctHexSiPentH
将LiH(10.7g,1.3mol,4.6当量)悬浮于200mL无水THF中。向悬浮液中加入OctHexSiPentCl(102g,0.31mmol,1.0当量),加热至90℃持续16小时,并另外在120℃下加热18小时。反应混合物的GC-MS分析证明所有氯取代基对氢化取代基的100%转化率。通过过滤将液相与LiCl分离,并通过分馏分离出76g的OctHexSiPentH(0.26mol,86%产率,沸点:120℃,10
1
29
13
i
将OctHexSiPentH(8.2g,0.027mol,1.0当量)、20mL的无水二甘醇二甲醚和7.8mL(0.068mol,2.5当量)的1-庚烯添加到Schlenk烧瓶中的200mg(2.5重量%)催化剂(B770011)中。加热至回流(100℃)60小时后,反应混合物的GC-MS分析证明所需产物以45%形成。为了将氢硅烷完全转化为相应的四烷基硅烷,将反应混合物转移到安瓿中,并与额外当量的1-庚烯(0.027mol)和0.5mL Karstedt催化剂混合。将反应混合物冷却至-196℃,在真空下密封安瓿,并将其置于150℃下的干燥箱中60小时。然后打开安瓿,蒸馏出所有挥发物,并通过真空分馏分离出4.0g的OctHexSiPentHept(0.01mol,37%产率,沸点:140℃(10
1
29
13
j
将OctHexSiPentH(8.0g,0.027mol,1.0当量)、20mL的无水二甘醇二甲醚和12.7mL(0.068mol,2.5当量)的1-癸烯添加到Schlenk烧瓶中的200mg(2.5重量%)催化剂(B770011)中。加热至回流(100℃)60小时后,反应混合物的
1
29
13
k)OctHexSiPentH+1-十六烯→OctHexSiPentHexdec
将OctHexSiPentH(8.0g,0.027mol,1.0当量)、20mL的无水二甘醇二甲醚和9.4mL(0.068mol,2.5当量)的1-十六烯添加到Schlenk烧瓶中的200mg(2.5重量%)催化剂(B770011)中。加热至回流(100℃)60小时后,反应混合物的
Si=2.8ppm,R
1
29
13
本发明的优选实施方案
在下文中,示出了本发明的优选实施方案。
实施方案1:
生产通式(I)的硅杂烃的方法
SiR
其中
R
R
并且其中R
所述方法包括
a)至少一个生产通式(II)的双官能单硅烷中间体的步骤
SiR
其中R
并且R
其是通过
-在再分配催化剂的存在下和任选地在一种或多种溶剂的存在下使通式(III)的有机基全氯单硅烷与通式(IV)的有机基全氢单硅烷进行再分配反应
SiR
其中R
并且R
SiR
其中R
并且R
或通过
-在再分配催化剂的存在下和任选地在一种或多种溶剂的存在下使通式(III)的有机基全氯单硅烷与原位形成的氢化产物进行再分配反应,所述原位形成的氢化产物是通过使通式(III)的单硅烷与有机金属氢化物供体或通式MH
