一种配合物及其制备方法和应用
文献发布时间:2024-04-18 19:58:26
技术领域
本发明涉及化工领域,具体的说,本发明涉及一种配合物及其制备方法和应用。
背景技术
1985年,日本的Ishihara等人首次成功合成出了间规聚苯乙烯,其熔点高达270℃,结晶速度极快(比等规聚苯乙烯高两个数量级)。间规聚苯乙烯除具有无规聚苯乙烯所有优点外,还拥有高弹性模量,高耐热性,强耐溶剂和耐化学试剂性等性能,可与工程塑料相媲美,而且拥有其它工程塑料所不具备的低密度和低介电常数等性能。在汽车工业的电器部件、家用电子产品和工业薄膜等领域具有广泛的应用前景。因此,间规聚苯乙烯的制备引起人们广泛的研究兴趣。
2004年,候召民教授课题组首次利用单茂稀土金属配合物催化苯乙烯聚合制备出了间规聚苯乙烯。2012年崔冬梅教授课题组开发出含有可配位氮原子、碳原子侧臂的限制几何构型稀土金属配合物,成功实现了苯乙烯的高间规选择性聚合。过去大量研究表明,催化体系中含有配位氧原子的Lewis碱,如四氢呋喃将导致稀土金属配合物催化苯乙烯间规聚合的活性大幅降低,甚至失活(Organometallics 2013,32,5,1445;ACS Catal.2016,6,176),因此制备稀土金属配合物时要选用配位数高、空间位阻大的配体,避免含有氧原子的Lewis碱配位。
Organometallics 2013,32,5,1445制备了一系列的半茂配体的稀土金属催化剂,催化体系中含有配位氧原子的Lewis碱,如氨基、四氢呋喃。ACS Catal.2016,6,176,合成了系列限制几何构型的稀土金属催化剂,结果发现随着稀土金属原子的尺寸减小,催化活性逐渐升高。上面所公开的技术均存在如下缺点:催化体系中含有配位氧原子的Lewis碱,导致稀土金属配合物催化苯乙烯间规聚合的活性大幅降低,甚至失活。
发明内容
本发明的一个目的在于提供一种配合物;
本发明的另一目的在于提供所述配合物的制备方法;
本发明的再一目的在于提供一种催化体系;
本发明的又一目的在于提供一种间规聚苯乙烯的制备方法;
为达上述目的,一方面,本发明提供了一种式(I)所示的配合物:
其中,
R
R
Ln为稀土元素;
R
E为C、Si或Ge;
Y为四氢呋喃、乙醚、乙二醇二甲醚或甲苯;
m=1或2;n=0、1或2。
根据本发明一些具体实施方案,其中,本发明所述具有式I结构的配合物为eta5配位的稀土配合物。
根据本发明一些具体实施方案,其中,式I所示的配合物是一种具有限制几何构型结构的化合物。
根据本发明一些具体实施方案,其中,R
根据本发明一些具体实施方案,其中,R
根据本发明一些具体实施方案,其中,R
根据本发明一些具体实施方案,其中,所述稀土元素为Sc、Y、La、Ce、Pr、Nd、Sm、Eu、Gd、Tb、Dy、Ho、Er、Tm、Yb或Lu。
根据本发明一些具体实施方案,其中,R
根据本发明一些具体实施方案,其中,R
根据本发明一些具体实施方案,其中,Ln选自Sc、Y、La、Nd、Gd、Tb、Dy、Ho、Er、Tm、Yb或Lu。
根据本发明一些具体实施方案,其中,Ln选自Sc、Y、La、Nd、Gd、Er、Tm或Lu。
根据本发明一些具体实施方案,其中,R
根据本发明一些具体实施方案,其中,R
根据本发明一些具体实施方案,其中,Y为四氢呋喃。
