一种PSD-95抑制剂及其用途
文献发布时间:2024-04-18 19:58:21
技术领域
本发明属于生物医药领域,具体涉及一种PSD-95抑制剂及其用途。
背景技术
N-甲基-D-天冬氨酸受体(N-methyl-D-aspartate receptor,NMDAR)在中枢神经系统中发挥着重要的作用,参与学习与记忆的形成,参与中枢神经系统的发育的突触的生成、可塑性的形成与谷氨酸介导的神经系统毒性。NMDA受体是兴奋性毒性(即谷氨酸介导的神经毒性)的主要介体,其与神经退行性疾病和急性脑损伤有关。可作为某些神经系统疾病的治疗靶点,如脑梗塞、神经病理性疼痛、癫痫及精神分裂症等。
突触后致密物(Postsynaptic Density,PSD)是兴奋性突触后膜超级信号分子复合体,是突触发挥传递功能的重要物质。根据PSD分子量的重量大小,可以将其分为4类:PSD-95、PSD-93、突触相关蛋白97(SAP-97)与SAP102。
PSD-95是含量最丰富、最重要的脚手架蛋白,主要存在于成熟的兴奋性谷氨酸能突触内,其相对分子质量(Mr)为95000,为突触后膜上的受体活动和稳定性所必需,在突触可塑性中起重要作用,是介导与整合突触信息最重要的突触蛋白同时也是鸟苷酸相关激酶家族成员之一。PSD-95包括三个PDZ结构域、一个SH3结构域和一个鸟苷酸激酶样(GK)结构域,由连接区连接。PSD-95几乎完全位于神经元的突触后密度区,并参与锚定突触蛋白。它的直接和间接结合伴侣包括神经连接蛋白、nNOS、NMDA受体、AMPA受体和钾通道。
根据mRNA对基因的剪接的不同,目前已知有10多种nNOS(Neuronal nitric oxidesynthase,神经一氧化氮合酶)剪接体,其分子量为160.8KD。nNOS含有C末端的还原与N末端的氧化两个结构域。其中N末端有两个不重叠的结合区:(1)PDZ区:由1~99个氨基酸组成,参与活性nNOS二聚体的形成;(2)β-finger结构:由100-300个氨基酸组成,含有特定的氨基酸序列-ETTF-,可以与其他蛋白质的PDZ结合而发挥其功能。通过PDZ-PDZ结构域或C末端PDZ反应可将nNOS锚定于质膜或细胞溶质蛋白。PSD-95能将nNOS与NMDA受体相连,通过NMDA受体的激活从而有效的活化nNOS。
脑卒中的特征是局部缺血区域、脑出血区域和/或创伤区域的神经元细胞死亡。而由于脑缺血引起的神经元死亡或损伤是一个损伤级联反应过程,脑缺血后组织血液灌注下降,兴奋性神经递质增加,激活NMDA和AMPA受体,引起离子通道开放,钙离子内流,激活大量的酶引发信号级联反应,造成多途径的神经细胞损伤。当发生脑缺血的情况时,NMDA(N-甲基-D-天冬氨酸受体)受体过度激活,通过阻断NMDA/PSD-95/nNOS途径可以阻止NO病理性释放。同时,据研究显示,阻断NMDA/PSD-95的偶联可能产生不可预测的生理反应,尽管NMDA受体的拮抗剂通过阻止谷氨酸介导的离子通量而有效降低兴奋性毒性,但它们也阻止了一些在生理学上重要的过程。其下游的PSD-95通过与多种蛋白的相互作用,引发一系列缺血性损伤,是脑缺血损伤的关键性位点,同时也是药物治疗的潜在靶点,而阻断nNOS与PSD95之间的偶联对于阻止NO病理性释放更有针对性,因此是预防和治疗缺血性脑卒中等受神经元损伤影响的疾病更理想的靶点。因此PSD-95抑制剂的研发对于包括脑卒中在内的多种兴奋性神经毒性引起的神经系统损伤具有很大的药用意义。