在通式(III)中,R
并且R
在通式MH
所述有机金属氢化物供体选自二异丁基氢化铝、Me
或通过
-氯化反应,所述氯化反应包括在至少一种催化剂的存在下、任选地在一种或多种溶剂的存在下使通式(IV)的有机基全氢单硅烷与四氯硅烷(SiCl
SiR
其中R
并且R
或通过
-通式(IV)的有机基全氢单硅烷的选择性部分氯化反应
SiR
其中R
并且R
其是通过使所述化合物与HCl/醚试剂任选地在一种或多种其它溶剂的存在下反应,
和
b)至少一个以下步骤:使由步骤(a)获得的通式(II)的双官能单硅烷中间体或HSiCl
SiR
其中R
R
或以获得式R
和
c)通过步骤b)中获得的通式(V)的化合物的氢化反应生产通式(VI)的中间体的步骤
SiR
其中在通式(V)和(VI)中
R
R
并且R
或通过式R
和
d)使从步骤c)获得的通式(VI)或R
SiR
其中所述中间体优选为其中R
实施方案2
根据实施方案1的方法,其中通式(I)的硅杂烃产物的硅中心处的四个有机基取代基R
实施方案3
根据实施方案1和2的方法,其中通式(I)的硅杂烃产物的硅中心处的四个有机基取代基R
实施方案4
根据实施方案1至3的方法,其中通式(I)的硅杂烃产物的R
实施方案5
根据前述实施方案中任一个的方法,其中通式(I)的硅杂烃产物的R
实施方案6
根据前述实施方案中任一个的方法,其中通式(I)的硅杂烃产物的取代基R
实施方案7
根据前述实施方案中任一个的方法,其中通式(I)的硅杂烃产物的取代基R
实施方案8
根据前述实施方案中任一个的方法,其中通式(I)的硅杂烃产物的取代基R
实施方案9
根据前述实施方案中任一个的方法,其中通式(I)的硅杂烃产物的取代基R
实施方案10
根据实施方案1至5或7至9的方法,其中通式(I)的硅杂烃产物的硅中心处的所有四个有机基取代基R
实施方案11
根据前述实施方案中任一个的方法,其中通式(I)的硅杂烃产物选自由Me
实施方案12
根据前述实施方案中任一个的方法,其中步骤a)中通式(II)的双官能单硅烷中间体是下式的化合物,
SiR
其中R
优选地R
实施方案13
根据前述实施方案中任一个的方法,其中步骤a)中通式(II)的双官能单硅烷中间体是下式的化合物,
SiR
其中R
优选地R
实施方案14
根据前述实施方案中任一个的方法,其中步骤a)中通式(II)的双官能单硅烷中间体是下式的化合物
SiR
其中R
实施方案15
根据前述实施方案中任一个的方法,其中步骤a)中通式(II)的双官能单硅烷中间体选自由MeSiHCl
实施方案16
根据前述实施方案中任一个的方法,其中通式(III)的步骤a)的起始材料是通式R
实施方案17
根据前述实施方案中任一个的方法,其中通式(IV)的步骤a)的起始材料是通式R
实施方案18
根据前述实施方案16和17的方法,其中在步骤a)的反应中应用的通式(III)和(IV)的起始材料中的一种或两种是从HSiCl
