2,3-双卤取代马来酸酯型内给电子体及其制备方法与应用
文献发布时间:2024-04-18 19:58:21
技术领域
本发明涉及聚丙烯催化剂内给电子体的设计合成与应用,具体涉及一种2,3-双取代马来酸酯型内给电子体及其制备方法与应用。
背景技术
邻苯二甲酸二酯类化合物是丙烯聚合Ziegler-Natta(Z-N)催化剂主要使用的内给电子体。尽管这类化合物作为内给电子体使用对于获得高活性高等规度的聚丙烯有利,但是获得的聚丙烯中残存的无法清除的邻苯二甲酸二酯类化合物依然会对人体造成损害。目前该类化合物在欧盟已经限制使用,可以预见在不久的将来,该类化合物在国内的使用也会受到限制。因此研发清洁型丙烯聚合Z-N催化剂专用内给电子体势在必行。
马来酸及其衍生物广泛应用于药物合成,例如N,N-二甲基-γ-(4-氯苯基)-2-吡啶丙胺顺丁烯二酸盐,俗名“扑尔敏”,是常见的抗过敏药物;(±)-3,4,5-三甲氧基苯甲酸(2-二甲胺基-2-苯基)丁酯马来酸盐,俗名“马来酸曲美布汀片”,是常见的胃肠动力抑制药;(E)-5-甲氧基-4'-三氟甲基苯戊酮-氧-(2-氨乙酰基肟)马来酸盐,药品名“马来酸氟伏沙明”,是治疗抑郁症或强迫症的药物,等等。以上种种事实表明:马来酸及其衍生物具有较好的安全性,尚无研究表明该类化合物对人体造成伤害的报道。
中国专利CN201210480335.9公布了采用马来酸二酯作为内给电子体制备高活性高等规度聚丙烯催化剂的技术。
该专利采用2,3-双烷基取代的马来酸二酯作为内给电子体时,催化剂的活性最高能够达到28.3kg PP/(g.Cat.h),聚丙烯的等规度可以达到98.7%。
但是该专利仍然存在两方面缺点:(1)2,3-双烷基取代的马来酸二酯合成技术难度较大,成本较高;(2)催化剂的活性较低,无法满足新时代背景下对聚丙烯低灰分的新要求。
发明内容
有鉴于此,本发明提供了2,3-双卤取代马来酸酯型内给电子体及其制备方法与应用。具体的,本发明提供如下技术方案:
一种2,3-双卤取代马来酸酯型内给电子体,其具有如下式I所示的结构式:
其中,X
根据本发明,所述R
根据本发明,所述内给电子体选自2,3-二氟马来酸二丙酯、2,3-二氟马来酸二正丁酯、2,3-二氟马来酸二异丁酯、2,3-二氟马来酸二正戊酯、2,3-二氟马来酸二异戊酯、2,3-二氯马来酸二丙酯、2,3-二氯马来酸二正丁酯、2,3-二氯马来酸二异丁酯、2,3-二氯马来酸二正戊酯、2,3-二氯马来酸二异戊酯、2,3-二溴马来酸二丙酯、2,3-二溴马来酸二正丁酯、2,3-二溴马来酸二异丁酯、2,3-二溴马来酸二正戊酯、2,3-二溴马来酸二异戊酯、2,3-二碘马来酸二丙酯、2,3-二碘马来酸二正丁酯、2,3-二碘马来酸二异丁酯、2,3-二碘马来酸二正戊酯、2,3-二碘马来酸二异戊酯、2-氟-3-氯马来酸二正丁酯、2-氟-3-溴马来酸二正丁酯、2-氟-3-碘马来酸二正丁酯、2-氯-3-溴马来酸二正丁酯、2-氯-3-碘马来酸二正丁酯、2-溴-3-碘马来酸二正丁酯、2-氟-3-氯马来酸二异丁酯、2-氟-3-溴马来酸二异丁酯、2-氟-3-碘马来酸二异丁酯、2-氯-3-溴马来酸二异丁酯、2-氯-3-碘马来酸二异丁酯或2-溴-3-碘马来酸二异丁酯。