此外,研究显示兴奋性神经递质NMDA在焦虑,癫痫和多种神经退行性疾病如阿尔茨海默氏病、肌萎缩性侧索硬化症(ALS)、帕金森氏病或亨廷顿氏病等中都发挥重要作用。例如,研究显示中枢的谷氨酸能系统过度兴奋可引发焦虑,而NMDA受体(NMDAR)负责谷氨酸兴奋性神经毒性的主要部分。癫痫的发作包含起动、发作性放电的维持与扩展以及发作性放电的抑制3个不同而连续的病理生理过程,在该过程中兴奋性神经递质如谷氨酸、天门冬氨酸起重要作用。在阿尔兹海默症中,PSD-95通过GluR6-PSD-95-MLK3通路参与导致其的神经毒性机制。此外,在亨廷顿氏病中,PSD-95是NMDA受体和huntingtin突变体神经毒性的介体。因此PSD-95抑制剂的研发对于上述疾病的治疗、改善和预防也具有重要意义。
Nerinetide(NA-1,TAT-NR2B9c,序列:YGRKKRRQRRRKLSSIESDV)是一种PSD-95抑制剂,从而破坏PSD-95与NMDA受体和神经元型一氧化氮合酶(nNOS)的结合,以及降低由脑缺血诱发的兴奋毒性。在临床前缺血性脑卒中模型中可减少脑缺血再灌注的梗死面积,并改善其功能预后。Nerinetide无严重的副作用,在疑似中风或其他缺血性或出血性状况但还尚未根据本领域已认可的标准作出确认不存在出血的诊断时可以施用Nerinetide。但Nerinetide在体内半衰期短,患者每天需要大剂量用药,患者顺应性差,临床费用高,且此化合物因与PSD-95中PDZ1-2的低亲和力可能使其成为无效的非选择性化合物。为满足临床需求,仍然需要开发更多的PSD-95抑制剂。
多肽是组成机体内各种细胞功能的生物活性物质,具有相对分子质量小、特异性高、易吸收、易合成及改造、可以提高机体免疫力、安全性高等特点,在肿瘤的临床治疗上具有较高的应用价值。噬菌体展示技术(phage display)是利用丝状噬菌体展示蛋白和多肽,从大量变异体中提取所需性质的多肽或蛋白。
利用噬菌体展示技术这种基于定向进化的高通量筛选技术,大大拓展了定向进化技术在多肽改造筛选方面的应用。
发明内容
为了解决现有技术中的不足,本发明公开一种PSD-95抑制剂及其用途,所述PSD-95抑制剂能够非典型地结合PSD-95的PDZ1和/或PDZ2结构域,从而抑制其与nNOS的蛋白质-蛋白质相互作用。
一方面,本发明提供一种多肽种多肽或其药学上可接受的盐,所述多肽氨基酸序列包含SEQ ID NO:1~SEQ ID NO:15氨基酸序列之一或与SEQ ID NO:1~SEQ ID NO:15具有至少80%序列同一性的氨基酸序列或经修饰的SEQ ID NO:1~SEQ ID NO:15所示的氨基酸序列的衍生物。
在一些实施方式中,所述多肽氨基酸序列包含SEQ ID NO:1~SEQ ID NO:15氨基酸序列之一或与SEQ ID NO:1~SEQ ID NO:15具有至少90%序列同一性的氨基酸序列或经修饰的SEQ ID NO:1~SEQ ID NO:15所示的氨基酸序列的衍生物。
在一些实施方式中,所述多肽氨基酸序列包含SEQ ID NO:1~SEQ ID NO:15氨基酸序列之一或与SEQ ID NO:1~SEQ ID NO:15具有至少95%序列同一性的氨基酸序列或经修饰的SEQ ID NO:1~SEQ ID NO:15所示的氨基酸序列的衍生物。
