一种邻羧基苯甲醛的制备方法
文献发布时间:2024-04-18 19:58:21
技术领域
本发明涉及有机合成技术领域,特别涉及一种邻羧基苯甲醛的制备方法。
背景技术
AZD2281(Ku-0059436),又名奥拉帕尼(Olaparib),其结构如下式所示:
Olaparib是一种口服有效的PARP抑制剂,抑制PARP-1和PARP-2的IC50分别为5和1nM。当Olaparib浓度为30-100nM作用于SW620细胞,使PARP-1失活。与BRCA1和BRCA2充足细胞系(Hs578T,MDA-MB-231,T47D)相比,BRCA1缺陷细胞系(MDA-MB-463和HCC1937)对Olaparib过分敏感Olaparib抑制PARP,阻断碱基切除修复,导致KB2P细胞对Olaparib强烈敏感。结果导致在DNA复制时由单链断裂转变为双链断裂,由此激活BRCA2依赖的重组途径。
奥拉帕尼目前主要应用于用于治疗BRCA基因突变的肿瘤,比如卵巢癌,乳腺癌,前列腺癌。PARP酶涉及细胞的正常功能,如DNA的转录和DNA修复。美国药监局临床研究显示奥拉帕尼无论是作为单药治疗或联合铂类为基础的化疗,均能够抑制试管中肿瘤细胞的生长,和减少人类肿瘤的生长。而奥拉帕尼(Olaparib)在工业生产中一个重要的起始物料中间体则正是邻羧基苯甲醛。
现有的邻羧基苯甲醛的制备方法通常采用以邻二甲苯为原料,经二溴化锰催化在双氧水高温高压作用下合成路线,但是该路线对设备要求过高,且用到的双氧水对热稳定性不足,受热容易迅速分解成氧气进而引发爆炸或者火灾,放大操作不安全,总收率也较低,会得到相当数量的邻苯二甲酸副产物。又或者以邻醛基苯甲腈为原料,经硫酸酸性条件水解氰基得到目标产物,但是该路线起始物料价格昂贵,后处理会产生很多强酸性工业废水。又或者苯酞为起始物料,经液溴或者NBS进行自由基取代反应得到3-溴苯酞中间体,再水解得到目标产物,但是该反应需要使用到或者反应中会生成液溴这种有毒且易挥发的物质,对生产人员来说会是一个不小的安全隐患成本高,反应使用的到的溶剂,如氯仿或者四氯化碳也会对肝脏造成巨大损伤。
所以,现在有必要对现有技术进行改进,以提供更可靠的方案。
发明内容
本发明所要解决的技术问题在于针对上述现有技术中的不足,提供一种邻羧基苯甲醛的制备方法。
为解决上述技术问题,本发明采用的技术方案是:一种邻羧基苯甲醛的制备方法,包括以下步骤:
S1、将苯酞在极性质子溶剂中与过一硫酸氢钾复合盐、碱反应,制得中间体;
S2、将步骤S1所得中间体用强质子酸水溶液水解,得到产物邻羧基苯甲醛。
优选的是,步骤S1中的碱为乙醇钠、叔丁醇钾、特戊醇钠、甲醇钠中的任意一种。
优选的是,步骤S1中的所用极性质子溶剂为甲醇、乙醇、异丙醇、叔丁醇中的任意一种。
优选的是,步骤S1中的碱与苯酞的摩尔比为4.5:1~4.05:1。
优选的是,步骤S1中的极性质子溶剂与苯酞的体积比为5~15:1,步骤S1中的过一硫酸氢钾复合盐与苯酞的的摩尔比为1.5:1~1.05:1。
优选的是,步骤S2中的强质子酸为硫酸、盐酸、三氟乙酸中的任意一种。
优选的是,步骤S2中的强质子酸水溶液的质量分数为10%~40%,强质子酸水溶液的体积为苯酞的5~8倍。
优选的是,所述的邻羧基苯甲醛的制备方法,还包括以下步骤:
S3、将步骤S2得到的产物用氢氧化钠水溶液调节pH为3-4,再用亚硫酸钠饱和溶液淬灭多余的过一硫酸氢钾复合盐,降温重结晶,过滤,真空干燥,得到最终产品邻羧基苯甲醛。