具体的,所述内给电子体选自如下式I-1~I-5所示化合物中的一种:
本发明还提供上述2,3-双卤取代马来酸酯型内给电子体的合成方法,所述方法包括:
1)将马来酸酐和卤化试剂混合,进行卤化反应,制备中间产物;
2)在催化剂存在下,将步骤1)的中间产物与醇类化合物反应,制备得到式I所示的2,3-双卤取代马来酸酯型内给电子体。
根据本发明,步骤1)的反应式如下所示:
根据本发明,步骤2)的反应式如下所示:
根据本发明,步骤1)中,所述的卤化试剂选自氢氟酸、氯化亚砜、溴、碘化钾中的一种或多种。
根据本发明,步骤1)中,马来酸酐和卤化试剂的摩尔比为1:(6~20),优选为1:(8~17)。
根据本发明,步骤1)中,所述马来酸酐可以先分散于有机碱中,所述有机碱为三乙胺、吡啶中的至少一种;所述马来酸酐与有机碱的摩尔比为1:(2~10),示例性为1:2、1:4、1:6、1:8或1:10。
根据本发明,步骤1)中,所述卤化反应先在-10~0℃反应30min~2h,再在50~90℃加热反应30min~2h。
根据本发明,步骤1)还包括后处理步骤,所述后处理步骤包括:卤化反应结束后,除去反应体系的未反应的卤化试剂,并将剩余的固体采用甲苯溶解,除去副产物,蒸干得到纯化的中间产物。
根据本发明,步骤1)中,反应结束后,还包括蒸馏、分离、洗涤、干燥等后处理过程。
根据本发明,步骤2)中,所述醇类化合物为R
根据本发明,步骤2)中,所述催化剂选自浓硫酸、三氟甲磺酸、对甲基苯磺酸中的一种或多种。
根据本发明,步骤2)中,中间产物、醇类化合物、催化剂的质量体积比为(5-25)g:(30-50)mL:1mL,优选为(7-20)g:(35-45)mL:1mL。
根据本发明,步骤2)中,所述反应可以在溶剂中进行,所述溶剂为苯或甲苯;步骤2)中,反应溶液的浓度为0.1g/mL~5g/mL。
根据本发明,步骤2)中,反应结束后,还包括采用层析柱分离纯化等后处理过程。
本发明还提供上述2,3-双卤取代马来酸酯型内给电子体的应用,应用于催化剂体系中。
根据本发明,所述内给电子体用于烯烃聚合用催化剂体系;具体为固体催化剂体系。
本发明还提供一种烯烃聚合用催化剂固体组分,所述催化剂固体组分包括所述2,3-双卤取代马来酸酯型内给电子体。
根据本发明,所述催化剂固体组分中,以质量百分比为100%计,所述内给电子体的质量百分含量为7wt.%-10wt.%,优选为7wt.%-9wt.%。
根据本发明,所述催化剂固体组分还包括钛活性组分,所述钛活性组分的质量(以钛元素计算)百分数为0.5wt.%-2.5wt.%,优选为1.5wt.%-2.5wt.%。
根据本发明,所述钛活性组分为钛化合物,具有如下式(II)所示的结构:
TiX
其中X
p为0-4的整数;
R
示例性地,R