在一些实施方案中,所述多肽氨基酸序列如SEQ ID NO:1~SEQ ID NO:15所示,
SAAGVVCYYWKGNIDFCHTRGGSGS(SEQ ID NO:1)、
SAAGIFCEMWRGAVDFCYEWKGSGS(SEQ ID NO:2)、
SAAGCTWVTHFDHVTWTEKVCGSGS(SEQ ID NO:3)、
SAAGCRIIETDVDTFITTLICGSGS(SEQ ID NO:4)、
SAAGCKWITHFEEHVWFEMVCGSGS(SEQ ID NO:5)、
SAAGCYYITTFMEAIVTEERCGSGS(SEQ ID NO:6)、
SAAGCRYHTQFSRTVWTMWICGSGS(SEQ ID NO:7)、
SAAGCHKESIIGAQIQTIVVCGSGS(SEQ ID NO:8)、
SAAGCQFETYFGEEVSTIWSCGSGS(SEQ ID NO:9)、
SAAGCKQVTLFWIDVTTYYICGSGS(SEQ ID NO:10)、
SAAGCYRETTFGIRILESWHCGSGS(SEQ ID NO:11)、
SAAGMVCETMMDTWVTVCWRRGSGS(SEQ ID NO:12)、
SAAGCRTWTHFIQIIITEQVCGSGS(SEQ ID NO:13)、
SAAGCQTWTTFEIFVWSELVCGSGS(SEQ ID NO:14)、
SAAGCKQVTLFWIDVTTYYICGSGS(SEQ ID NO:15)。
在一些实施方案中,所述经修饰的氨基酸序列的衍生物选自以下的一种或更多种修饰:N端和/或C端修饰;以一个或多个天然和/或非天然氨基酸残基取代一个、或多个氨基酸残基。
在一些实施方案中,所述修饰包括氨基化、羟基化、羧基化、羰基化、酰胺化、烷基化、磷酸化、糖基化、环化、生物素化、乙酰化、酯化、荧光基团修饰、聚乙二醇PEG修饰、固定化修饰。
本文定义的多肽可以是所述多肽的药学上可接受的盐或前药的形式。
在一些实施方案中,所述药学可接受的盐可选自三氟乙酸盐、醋酸盐、盐酸盐和磷酸盐。
在一些实施方案中,所述多肽通过采用固相合成法得到。
在一些实施方案中,所述多肽通过噬菌体展示技术筛选获得。
另一方面,本发明提供一种多核苷酸,其编码上述的多肽。
另一方面,本发明提供一种药物组合物,其包含上述的多肽或其药学上可接受的盐或上述的多核苷酸及药学可接受的载体、赋形剂和/或稀释剂。
在一些实施方案中,本申请的多肽可以以药物组合物的形式被施用。药物组合物可以通过常规的混合、溶解、制粒、制锭、研磨、乳化、包封、捕获或冻干方法制造。可以使用一种或多种生理学可接受的便于将本申请的化合物加工成可药用制剂的载体、稀释剂、赋形剂或辅料,以常规方式配制药物组合物。适当的配制依赖于选择的施用途径。
在一些实施方案中,施用可以是肠胃外、静脉内、经口、皮下、动脉内、颅内、鞘内、腹膜内、局部、鼻内或肌内的。
另一方面,本发明提供一种上述的多肽或其药学可接受的盐、多核苷酸或药物组合物在制备用于治疗、改善或预防个体中的PSD-95功能异常引起的疾病的药物中的用途。
在一些实施方案中,所述PSD-95功能异常引起的疾病可选自脑卒中、中风、神经退行性疾病、焦虑或癫痫、神经病理性疼痛。
在一些实施方案中,其中所述脑卒中选自缺血性卒中或出血性脑卒中。