实施方案19
根据前述实施方案1至18的方法,其中根据通式(III)的步骤a)的起始材料是MeSiCl
实施方案20
根据前述实施方案中任一个的方法,其中根据通式(IV)的步骤a)的起始材料是MeSiH
实施方案21
根据前述实施方案中任一个的方法,其中至少一种通式(II)的中间体是通过如以上所定义的通式(III)的化合物和通式(IV)的化合物的再分配反应获得的,其中所述再分配催化剂选自一种或多种选自由以下组成的组的化合物:
-鏻卤化物,优选鏻氯化物R
-膦类R
-胺类R
-N-杂环胺,优选甲基咪唑,诸如2-甲基咪唑、4-甲基咪唑和1-甲基咪唑,以及
-铵卤化物,优选式R
实施方案22
根据前述实施方案中任一个的方法,其中至少一个步骤a)在溶剂的存在下进行,其中溶剂选自由醚、烷烃或芳族溶剂组成的组,更优选地选自由THF、1,4-二噁烷、二甘醇二甲醚、四甘醇二甲醚、己烷和苯组成的组,最优选地该溶剂是THF。
实施方案23
根据前述实施方案中任一个的方法,其中至少一个步骤a)中的反应温度在0℃至180℃,优选20℃至160℃,最优选60℃至120℃的范围内。
实施方案24
根据前述实施方案中任一个的方法,其中至少一个步骤a)中的再分配配对物选自配对MeSiCl
实施方案25
根据前述实施方案中任一个的方法,其中步骤a)中的至少一种通式(II)的中间体是通过在再分配催化剂的存在下通式(III)的化合物和原位形成的氢化产物的再分配反应获得的,所述原位形成的氢化产物通过使一种或多种通式(III)的单硅烷与有机金属氢化物供体或通式MH
-R
-膦类R
-胺类R
-N-杂环胺,优选甲基咪唑,更优选2-甲基咪唑、4-甲基咪唑和1-甲基咪唑,以及
-铵化合物,诸如R
实施方案26
根据前述实施方案中任一个的方法,其中涉及全氯化起始材料的原位还原的再分配反应中的溶剂选自醚溶剂,更优选THF、二甘醇二甲醚、1,4-二噁烷、三甘醇二甲醚、四甘醇二甲醚、DME,最优选THF、1,4-二噁烷、二甘醇二甲醚,并且反应温度在0℃至180℃、优选20℃至160℃、且最优选60℃至120℃的范围内。
实施方案27
根据前述实施方案中任一个的方法,其中通式(III)的化合物选自由MeSiCl
实施方案28
根据前述实施方案中任一个的方法,其中在步骤a)中,至少一种通式(II)的中间体是在通式(IV)的化合物的通过使该化合物与HCl/醚试剂反应的选择性部分氯化反应中获得的,其中所述HCl/醚试剂优选选自THF/HCl、二乙醚/HCl、二甘醇二甲醚/HCl、1,4-二噁烷/HCl、二丁醚/HCl,更优选选自二甘醇二甲醚/HCl、二乙醚/HCl、1,4-二噁烷/HCl、二丁醚/HCl,并且最优选选自二乙醚/HCl或二甘醇二甲醚/HCl。
实施方案29
根据前述实施方案中任一个的方法,其中至少一种通式(II)的中间体是在通式(IV)SiR
实施方案30