根据本发明,所述钛活性组分可以是四烷氧基钛,如四甲氧基钛、四乙氧基钛、四正丙氧基钛、四异丙氧基钛、四正丁氧基钛、四异丁氧基钛、四环己氧基钛和四苯氧基钛;四卤化钛,如四氯化钛、四溴化钛、四碘化钛;烷氧基三卤化钛,如甲氧基三氯化钛,乙氧基三氯化钛、正丙氧基三氯化钛、正丁氧基三氯化钛、和乙氧三溴化钛;二烷氧基二卤化钛,如二甲氧基二氯化钛、二乙氧基二氯化钛、二异丙氧基二氯化钛、二丙氧基二氯化钛和二乙氧基二溴化钛;三烷氧基单卤化钛,如三甲氧基氯化钛、三乙氧基氯化钛、三异丙氧基氯化钛、三正丙氧基氯化钛和三正丁氧基氯化钛等中的至少一种。其中,优选含卤素的钛化合物,尤其是四氯化钛。
根据本发明,所述催化剂固体组分的制备原料还可以包括卤化镁、一元醇化合物和钛酸酯。
根据本发明,所述卤化镁选自氯化镁,溴化镁和碘化镁中的一种,优选无水氯化镁。
根据本发明,所述一元醇化合物选自C
根据本发明,所述钛酸酯为四钛酸酯化合物,通式为Ti(OR)
本发明还提供一种催化剂体系,所述催化剂体系包括所述烯烃聚合用催化剂固体组分。
本发明还提供一种烯烃聚合的方法,所述方法采用上述催化剂体系。
根据本发明的优选技术方案,所述烯烃可以选自乙烯、丙烯、丁烯、1-己烯等α-烯烃。
根据本发明的另一优选技术方案,所述聚合包括均聚和共聚。
术语解释与说明
术语“C
术语“C
“C
术语“C
术语“C
本发明的有益效果
(1)采用本发明提供的2,3-双卤取代马来酸酯作为内给电子体,能够获得超高活性的聚丙烯催化剂(活性>36kg PP/(g.Cat.h)),相比于2,3-双烷基取代马来酸酯或者2,3-双芳基取代马来酸酯,催化剂的性能更为优异。
(2)采用廉价易得的马来酸酐为原料,经过两步反应即可高效实现得2,3-双卤取代马来酸酯的合成,方法简单易行,成本较低。
附图说明
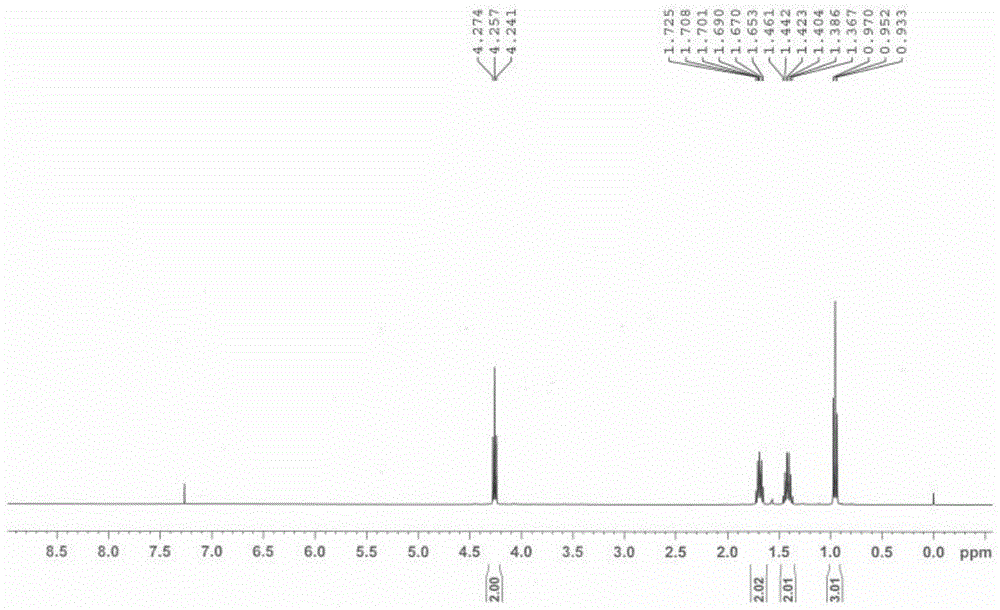
图1是实施例1中2,3-二氯马来酸二正丁酯的核磁氢谱图。
图2是实施例1中2,3-二氯马来酸二正丁酯的核磁碳谱图。
图3是实施例1中2,3-二氯马来酸二正丁酯的质谱图。
图4是实施例1中催化剂的SEM图。
具体实施方式
如上所述,本发明还提供上述烯烃聚合用催化剂固体组分的制备方法,所述方法包括:
(S1)醇合物溶液的制备:
将卤化镁、一元醇化合物、钛酸酯和式I所示的2,3-双卤取代马来酸酯型内给电子体在烷烃溶剂中混合、反应,得到稳定均匀的醇合物溶液;
(S2)催化剂固体组分的制备:
在惰性氛围下,将步骤(S1)中醇合物溶液与钛活性组分混合,当体系温度升为80~135℃,加入2,3-双卤取代马来酸酯型内给电子体反应,制得烯烃聚合用催化剂固体组分。
根据本发明,所述制备方法还包括步骤(S3),步骤(S2)反应结束后,滤出液体,再加入钛活性组分,并在80~135℃温度下继续反应,制得烯烃聚合用催化剂固体组分。本发明步骤(S3)中加入钛活性组分,可以提高催化剂固相组分中钛的含量。
根据本发明,步骤(S1)中,所述一元醇化合物、卤化镁和2,3-双卤取代马来酸酯型内给电子体的摩尔比为(1~10):1:(0.01~0.2),优选为(2~8):1:(0.05~0.15),更优选(3~6):1:(0.08~0.15)。
根据本发明,步骤(S1)中,钛酸酯和2,3-双卤取代马来酸酯型内给电子体的摩尔比为1:(1:5),示例性地为1:1。
根据本发明,步骤(S1)中,烷烃溶剂和卤化镁的比例为(0.5~20):1,单位为mL/g,优选(1~15):1,更优选(3~10):1。