根据本发明一些具体实施方案,其中,n=0或1。
本发明中对上述取代基的选择没有其他特别限制,R
本发明对具有式II结构的环戊二烯基及其衍生物、具有式III结构的茚基及其衍生物和具有式IV结构的芴基及其衍生物没有特别限制,以本领域技术人员熟知的环戊二烯基及其衍生物、茚基及其衍生物或芴基及其衍生物即可。
本发明所述溶剂分子为配位在稀土配合物上。
本发明对上述具有式I结构的配合物中取代基的选取和组合没有特别限制。在不矛盾的前提下,各具体实施方案可以任意组合。
根据本发明一些具体实施方案,其中,式(I)所示的配合物选自:
其中,化合物1~化合物12所示的配合物为R4是烷硅或烷胺基的稀土配合物,化合物13~化合物17所示的配合物为R4是烯丙基的稀土配合物,化合物18~化合物20所示的配合物为R4是硼氢基的稀土配合物。
另一方面,本发明还提供了本发明任意一项所述式(I)所示的配合物的制备方法,其中,所述方法包括如下步骤:
(1)将具有式(V)结构的配体、RLi和有机溶剂混合后反应得到第一反应混合物;所述有机溶剂为四氢呋喃、乙醚、乙二醇二甲醚或甲苯;
(2)将步骤(1)得到的第一反应混合物和稀土卤化物进行反应得到第二反应混合物;
(3)将步骤(2)得到的第二反应混合物与含取代基化合物进行反应,得到式(I)所示的配合物;
其中,R
所述含取代基化合物选自含有C
根据本发明一些具体实施方案,其中,所述RLi的R选自甲基、乙基、丙基、异丙基、正丁基、仲丁基、叔丁基、硅氨基、二甲氨基、二乙胺基、二丙胺基、N,N-二甲氨基苯基、三甲硅基甲基、双三甲硅基甲基、邻-甲巯基苯基、邻-二甲膦基苯基、四氢硼基、甲氧基、乙氧基、异丙氧基、正丙氧基、正丁氧基仲丁氧基和叔丁氧基中的一种或多种的组合。
根据本发明一些具体实施方案,其中,所述RLi的R选自正丁基、N,N-二甲氨基苯基、三甲硅基甲基和双三甲硅基甲基中的一种或多种的组合。
根据本发明一些具体实施方案,其中,所述RLi的R为正丁基。
根据本发明一些具体实施方案,其中,
含有烯丙基的化合物为烯丙基格氏试剂或烯丙基衍生物格氏试剂;
和含有硼氢基的化合物
根据本发明一些具体实施方案,其中,所述烯丙基格氏试剂为C
根据本发明一些具体实施方案,其中,所述取代苯基为被C
根据本发明一些具体实施方案,其中,含有C
根据本发明一些具体实施方案,其中,
所述RLi与具有式V结构的配体的摩尔比为(1-1.2):1;
所述稀土卤化物与具有式V结构的配体的摩尔比为(1-1.2):1;
所述含取代基化合物与具有式V结构的配体的摩尔比为(2~2.4):1。
根据本发明一些具体实施方案,其中,所述RLi与具有式V结构的配体的摩尔比为(1:1.15):1。
根据本发明一些具体实施方案,其中,所述稀土卤化物与具有式V结构的配体的摩尔比为(1.05~1.15):1。
根据本发明一些具体实施方案,其中,所述含取代基化合物与具有式V结构的配体的摩尔比为(2~2.2):1。
根据本发明一些具体实施方案,其中,有机溶剂的体积与具有式(V)结构的配体的物质量的比例为(4~6)L:1mol。
根据本发明一些具体实施方案,其中,有机溶剂的体积与具有式(V)结构的配体的物质量的比例为(4.5~5.5)L:1mol。
根据本发明一些具体实施方案,其中,步骤(1)是在无水无氧的条件下进行反应。