优选的是,所述的邻羧基苯甲醛的制备方法,包括以下步骤:
S1、将苯酞在极性质子溶剂中与过一硫酸氢钾复合盐、碱反应6-12h,制得中间体;
S2、将步骤S1所得中间体加入强质子酸水溶液中,在50-70℃下反应1-3h,得到产物;
S3、将步骤S2得到的产物用氢氧化钠水溶液调节pH为3-4,再用亚硫酸钠饱和溶液淬灭多余的过一硫酸氢钾复合盐,浓缩掉甲醇后,降温至5-10℃进行重结晶,过滤,所得固体物真空干燥,得到最终产品邻羧基苯甲醛。
优选的是,所述的邻羧基苯甲醛的制备方法,包括以下步骤:
S1、将苯酞加入甲醇溶剂中,然后加入甲醇钠,投料过程中注意控制温度不要超过10℃,投料完毕0℃下反应30min;
然后自然升温至室温反应30min,再冰浴降温至0℃以下,分批加入过一硫酸氢钾复合盐,投料过程中注意控制温度不要超过10℃,投料完成后自然升温至室温,反应8h,制得中间体;
S2、将步骤S1所得中间体温降至-2至2℃,然后滴加质量分数为30%的硫酸水溶液,加料过程中控制温度不要超过20℃,加完升温至65℃反应2h;
S3、将步骤S2得到的产物用氢氧化钠水溶液调节pH为3-4,再用亚硫酸钠饱和溶液淬灭多余的过一硫酸氢钾复合盐,浓缩掉甲醇后,降温至5-10℃进行重结晶,过滤,所得固体物50℃下真空干燥,得到最终产品邻羧基苯甲醛。
本发明的有益效果是:
本发明提供的邻羧基苯甲醛的制备方法能够有效降低合成成本,避免了危险和有毒的试剂的使用,有效减少工业三废的产生,本发明的工艺简单,操作便捷,原料廉价易得,产率高,有利于大规模工业化生产。
附图说明
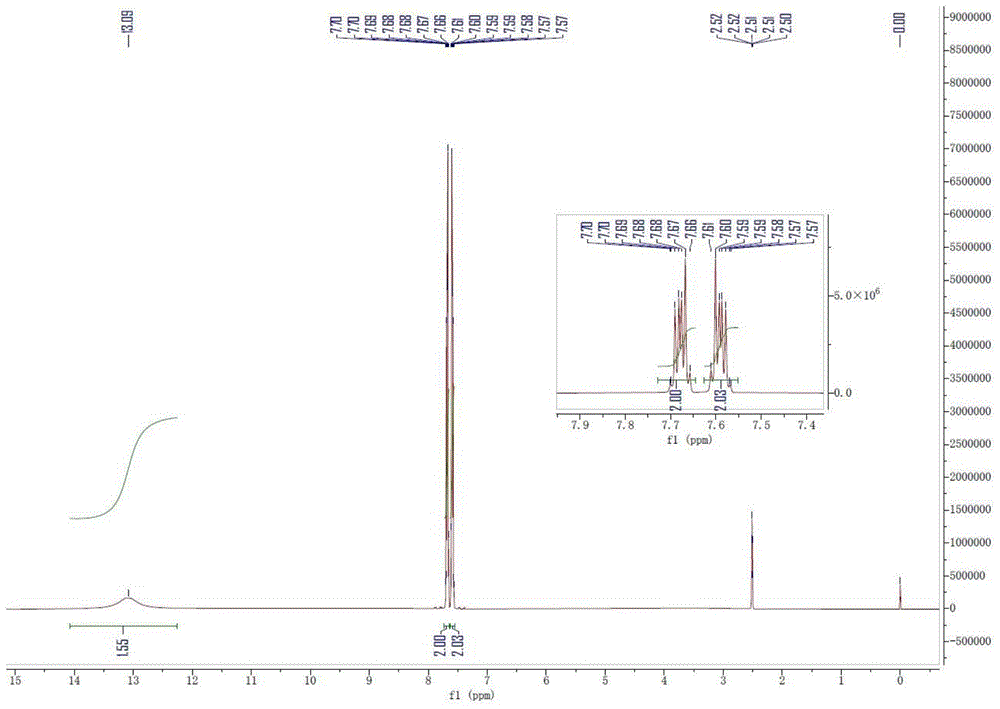
图1为实施例1制备的邻羧基苯甲醛的核磁氢谱;
图2为实施例1制备的邻羧基苯甲醛的核磁碳谱。
具体实施方式
下面结合实施例对本发明做进一步的详细说明,以令本领域技术人员参照说明书文字能够据以实施。
应当理解,本文所使用的诸如“具有”、“包含”以及“包括”术语并不排除一个或多个其它元件或其组合的存在或添加。
下列实施例中所使用的试验方法如无特殊说明,均为常规方法。下列实施例中所用的材料试剂等,如无特殊说明,均可从商业途径得到。下列实施例中未注明具体条件者,按照常规条件或者制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
本发明提供一种邻羧基苯甲醛的制备方法,采用过一硫酸氢钾复合盐碱性条件下氧化,再酸性条件下水解得到最终产物,具体包括以下步骤:
S1、将苯酞在极性质子溶剂中与过一硫酸氢钾复合盐、碱反应,制得中间体,此中间体不需要分离纯化;
S2、将步骤S1所得中间体用强质子酸水溶液水解,得到产物邻羧基苯甲醛。
反应路线如下:
反应机理如下:
在优选的实施例中,步骤S1中的碱为乙醇钠、叔丁醇钾、特戊醇钠、甲醇钠中的任意一种,更优选为甲醇钠。
在优选的实施例中,步骤S1中的所用极性质子溶剂为甲醇、乙醇、异丙醇、叔丁醇中的任意一种,更优选为甲醇。
在优选的实施例中,步骤S1中的反应时间可以为6-12h,例如可以为6、8、10、12h或它们两两之间构成的任意数值,更优选为8h。
在优选的实施例中,步骤S1中的碱与苯酞的摩尔比为4.5:1~4.05:1,例如可以为4.5:1、4.3:1、4.2:1、4.1:1、4.05:1或它们两两之间构成的任意数值,更优选为4.2:1;。