根据本发明,步骤(S1)中,所述烷烃溶剂选自C
根据本发明,步骤(S1)中,反应温度为40~200℃,优选60~180℃,更优选80~150℃;反应时间为1~6小时。
根据本发明,步骤(S2)中加入的钛活性组分与步骤(S3)中加入的钛活性组分的体积比为(0.6~1):1。
根据本发明,步骤(S2)和步骤(S3)中,钛活性组分的加入总体积与醇合物溶液的体积比为(1~4):1。
根据本发明,步骤(S2)中,2,3-双卤取代马来酸酯型内给电子体的加入量与卤化镁摩尔比为(0.01~0.2):1,优选为(0.06-0.15):1。
根据本发明,步骤(S3)中,加入钛活性组分后,在80~135℃温度下继续反应1~4小时。
根据本发明,所述烯烃聚合用固体催化剂的制备方法中,两次加入的2,3-双卤取代马来酸酯型内给电子体的总量与卤化镁的摩尔比为(0.05~0.25):1。
根据本发明,步骤(S2)或步骤(S3)还包括后处理步骤:反应结束后滤出反应液,并采用己烷洗涤,干燥后得到催化剂固体组分。
作为本发明示例性的实施方案,所述烯烃聚合用催化剂固体组分的制备方法具体包括:
(S1)醇合物溶液的制备:
将卤化镁、一元醇化合物、钛酸酯和式I所示的2,3-双卤取代马来酸酯型内给电子体在烷烃溶剂中充分反应,其中一元醇化合物与卤化镁的摩尔比为(1~10):1;2,3-双卤取代马来酸酯型内给电子体和卤化镁的摩尔比为(0.01~0.2):1;钛酸酯用量和2,3-双卤取代马来酸酯型内给电子体摩尔量相等;烷烃溶剂和卤化镁的比例为(0.5~20):1(mL/g),在40~200℃反应1~6小时,得到稳定均匀醇合物溶液;
(S2)催化剂固体组分的制备:将上述制备的醇合物溶液加到经氮气充分置换,装有-25~40℃的钛活性组分的反应器中充分接触后,开始升温,当温度升至80~135℃时,加入与卤化镁摩尔比为(0.01~0.2):1的2,3-双卤取代马来酸酯型内给电子体,并在该温度下反应1~4小时,反应结束后,滤出液体,再加入钛活性组分,在80~135℃温度下继续反应1~4小时,过滤出液体,用溶剂洗涤,干燥,制得钛催化剂固体组分,其中,钛活性组分的加入总量和醇合物溶液的体积比为(1~4):1;
其中,两次加入的2,3-双卤取代马来酸酯型内给电子体的总量与卤化镁摩尔比为(0.05~0.25):1。
下文将结合具体实施例对本发明的技术方案做更进一步的详细说明。应当理解,下列实施例仅为示例性地说明和解释本发明,而不应被解释为对本发明保护范围的限制。凡基于本发明上述内容所实现的技术均涵盖在本发明旨在保护的范围内。
除非另有说明,以下实施例中使用的原料和试剂均为市售商品,或者可以通过已知方法制备。
1、实施例1-5和对比例1-2中,聚合活性通过如下方法进行计算:
聚合活性的单位为g PP/g Cat.h。
2、实施例1-5和对比例1-2中,聚丙烯的等规度采用正庚烷抽提法测定:使用沸腾的正庚烷抽提6小时,干燥后测定不溶物占总重量的百分比。
3、采用光电子能谱(EDS)对催化剂组分钛含量进行分析;采用气相色谱(GC)法对催化剂组分内给电子体含量进行分析。