根据本发明一些具体实施方案,其中,步骤(1)所述反应的时间为0.8~1.5小时;优选为0.8~1.2小时;更优选为1小时。
根据本发明一些具体实施方案,其中,步骤(1)所述反应的温度为-78℃~40℃;优选为-50℃~30℃;更优选为-10℃~20℃。
根据本发明一些具体实施方案,其中,步骤(1)的有机溶剂选自四氢呋喃、吡啶、正己烷或乙二醇二甲醚。
根据本发明一些具体实施方案,其中,步骤(1)的有机溶剂为四氢呋喃。
根据本发明一些具体实施方案,其中,步骤(2)是在第二有机溶剂的存在下将步骤(1)得到的第一反应混合物和稀土卤化物进行反应得到第二反应混合物。
根据本发明一些具体实施方案,其中,步骤(2)的第二有机溶剂选自四氢呋喃、吡啶、正己烷或乙二醇二甲醚。
根据本发明一些具体实施方案,其中,步骤(2)的第二有机溶剂为正己烷。
根据本发明一些具体实施方案,其中,步骤(2)的稀土卤化物为稀土的三氯化物。
根据本发明一些具体实施方案,其中,步骤(2)的反应的反应温度为10℃-40℃。
根据本发明一些具体实施方案,其中,步骤(2)的反应的反应时间为3-5小时。
根据本发明一些具体实施方案,其中,步骤(2)的反应的反应时间为3.5-4.5小时。
根据本发明一些具体实施方案,其中,步骤(2)的反应的反应时间为4小时。
根据本发明一些具体实施方案,其中,步骤(3)的反应的反应温度为10℃-40℃;优选为20-30℃。
根据本发明一些具体实施方案,其中,步骤(3)的反应的反应时间为10~14小时。
根据本发明一些具体实施方案,其中,步骤(3)的反应的反应时间为11~13小时。
根据本发明一些具体实施方案,其中,步骤(3)的反应的反应时间为12小时。
根据本发明一些具体实施方案,其中,步骤(3)包括将步骤(2)得到的第二反应混合物与含取代基化合物进行反应,反应完毕后,去除溶剂并用甲苯萃取和浓缩后,得到式(I)所示的配合物。
本发明对去除溶剂的方法没有特别限制,以本领域技术人员熟知的去除溶剂的方法即可;本发明对甲苯萃取的条件没有特别限制,以本领域技术人员熟知的甲苯萃取的条件即可;本发明对浓缩的方法没有特别限制,以本领域技术人员熟知的浓缩的方法即可。
本发明的配合物的制备方法简单,收率高达50~95%。
本发明对上述具有式V结构的配体的来源没有特别限制,以本领域技术人员熟知的合成方法制备即可,优选参照以下文献进行制备,(H.Miao,S.Wang,S.Zhou,Y.Wei,Z.Zhou,H.Zhu,S.Wu,H.Wang,Inorganica Chimica Acta,2010,363,1325-1331)。
再一方面,本发明还提供了一种催化体系,其中,所述催化体系至少含有本发明任意一项所述的配合物。
根据本发明一些具体实施方案,其中,所述催化体系含有本发明任意一项所述的配合物作为主催化剂、以及至少一种助催化剂。
根据本发明一些具体实施方案,其中,所述助催化剂选自铝氧烷化合物、或铝氧烷化合物和烷基铝化合物的组合物、或有机硼盐和烷基铝化合物的组合物。
根据本发明一些具体实施方案,其中,所述铝氧烷化合物为烷基铝氧烷。
根据本发明一些具体实施方案,其中,所述烷基铝氧烷的烷基为甲基、乙基、丙基、异丙基、正丁基、仲丁基或叔丁基。