在优选的实施例中,步骤S1中的极性质子溶剂的体积为苯酞的5~15倍,例如可以为5、7、9、11、13、15倍或它们两两之间构成的任意数值,更优选为10倍。
在优选的实施例中,步骤S1中的过一硫酸氢钾复合盐与苯酞的的摩尔比为1.5:1~1.05:1,例如可以为1.5:1、1.3:1、1.2:1、1.1:1、1.05:1或它们两两之间构成的任意数值,更优选为1.2:1。
在优选的实施例中,步骤S2中的强质子酸为硫酸、盐酸、三氟乙酸中的任意一种,更优选为硫酸。
在优选的实施例中,步骤S2中的强质子酸水溶液的质量分数为10%~40%,例如可以为10%、20%、30%、40%或它们两两之间构成的任意数值,更优选为30%。
在优选的实施例中,强质子酸水溶液的体积为苯酞的5~8倍,例如可以为5、6、7、8倍或它们两两之间构成的任意数值,更优选为6倍。
在优选的实施例中,步骤S2中的反应温度可以为50-70℃,例如可以为50℃、60℃、65℃、70℃或它们两两之间构成的任意数值,更优选为65℃。
在优选的实施例中,步骤S2中的反应时间可以为1-3h,例如可以为1、1.5、2、2.5、3h或它们两两之间构成的任意数值,更优选为2h。
在优选的实施例中,该邻羧基苯甲醛的制备方法还包括以下步骤:
S3、将步骤S2得到的产物用氢氧化钠水溶液调节pH为3-4,再用亚硫酸钠饱和溶液淬灭多余的过一硫酸氢钾复合盐,降温重结晶,过滤,真空干燥,得到最终产品邻羧基苯甲醛。
在优选的实施例中,的邻羧基苯甲醛的制备方法包括以下步骤:
S1、将苯酞在极性质子溶剂中与过一硫酸氢钾复合盐、碱反应6-12h,制得中间体;
S2、将步骤S1所得中间体加入强质子酸水溶液中,在50-70℃下反应1-3h,得到产物;
S3、将步骤S2得到的产物用氢氧化钠水溶液调节pH为3-4,再用亚硫酸钠饱和溶液淬灭多余的过一硫酸氢钾复合盐,浓缩掉甲醇后,降温至5-10℃进行重结晶,过滤,所得固体物真空干燥,得到最终产品邻羧基苯甲醛。
以上为本发明的总体构思,以下在其基础上提供详细的实施例,以对本发明作进一步说明。
实施例1
一种邻羧基苯甲醛的制备方法,包括以下步骤:
步骤1、制备中间体:
向20L三口瓶内加入6.7L甲醇,再加入原料苯酞(670g,5mol),加完降温至0℃以下,分批缓慢加入甲醇钠(1134g,21mol),投料过程中注意控制温度不要超过10℃,投料完毕保温0℃反应30min,然后自然升温至室温反应30min。再冰浴降温至0℃以下,缓慢分批加入过一硫酸氢钾复合盐(3689g,6mol),加料过程中注意控制温度不要超过10℃,滴加完毕撤去冰浴,自然升温至室温反应8h。TLC反应完全后无需分离纯化,直接进行下一步操作;
步骤2、制备邻羧基苯甲醛
将步骤1所得的产物溶液进行降温,使内温降至0℃左右,向产物溶液内缓慢滴加4.02L 30wt%硫酸水溶液,加料过程中注意控制温度不要超过20℃,加完升温至65℃反应2h;
TLC反应完全后,用5wt%氢氧化钠水溶液调节pH为3-4,再加入126g亚硫酸钠淬灭多余的过一硫酸氢钾,浓缩掉甲醇后,缓慢将体系温度降至5-10℃,有白色固体析出,过滤,滤饼用冰水淋洗除盐,50℃真空干燥得到邻羧基苯甲醛白色固体685克,收率91.25%,产物的核磁谱如图1(核磁氢谱谱图)和图2(核磁碳谱谱图)所示,核磁数据如下:
1
δ13.09(s,2H),7.66-7.70(m,2H),7.57-7.61(m,2H).