根据前述实施方案中任一个的方法,其中在至少一个步骤a)中,在至少一种催化剂存在下与HCl/醚试剂或与SiCl
实施方案31
根据前述实施方案中任一个的方法,其中在至少一种催化剂存在下与HCl/醚试剂或与SiCl
实施方案32
根据前述实施方案中任一个的方法,其中使用Rh或Pt基催化剂,更优选使用固定在载体上的Pt催化剂,甚至更优选使用固定在二氧化硅上的Pt催化剂,最优选使用包含共价键合到二氧化硅载体的含金属的硅氧烷聚合物基质的固定在二氧化硅上的Pt催化剂,特别是包封在共价键合到二氧化硅载体的硅氧烷聚合物基质中的Pt纳米颗粒,进行至少一个金属催化的氢化硅烷化步骤(b)。
实施方案33
根据前述实施方案中任一个的方法,其中进行步骤b)的通式(II)的双官能单硅烷中间体选自R
实施方案34
根据前述实施方案中任一个的方法,其中步骤b)的氢化硅烷化反应中的含有至少一个C-C双键或C-C三键的化合物选自由烯烃、环烯烃、多烯烃、炔烃、环炔烃、多炔烃组成的组,优选烯烃、环烯烃、炔烃、环炔烃,更优选烯烃、环烷烃、炔烃,甚至更优选烯烃,最优选单不饱和末端烯烃。
实施方案35
根据前述实施方案中任一个的方法,其中至少一个步骤b)在0℃至180℃、优选20℃至140℃、最优选60℃至100℃范围内的温度下进行,并且其中进一步优选不使用另外的溶剂或溶剂选自THF、二甘醇二甲醚、1,4-二噁烷、苯或甲苯,优选选自THF、二甘醇二甲醚或1,4-二噁烷,更优选选自THF或1,4-二噁烷,最优选地该溶剂是THF。
实施方案36
根据前述实施方案中任一个的方法,其中在步骤c)中,通式(V)的中间体是通过与有机金属氢化物供体试剂或其中M和x如以上所定义的通式MH
实施方案37
根据前述实施方案中任一个的方法,其中步骤d)的氢化硅烷化反应的催化剂选自Rh或Pt基催化剂,更优选选自固定在载体上的Pt催化剂,甚至更优选选自固定在二氧化硅上的Pt催化剂,最优选选自包含共价键合到二氧化硅载体的含金属的硅氧烷聚合物基质的固定在二氧化硅上的Pt催化剂,特别是包封在共价键合到二氧化硅载体的硅氧烷聚合物基质中的Pt纳米颗粒。
实施方案38
根据前述实施方案中任一个的方法,其中进行步骤d)的氢化硅烷化反应的含有一个或多个C-C双键或C-C三键的化合物选自由烯烃、环烯烃、多烯烃、炔烃、环炔烃、多炔烃组成的组,优选烯烃、环烯烃、炔烃、环炔烃,更优选烯烃、环烷烃、炔烃,甚至更优选烯烃,最优选单不饱和末端烯烃。
实施方案39
根据前述实施方案中任一个的方法,其中步骤c)的氢化反应和步骤d)的氢化硅烷化反应在一步程序中进行。
附图说明
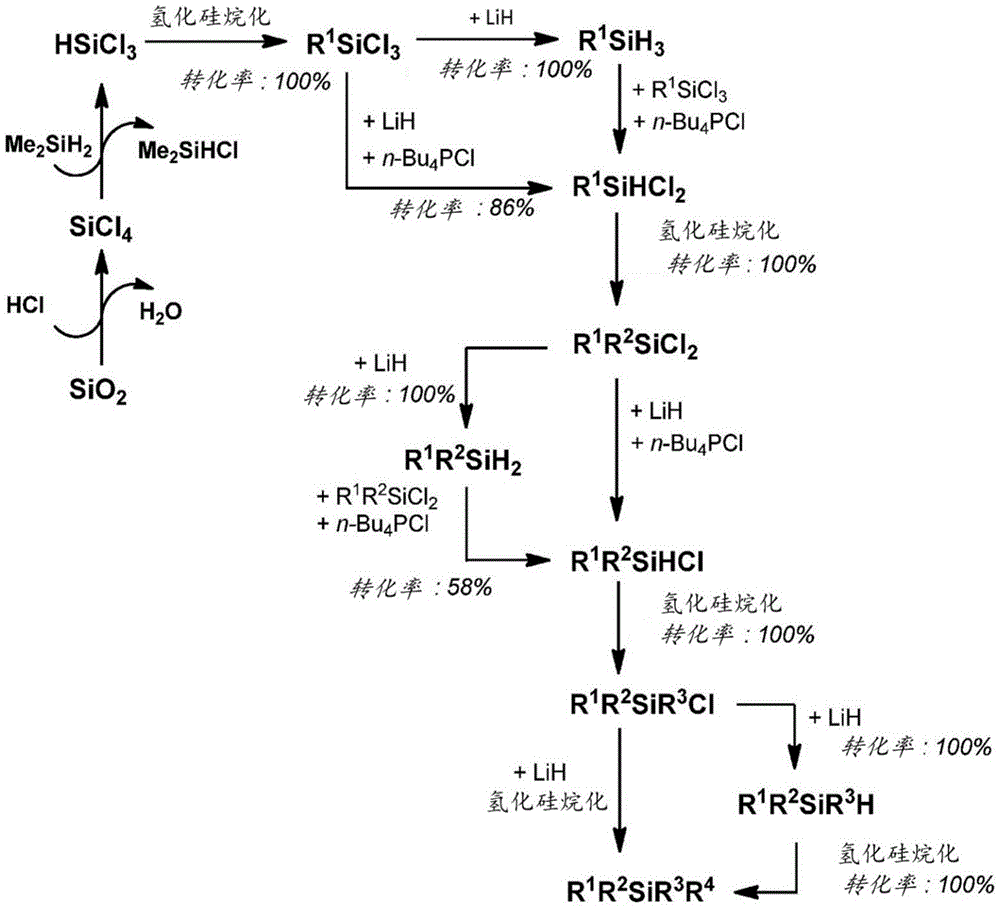
在图1中,显示了从R
图2显示了从MeSiH
图3显示了从Me
- 一种聚硅氧烷/全氢聚硅氮烷杂化聚合物及其合成方法
- N-烷基氮杂硅环戊烷合成方法及N-烷基氮杂硅环戊烷
- 一种氮杂环丁硅前体化合物的合成方法及以其合成六元硅氮杂环化合物的方法