根据本发明一些具体实施方案,其中,所述铝氧烷化合物为甲基铝氧烷、干燥的甲基铝氧烷或修饰的甲基铝氧烷。
根据本发明一些具体实施方案,其中,所述铝氧烷化合物为甲基铝氧烷或修饰的甲基铝氧烷。
本发明对所述干燥的甲基铝氧烷没有特别限制,以本领域技术人员熟知的关于干燥的甲基铝氧烷的定义即可,即甲基铝氧烷中不包含甲基铝。
本发明对所述修饰的甲基铝氧烷没有特别限制,以本领域技术人员熟知的关于修饰的甲基铝氧烷的定义即可,即甲基铝氧烷中包含异丁基铝。
根据本发明一些具体实施方案,其中,所述有机硼盐选自[Ph
根据本发明一些具体实施方案,其中,所述烷基铝化合物为烷基铝、氢化烷基铝或氯化烷基铝。
根据本发明一些具体实施方案,其中,所述有机硼盐与所述配合物的摩尔比为(0.5~10.0):1。
根据本发明一些具体实施方案,其中,所述有机硼盐与所述配合物的摩尔比为(1.0~5.0):1。
根据本发明一些具体实施方案,其中,所述有机硼盐与所述配合物的摩尔比为(1.0~3.0):1。
根据本发明一些具体实施方案,其中,所述烷基铝化合物与所述配合物的摩尔比为(1~2000):1。
根据本发明一些具体实施方案,其中,所述烷基铝化合物与所述配合物的摩尔比为(1~100):1。
根据本发明一些具体实施方案,其中,所述烷基铝化合物与所述配合物的摩尔比为(5~50):1。
根据本发明一些具体实施方案,其中,所述铝氧烷化合物(甲基铝氧烷)与所述配合物的摩尔比为(1~2000):1。
根据本发明一些具体实施方案,其中,所述铝氧烷化合物(甲基铝氧烷)与所述配合物的摩尔比为(1~100):1。
根据本发明一些具体实施方案,其中,所述铝氧烷化合物(甲基铝氧烷)与所述配合物的摩尔比为(5~50):1。
根据本发明一些具体实施方案,其中,所述烷基铝化合物选自烷基铝、氢化烷基铝和氯化烷基铝中的一种或多种的组合。
根据本发明一些具体实施方案,其中,烷基铝中的烷基为甲基、乙基、丙基、异丙基、正丁基、仲丁基或叔丁基。
根据本发明一些具体实施方案,其中,所述烷基铝化合物选自三甲基铝、三乙基铝、三正丙基铝、三正丁基铝、三异丙基铝、三异丁基铝、三戊基铝、三己基铝、三环己基铝、三辛基铝、三苯基铝、三对甲苯基铝、三苄基铝、乙基二苄基铝、乙基二对甲苯基铝、二乙基苄基铝、二甲基氢化铝、二乙基氢化铝、二正丙基氢化铝、二正丁基氢化铝、二异丙基氢化铝、二异丁基氢化铝、二戊基氢化铝、二己基氢化铝、二环己基氢化铝、二辛基氢化铝、二苯基氢化铝、二对甲苯基氢化铝、二苄基氢化铝、乙基苄基氢化铝、乙基对甲苯基氢化铝、二甲基氯化铝、二乙基氯化铝、二正丙基氯化铝、二正丁基氯化铝、二异丙基氯化铝、二异丁基氯化铝、二戊基氯化铝、二己基氯化铝、二环己基氯化铝、二辛基氯化铝、二苯基氯化铝、二对甲苯基氯化铝、二苄基氯化铝、乙基苄基氯化铝、乙基对甲苯基氯化铝、甲基铝氧烷、乙基铝氧烷、正丙基铝氧烷和正丁基铝氧烷中的一种或多种的组合。
根据本发明一些具体实施方案,其中,所述烷基铝化合物选自甲基铝、三甲基铝、三乙基铝、三异丁基铝、甲基铝氧烷、氢化二异丁基铝和氯化二乙基铝中的一种或多种的组合。
又一方面,本发明还提供了一种间规聚苯乙烯的制备方法,其中,所述制备方法包括以苯乙烯作为反应单体,在本发明任意一项所述的催化体系存在下进行聚合反应,得到所述间规聚苯乙烯。