在一些实施方案中,其中所述神经退行性疾病选自阿尔茨海默氏病、肌萎缩性侧索硬化症、帕金森氏病或亨廷顿氏病。
如本文所用,术语“噬菌体展示技术”是由Smith等人于1985年提出,并于1989年成功构建。噬菌体展示是一种选择技术,将多肽或蛋白质与噬菌体外壳蛋白融合并展示在病毒粒子表面。噬菌体展示技术是将编码外源多肽或蛋白质的DNA序列通过基因工程插入到噬菌体基因,从而在噬菌体衣壳蛋白表面展示多肽或蛋白质的技术。大量展示有不同随机多肽的噬菌体可构成噬菌体文库,用于针对特定靶标的筛选,以探究多肽与靶标之间的相互作用。在生物学领域,该技术被广泛的应用于蛋白质与蛋白质、蛋白质与多肽、蛋白质与DNA等分子相互作用的研究中。
噬菌体展示技术的原理,是将一段外源基因插入到噬菌体外壳蛋白结构基因的适当位置,在阅读框正常且不影响外壳蛋白正常功能的情况下,外源基因会随着外壳蛋白的表达而表达,从而使多肽或蛋白以融合蛋白的形式展现在噬菌体表面。被展示的蛋白可以保持相对独立的空间结构和生物活性,有利于靶蛋白的结合,因而可以利用靶蛋白快速的进行噬菌体展示文库的筛选。在展示文库构建完成后,以靶蛋白为固定相,和展示文库共同孵育一段时间,洗去未结合噬菌体,再以竞争受体洗脱吸附的噬菌体。洗脱得到的噬菌体感染宿主菌繁殖扩增,然后进行下一轮洗脱。经过3~5轮的“吸附-洗脱-扩增”后(对于某些亲和力弱的抗体需经过更多轮的洗脱),便可以得到能与靶蛋白特异性结合的噬菌体的高度富集。该技术的显著特点是建立了基因型和表现型之间的对应关系。
术语“氨基酸”是指含有氨基和羧基的分子。合适的氨基酸包括但不限于天然存在的氨基酸的D-和L-异构体,以及通过有机合成或其它代谢途径制备的非天然存在的氨基酸。如本文所用,术语氨基酸包括但不限于α-氨基酸、天然氨基酸、非天然氨基酸和氨基酸类似物。
术语“天然存在的氨基酸”是指在自然界中合成的肽中常见的20种L-氨基酸中的任何一种,即丙氨酸(Ala或A)、精氨酸(Arg或R)、天冬酰胺(Asn或N)、天冬氨酸(Asp或D)、半胱氨酸(Cys或C)、谷氨酸(Glu或E)、谷氨酰胺(Glu或Q)、甘氨酸(Gly或G)、组氨酸(His或H)、异亮氨酸(Ile或I)、亮氨酸(Leu或L)、赖氨酸(Lys或K)、甲硫氨酸(Met或M)、苯丙氨酸(Phe或F)、脯氨酸(Pro或P)、丝氨酸(Ser或S)、苏氨酸(Thr或T)、色氨酸(Trp或W)、酪氨酸(Tyr或Y)和缬氨酸(Val或V)的L-异构体。
术语“肽”或“多肽”的含义是被本专业领域的技术人员所熟知的。通常情况下,肽或多肽是两个或多个氨基酸由酰胺键链接,酰胺键则由一个氨基酸的氨基与相邻氨基酸的羧基构成。本文所述的多肽可包含天然存在的氨基酸或者非天然存在的氨基酸。可被修饰成其类似物,衍生物,功能模拟物,伪肽等诸如此类包含至少两个氨基酸的化合物。除非指明N-端或C-末端具有特定的修饰,否则一个包含特定氨基酸序列的多肽,则包括不加修饰的和修饰的氨基和/或羧基末端,这是被本领域的专业技术人员所熟知的。一个特定的氨基酸序列的多肽可以包括修饰的氨基酸和/或额外的氨基酸,除非N-和/或C-末端包含妨碍进一步添加氨基酸的修饰。这样的修改包括,例如,N-末端的乙酰化和/或C-末端的酰胺化。