实施例1
(1)2,3-二氯马来酸二正丁酯I-1的合成:
取250mL干燥好的三口烧瓶,加入9.8g(0.1mol)马来酸酐和16.6mL(0.2mol)吡啶,置换氮气;在冰水浴下加入100mL(1.38mol)氯化亚砜,在冰水浴下反应1小时,然后加热到75℃回流40分钟。后处理:蒸除未参与反应的氯化亚砜,将残留固体溶解到甲苯中,过滤除去副产物吡啶盐酸盐,蒸干溶剂获得中间产物10.9g,收率为66%。
取250mL单口烧瓶加入上述中间产物、40ml正丁醇和100mL甲苯,加入1mL浓硫酸作为催化剂,加热(例如120℃)回流分水,TLC点板监测反应,待原料完全转化后,反应结束。采用层析柱对产物进行分离纯化,获得2,3-二氯马来酸二正丁酯15.5g,收率为79%。
(2)催化剂的制备:将5g无水氯化镁、18.9g异辛醇、2.2g(7.4mmol)2,3-二氯马来酸二正丁酯、2.65g钛酸四丁酯和30mL干燥的癸烷加入到反应瓶中,在氮气保护下于130℃反应4小时,使无水氯化镁充分溶解,得到稳定均匀的醇合物溶液,将反应体系缓慢降至室温。将该溶液在1小时内滴加到经氮气充分置换且装有-20℃下200mL四氯化钛的反应器中。滴加完毕后,经过3.5小时升温110℃,加入1.3g(4.4mmol)2,3-二氯马来酸二正丁酯,在此温度下反应2小时。反应结束后过滤出液体,重新加入200ml四氯化钛,在110℃反应2小时。反应结束后滤出反应液,用干燥过的己烷洗涤6次,干燥后得到催化剂。
(3)丙烯聚合:加入20mL三乙基铝溶液到5L反应釜内进行除杂;再向反应釜内加入20mg上述催化剂、1.0mL二异丙基二甲氧基硅烷溶液(外给电子体中硅元素和钛活性组分中钛元素的摩尔比为20:1)和10mL三乙基铝溶液(Al/Ti=100,mol比);打开氢气进釜阀门,加入0.1MPa的氢气;随后加入1200g丙烯液体,开启搅拌(12Hz)和反应釜自动控温(反应温度为70℃,反应的下限温度为69.5℃,上限温度为70.5℃)。当反应温度首次达到68.5℃时,开始反应计时,反应时间为60min。停止反应釜自动控温,放空釜内没有反应完的丙烯,然后开釜放料,得到聚丙烯产品。实验结果见表1。
图1是实施例1中2,3-二氯马来酸二正丁酯的核磁氢谱图。
图2是实施例1中2,3-二氯马来酸二正丁酯的核磁碳谱图。
图3是实施例1中2,3-二氯马来酸二正丁酯的质谱图。
图4是实施例1中催化剂的SEM图。
实施例2
(1)2,3-二氯马来酸二异丁酯I-2的合成:
采用实施例1相同的方法,酸酐的醇解反应用异丁醇代替正丁醇进行反应,获得目标产物2,3-二氯马来酸二异丁酯,总收率为53%。
(2)丙烯聚合:采用实施例1相同的方法,以2,3-二氯马来酸二异丁酯替代2,3-二氯马来酸二正丁酯进行催化剂的制备,并进行丙烯聚合。实验结果见表1。
实施例3
(1)2,3-二溴马来酸二正丁酯I-3的合成:
采用实施例1相同的方法,采用溴替代氯化亚砜作为卤化试剂进行反应,获得目标产物2,3-二溴马来酸二正丁酯,总收率为51%。