13
δ169.17,133.30,131.25,128.82,40.57,40.36,40.15,39.94,39.73,39.52,39.31
实施例2
一种邻羧基苯甲醛的制备方法,包括以下步骤:
步骤1、制备中间体:
向20L三口瓶内加入3.35L叔丁醇钾,再加入原料苯酞(670g,5mol),加完降温至0℃以下,分批缓慢加入叔丁醇钾(2272g,20.25mol),投料过程中注意控制温度不要超过10℃,投料完毕保温0℃反应30min,然后自然升温至室温反应30min。再冰浴降温至0℃以下,缓慢分批加入过一硫酸氢钾复合盐(3228g,5.25mol),加料过程中注意控制温度不要超过10℃,滴加完毕撤去冰浴,自然升温至室温反应6h。TLC反应完全后无需分离纯化,直接进行下一步操作;
步骤2、制备邻羧基苯甲醛
将步骤1所得的产物溶液进行降温,使内温降至0℃左右,向产物溶液内缓慢滴加3.85L 30wt%硫酸水溶液,加料过程中注意控制温度不要超过20℃,加完升温至65℃反应2h;
TLC反应完全后,用5wt%氢氧化钠水溶液调节pH为3-4,再加入126g亚硫酸钠淬灭多余的过一硫酸氢钾,浓缩掉甲醇后,缓慢将体系温度降至5-10℃,有白色固体析出,过滤,滤饼用冰水淋洗除盐,50℃真空干燥得到邻羧基苯甲醛白色固体645克,收率85.92%。
实施例3
一种邻羧基苯甲醛的制备方法,包括以下步骤:
步骤1、制备中间体:
向20L三口瓶内加入10L乙醇,再加入原料苯酞(670g,5mol),加完降温至0℃以下,分批缓慢加入乙醇钠(1530g,22.5mol),投料过程中注意控制温度不要超过10℃,投料完毕保温0℃反应30min,然后自然升温至室温反应30min。再冰浴降温至0℃以下,缓慢分批加入过一硫酸氢钾复合盐(4605g,7.5mol),加料过程中注意控制温度不要超过10℃,滴加完毕撤去冰浴,自然升温至室温反应12h。TLC反应完全后无需分离纯化,直接进行下一步操作;
步骤2、制备邻羧基苯甲醛
将步骤1所得的产物溶液进行降温,使内温降至0℃左右,向产物溶液内缓慢滴加4.20L 30wt%硫酸水溶液,加料过程中注意控制温度不要超过20℃,加完升温至65℃反应2h;
TLC反应完全后,用5wt%氢氧化钠水溶液调节pH为3-4,再加入126g亚硫酸钠淬灭多余的过一硫酸氢钾,浓缩掉甲醇后,缓慢将体系温度降至5-10℃,有白色固体析出,过滤,滤饼用冰水淋洗除盐,50℃真空干燥得到邻羧基苯甲醛白色固体680克,收率90.58%。
尽管本发明的实施方案已公开如上,但其并不仅仅限于说明书和实施方式中所列运用,它完全可以被适用于各种适合本发明的领域,对于熟悉本领域的人员而言,可容易地实现另外的修改,因此在不背离权利要求及等同范围所限定的一般概念下,本发明并不限于特定的细节。
- 一种邻氟邻亚胺基苯甲酸中间体化合物及其制备方法和应用
- 一种基于催化氧化反应的间溴苯甲醛的制备方法
- 一种甲苯氧化合成苯甲醛的高稳定性催化剂及其制备方法
- 一种制备邻羧基苯甲醛的方法
- 一种邻羧基苯甲醛的制备方法