本发明的多肽可以通过改造修饰,形成多肽衍生物。正如本领域技术人员所熟知的,可以对多肽进行各种改造修饰。典型的改造修饰包含但不限于,N-末端乙酰化、C-末端酰胺化、d型氨基酸替换、非天然氨基酸替换、脂肪酸修饰或以上各种修饰改造的组合。本发明包括任何被众所周知的多肽的修饰改造。例如,多肽衍生物可以包括对于多肽的化学修饰,如烷基化、酰基化、氨基甲酰化、碘化或其他任何产生多肽衍生物的改造修饰。多肽的改造修饰可以包含改造过的氨基酸,例如,羟基脯氨酸或羧基谷氨酸,并且可以包括以非肽键相连的氨基酸。
对于本发明的多肽的其它修饰改造可采用非天然氨基酸对多肽中的天然氨基酸进行取代,非天然氨基酸包含但不限于,2-氨基脂肪酸(Aad)、3-氨基脂肪酸(βAad)、β-丙氨酸,β-氨基丙酸(βAla)、2-氨基丁酸(Abu)、4-氨基丁酸、哌啶羧酸(4Abu)、6-氨基己酸(Acp)、2-氨基庚酸(Ahe)、2-氨基异丁酸(Aib)、3-氨基异丁酸(βAib)、2-氨基庚二酸(Apm)、2,4-二氨基丁酸(Dbu)、锁链素(Des),2,2'-二氨基庚二酸(Dpm),2,3-二氨基丙酸(Dpr),N乙基甘氨酸(EtGly)、N-乙基天冬酰胺(EtAsn),羟赖氨酸(Hyl)、异羟赖氨酸(aHyl)、3-羟脯氨酸(3Hyp)、4-羟基脯氨酸(4Hyp)、异锁链素(Ide)、异-异亮氨酸(aIle)、N-甲基甘氨酸(MeGly)、N-甲基异亮氨酸(MeIle)、6-N-甲基赖氨酸(MeLys)、N-甲基缬氨酸(MeVal)、正缬氨酸(Nva)、正亮氨酸(Nle)和鸟氨酸(Orn)。当然,所有被修饰改造的α-氨基酸可以被相应的β-,γ-或ω-氨基羧酸所取代。
本发明的多肽可以使用本领域技术人员所熟知的方法制备,包括众所周知的化学合成的方法。因此,当多肽或其衍生物包含一个或多个非标准氨基酸,则极有可能是通过化学合成法制备而来。除了使用化学合成的方法制备多肽或其衍生物,还可以通过编码核酸表达来制备。这对于制备只含有天然氨基酸的多肽或其衍生物特别适用,在这种情况下可以使用众所周知的核酸编码多肽序列的制备方法(参见Sambrook etal.,MolecularCloning:ALa bora tory Manua l,Third Ed.,Cold Spring Ha rbor Laboratory,NewYork(2001);Ausubel et al.,Current Protocols in Molecular Biology,JohnWileyand Sons,Baltimore,MD(1999))。多肽可以在生物体中表达,并通过公知的纯化技术进行纯化。
术语“PDZ结构域”是指约90个氨基酸的模块蛋白质结构域,其特征是对脑突触蛋白PSD-95、果蝇(Drosophila)分隔连接蛋白Discs-Large(DLG)和上皮紧密连接蛋白Z01(Z01)具有显著(例如至少60%)的序列同一性。PDZ结构域也称作Discs-Large同源性重复(“DHRs”)和GLGF重复。PDZ结构域通常显示保留核心共有序列(Doyle,D.A.,1996,Cell 85:1067-76)。示例性的含PDZ结构域的蛋白质和PDZ结构域序列在美国申请No.10/714,537中公开。
与现有技术相比,本发明的有益效果为:
本发明所述的多肽具有相对分子质量小,其合成、表达及修饰等易于实现,且对PSD95与其受体的结合具有良好的抑制效率。
附图说明
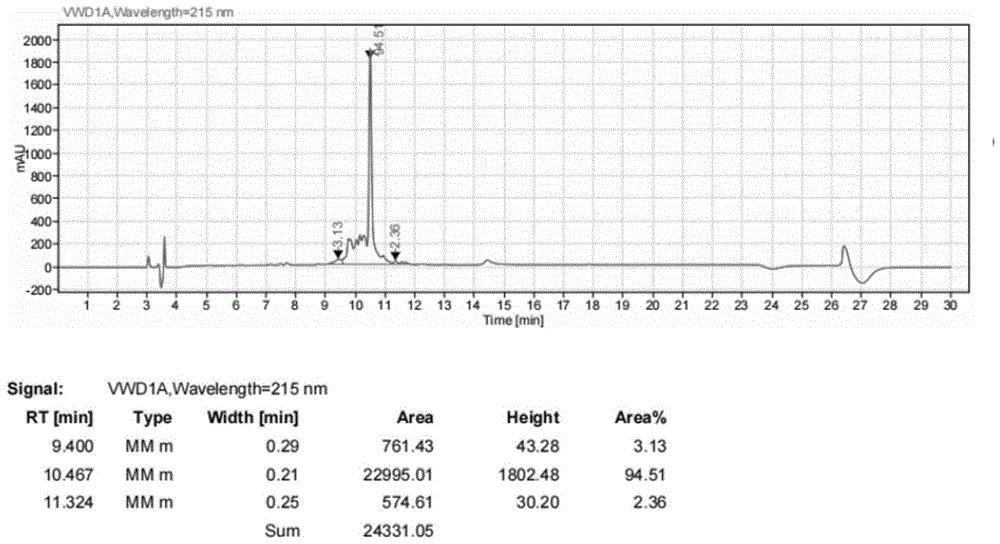