(2)丙烯聚合:采用实施例1相同的方法,以2,3-二溴马来酸二正丁酯替代2,3-二氯马来酸二正丁酯进行催化剂的制备,并进行丙烯聚合。实验结果见表1。
实施例4
(1)2,3-二溴马来酸二异丁酯I-4的合成:
采用实施例3相同的方法,酸酐的醇解反应用异丁醇代替正丁醇进行反应,获得目标产物2,3-二溴马来酸二异丁酯,总收率为61%。
(2)丙烯聚合:采用实施例1相同的方法,以2,3-二溴马来酸二异丁酯替代2,3-二氯马来酸二正丁酯进行催化剂的制备,并进行丙烯聚合。实验结果见表1。
实施例5
(1)2-氯-3-溴马来酸二正丁酯I-5的合成:
采用实施例1相同的方法,依次采用溴(6.9mol)和氯化亚砜(7.0mol)作为卤化试剂进行反应,最终获得目标产物2-氯-3-溴马来酸二正丁酯,总收率为61%。
(2)丙烯聚合:采用实施例1相同的方法,以2-氯-3-溴马来酸二正丁酯I-5为内给电子体进行催化剂制备,并进行丙烯聚合。实验结果见表1。
对比例1
采用实施例1相同的方法,以中国专利CN201210480335.9公布的2,3-二异丁基马来酸二正丁酯(即式I-6)作为内给电子体进行催化剂制备,并进行丙烯聚合。实验结果见表1。
对比例2
采用实施例1相同的方法,以2,3-二苯基马来酸二正丁酯(即式I-7)作为内给电子体进行催化剂制备,并进行丙烯聚合。实验结果见表1。
表1实验结果
表1中,酯含量指内给电子体2,3-双卤取代马来酸酯的含量。
从表1可知,本发明的双卤取代马来酸酯是一种性能良好的内给电子体,采用该化合物能够制备性能优异的催化剂。当马来酸酯2,3-位为卤素取代(实施例1-5)时,相比于烷基取代(对比例1)和芳基取代(对比例2),催化剂具备更为优异的催化活性(>36kg PP/(g.Cat.h)),获得的聚丙烯具备更为优异的等规度(>99%)。催化剂的高活性实际表现为聚合物中催化剂残留物(灰分的主要组成之一)含量低,因此高活性的催化剂对于获得低灰分的聚合物有利。以上实验结果表明:卤素取代的马来酸酯相比其他取代(烷基取代和芳基取代),具备更高的活性,更有利于获得的低灰分聚丙烯产品。
由实施例1-5可知,卤素取代的马来酸酯,无论是双氯取代(实施例1和2),双溴取代(实施例3和4),还是两种卤素同时取代(实施例5),所获得的催化剂均能表现出极佳的催化活性,获得的聚丙烯表现出极高的等规度。
综上所述,本发明提供的2,3-双卤取代马来酸酯具有简易的合成路线、低廉的合成成本,该类化合物作为内给电子体使用时,催化剂的性能明显优于双烷基取代马来酸酯和双芳基取代马来酸酯类化合物,而且催化剂的活性可以大于36kg PP/(g.Cat.h),获得的聚丙烯的等规度大于99%。该类内给电子体及其催化剂具备良好的工业化应用前景。
以上,对本发明的实施方式进行了示例性的说明。但是,本发明的保护范围不拘囿于上述实施方式。凡在本发明的精神和原则之内,本领域技术人员所作出的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 由氯代马来酸酯或氯代富马酸酯或其混合物制备取代或无取代-2,3-吡啶二羧酸酯的方法
- 具有卤代‑丙二酸酯内电子给体的催化剂组合物和由其制备的聚合物