图1:Seq ID No.1氨基酸序列对应化合物的HPLC图谱
图2:Seq ID No.1氨基酸序列对应化合物的Ms图谱
具体实施例
合成实施例
本公开提供的多肽化合物及其衍生物采用固相合成的方法合成。合成载体为Fmoc-Cys(Trt)-2-Chlotrityl Resin树脂。合成过程中,首先将Fmoc-Cys(Trt)-2-Chlotrityl Resin树脂在N,N-二甲基甲酰胺(DMF)中充分溶胀,然后该固相载体与活化后氨基酸衍生物重复缩合→洗涤→去保护Fmoc→洗涤→下一轮氨基酸缩合的操作以达到所要合成的多肽链长度,最后用三氟乙酸:水:三异丙基硅烷:苯甲硫醚(90:2.5:2.5:5:,v:v:v:v)的混合溶液与树脂反应将多肽从固相载体上裂解下来,再由冷冻甲基叔丁基醚沉降后得到直链前体的固体粗品。切割后的直链前体粗品在碱性溶液中进行二硫键氧化得到目标多肽粗品。多肽粗品在0.1%三氟乙酸的乙腈/水的体系由C-18反相制备色谱柱纯化分离后得到多肽及其衍生物的纯品。
实验试剂
以下仅列出Seq ID No.1的合成方法为例,其余序列可参照实施例1的方法,具体合成的氨基酸序列如表1所示。
实施例1:Seq ID No.1氨基酸序列对应化合物的合成
步骤1:耦连第一位氨基酸Fmoc-Cys(Trt)-OH
将84mg(0.1mmol)2-Chlorotrityl chloride树脂在DCM中充分溶胀1h。称取Fmoc-Cys(Trt)-OH(0.08mmol)和二异丙基乙胺(DIEA,0.32mmol)溶于5ml DCM中并加入树脂中,在室温反应2h。反应完成后加入封闭液(10ml)DCM:甲醇:DIEA(85:10:5,v:v:v)室温10min进行封闭。封闭后的树脂用DCM洗5次,DMF洗5次。
步骤2:直链前体肽链合成
SAAGVVCYYWKGNIDFCHTRGGSGS
将步骤1得到的树脂在DMF中充分溶胀1h,之后将依照直链前体序列从羧基端第二位I到氨基端的顺序合成。每一个耦连周期进行如下:
·20%哌啶/DMF(20%v/v,10mL)进行Fmoc-去保护两次,每次8min。
·DMF冲洗树脂6-8次直到中性pH。
·用DMF溶解0.5mmol Fmoc-AA,0.5mmol 6-氯苯并三氮唑-1,1,3,3-四甲基脲六氟磷酸酯(HCTU)和1mmol 4-甲基吗啉(NMM),加入树脂室温反应1h。
·下一个氨基酸耦连之前用DMF冲洗树脂4-6次。
直链多肽合成后用DMF冲洗树脂5次,DCM冲洗树脂5次。树脂在真空中抽干。
步骤3:直链前体肽链切割
将新鲜配制的切割鸡尾酒(10mL)三氟乙酸:水:三异丙基硅烷:苯甲硫醚(90:2.5:2.5:5,v:v:v:v)加入到步骤2所得树脂中,在室温下振荡反应2小时。反应结束后将反应溶液过滤,并用三氟乙酸洗涤树脂,与反应溶液合并,用4倍体积冷MTBE沉淀得到粗品。用MTBE洗涤粗品3次,放入真空中抽干。
步骤4:分子内二硫键形成
在步骤3得到的粗品中加入DMSO充分溶解(DMSO体积为反应体系总体积的20%),然后将溶解后的多肽溶液缓慢滴加至50%乙腈水溶液中,终浓度为1mg/ml,室温震荡16小时。LC-MS监测反应结果,反应结束后直接进行纯化制备。
步骤5:多肽的纯化制备
将多肽粗品用20%乙腈水溶液溶解后,经过0.45um膜过滤后用反相高效液相色谱系统进行分离,缓冲液为A(0.1%三氟乙酸,水溶液)和B(0.1%三氟乙酸,乙腈)。其中,色谱柱为BR-C18(赛分)反相色谱柱,纯化过程中色谱仪检测波长设定为230nm,流速为15mL/min,梯度为20-50%乙腈in 40min。收集产物相关馏分,HPLC鉴定纯度后将>95%的馏分合并,冻干,获得多肽纯品。
步骤6:检测与表征方法
将步骤5的多肽纯品通过分析性高效液相色谱和液相色谱/质谱联用确定纯度及目标物分子量。。所得氨基酸序列如表1所示。
表1实施例中所用的氨基酸序列
生物测试实施例
测试例1:ELISA测试多肽样品抑制Tat-NR2B9c-B与cMyc-PSD95 alpha 1-392的结合
1)主要实验材料
2)实验步骤:
采用ELISA(酶联免疫吸附测定)方法测试多肽样品对Tat-NR2B9c-B结合cMyc-PSD95 alpha 1-392的抑制作用。将cMyc-PSD95 alpha 1-392以0.37μg/ml,25ul/孔包被在384孔板(greiner 781097)上。对cMyc-PSD95 alpha 1-392包被的ELSIA板进行封闭后,将多肽样品倍比稀释至0~10μM(从10μM往下3倍梯度稀释共8个浓度)。用工作站转移多肽样品12.5μl/孔至384孔板中,瞬时离心去除气泡.再使用8孔排枪(10-100ul)移液12.5μl/孔12nM Tat-NR2B9c-B,瞬时离心去除气泡后37℃孵育1h。弃掉孔中液体,加入80μl pH=7.4的1×TBST洗涤液洗板3-5次,每次3~5min。控干检测板加入25μL/孔1:10000稀释的Streptavidin HRP,瞬时离心去除气泡后放入37℃培养箱孵育1h。再次洗板控干检测板后,每孔加入25μlTMB显色液,继续放入培养箱37℃孵育30min。最后每孔加入25μl终止液(1MHCL)终止反应。使用酶标仪Cytation5读取在450nm下的吸光度。
3)实验结果
应用ELISA方法进行测试本发明的多肽抑制Tat-NR2B9c-B与PSD95相关蛋白的结合作用,测得的结果通过得到的IC50值如表2所示;结果显示本发明的多肽对cMyc-PSD95alpha1-392与Tat-NR2B9C-B的结合具有较好的抑制活性。
表2:ELISA测试多肽对cMyc-PSD95 alpha 1-392与Tat-NR2B9C-B的结合IC50值
测试例2:FRET测试多肽样品抑制cMyc-PSD95 alpha 1-392与Tat-NR2B9C-B的结合
1)主要实验材料
2)实验步骤:
采用FRET(荧光共振能量转移)方法测试多肽样品对cMyc-PSD95 alpha 1-392结合Tat-NR2B9C-B的抑制作用。将多肽样品倍比稀释至0~10μM(从10μM往下3倍梯度稀释共8个浓度)。使用8孔排枪(1-10ul)移液4μl/孔4nM cMyc-PSD95 alpha 1-392至384孔白板(Thermo 264706),瞬时离心去除气泡。用工作站转移多肽样品4μl/孔至384孔板中,瞬时离心去除气泡。再次使用8孔排枪移液4μl/孔20nM Tat-NR2B9c-B至384孔白板,瞬时离心去除气泡。将配制好的0.02μg/ml EU-steptavidin与1ug/ml MouseAnti-C-MYC IgG SureLightAPC 1:1混合.使用8孔排枪移液8μl/孔EU-APC预混液至384孔白板,瞬时离心去除气泡。室温避光孵育2h,使用酶标仪cytation5读取320nM激发620nm和665nm下的发射荧光值。
3)实验结果
应用FRET方法进行测试本发明的多肽抑制Tat-NR2B9c-B与PSD95相关蛋白的结合作用,测得的结果通过得到的IC50值如表3所示;结果显示本发明的多肽对cMyc-PSD95alpha1-392与Tat-NR2B9C-B的结合具有较好的抑制活性。
表3:FRET测试多肽对cMyc-PSD95 alpha 1-392与Tat-NR2B9C-B的结合IC50值
测试例3:ELISA测试多肽样品抑制biotin-nNOS1-299与cMyc-PSD95 alpha 1-392的结合1)主要实验材料
2)实验步骤:
采用ELISA(酶联免疫吸附测定)方法测试多肽样品对biotin-nNOS1-299结合cMyc-PSD95 alpha 1-392的抑制作用。将cMyc-PSD95 alpha 1-392以10μg/ml,25ul/孔包被在384孔板(greiner 781097)上。对cMyc-PSD95 alpha 1-392包被的ELSIA板进行封闭后,将多肽样品倍比稀释至0~10μM(从10μM往下3倍梯度稀释共8个浓度)。用工作站转移多肽样品12.5μl/孔至384孔板中,瞬时离心去除气泡.再使用8孔排枪(10-100ul)移液12.5μl/孔10μg/ml 743biotin-nNOS1-299,瞬时离心去除气泡后37℃孵育1h。弃掉孔中液体,加入80μl pH=7.4的1×TBST洗涤液洗板3-5次,每次3~5min。控干检测板加入25μL/孔1:10000稀释的Streptavidin HRP,瞬时离心去除气泡后放入37℃培养箱孵育1h。再次洗板控干检测板后,每孔加入25μlTMB显色液,继续放入培养箱37℃孵育30min。最后每孔加入25μl终止液(1M HCL)终止反应。使用酶标仪Cytation5读取在450nm下的吸光度。
3)实验结果
应用ELISA方法进行测试本发明的多肽抑制biotin-nNOS1-299与PSD95相关蛋白的结合作用,测得的结果通过得到的IC50值如表4所示;结果显示本发明的多肽对biotin-nNOS 1-299与PSD95相关蛋白的结合具有较好的抑制活性。
表4:ELISA测试多肽抑制biotin-nNOS1-299与cMyc-PSD95 alpha 1-392相关蛋白结合的IC50值
本发明提供了一种PSD-95抑制剂的多肽及其应用,本领域技术人员可以借鉴本文内容,适当改进工艺参数实现。特别需要指出的是,所有类似的替换和改动对本领域技术人员来说是显而易见的,它们都被视为包括在本发明。本发明的方法及应用已经通过较佳实施例进行了描述,相关人员明显能在不脱离本发明内容、精神和范围内对本文的方法和应用进行改动或适当变更与组合,来实现和应用本发明技术。
- 一种新型BTK激酶抑制剂的盐酸盐及其制备方法与用途
- 一种食品香料作为黄曲霉抑制剂的用途
- 一种新型环保高性能包被抑制剂及其制备方法和用途
- 一种STAT3抑制剂的甲磺酸盐及其制备方法与用途
- 一种STAT3抑制剂的马来酸盐及其制备方法与用途
- PSD-95环肽抑制剂及其用途
- 一种PSD-95抑制剂