根据本发明一些具体实施方案,其中,所述苯乙烯与所述配合物的摩尔比为(200-10000):1。
根据本发明一些具体实施方案,其中,所述苯乙烯与所述配合物的摩尔比为(500~8000):1。
根据本发明一些具体实施方案,其中,所述聚合反应的温度为-60℃-80℃。
根据本发明一些具体实施方案,其中,所述聚合反应的温度为-30℃-50℃。
根据本发明一些具体实施方案,其中,聚合反应的时间为1-30min。
根据本发明一些具体实施方案,其中,聚合反应的时间为1-10min。
根据本发明一些具体实施方案,其中,所述制备方法包括以苯乙烯作为反应单体,在本发明任意一项所述的催化体系存在下,在有机溶剂中进行聚合反应,得到所述间规聚苯乙烯。
根据本发明一些具体实施方案,其中,制备间规聚苯乙烯所用的有机溶剂为C
根据本发明一些具体实施方案,其中,制备间规聚苯乙烯所用的有机溶剂为戊烷、己烷、甲苯或二甲苯。
根据本发明一些具体实施方案,其中,所述制备方法包括先将所述催化体系和有机溶剂混合,形成催化体系溶液,再加入反应单体进行聚合反应。
根据本发明一些具体实施方案,其中,所述催化体系溶液中,具有式I结构的配合物的摩尔浓度为0.2mmol/L-2.0mmol/L。
根据本发明一些具体实施方案,其中,所述催化体系溶液中,具有式I结构的配合物的摩尔浓度为0.5mmol/L-1.8mmol/L。
根据本发明一些具体实施方案,其中,所述聚合反应是在无水无氧条件下进行的。
本发明的聚合用催化体系催化苯乙烯间规聚合反应具有可控聚合的特征
本发明制备间规聚苯乙烯时采用本发明提供的配合物和一种或一种以上助催化剂的催化剂组合,配合物由于存在茂环(环戊二烯基及其衍生物、茚基及其衍生物和芴基及其衍生物)的eta5配位方式以及含氮芳香环的吸电子作用,并且还存在氧原子配位到中心金属,对中心金属空间具有限制作用。因此,在催化间规苯乙烯聚合时可以使苯乙烯单体选择性的插入,并且通过改变加料量,来获得具有不同分子量的间规聚苯乙烯。
实验结果表明,本发明提供的间规苯乙烯聚合物制备方法,在制备间规聚苯乙烯过程中,单体转化率最高可达100%,活性最高可达19980kgmol
本发明对所述无水无氧的条件没有特别限制,以本领域技术人员熟知的无水无氧的条件即可,在本发明中优选采用通入惰性气体或氮气的方法来得到无氧的条件,更优选为通入氮气来得到无氧的条件。
本发明所述的制备方法制备得到的间规聚苯乙烯的间规度为80-100%。
根据本发明一些具体实施方案,其中,所述间规聚苯乙烯的数均分子量为8.4万-93.5万。
根据本发明一些具体实施方案,其中,所述间规聚苯乙烯的分子量分布最低可达1.21。
根据本发明一些具体实施方案,其中,所述间规聚苯乙烯的熔点为266-273℃。
综上所述,本发明提供了一种配合物及其制备方法和应用。本发明的技术方案具有如下优点:
本发明提供了一种间规聚苯乙烯的催化剂,是一种具有式I结构的稀土配合物,在助催化剂和有机溶剂的作用下,可将苯乙烯单体进行聚合反应,得到间规聚苯乙烯。
间规聚苯乙烯的合成,市场需求巨大,目前,国内外无规聚苯乙烯类产品产能已经严重过剩,无规聚苯乙烯产品技术门槛低,附加值低,如果企业要生产高附加值的产品,就必须进行技术升级,寻找具有市场前景的新型聚合物品种.本发明提供的间规苯乙烯聚合物制备方法,在制备间规聚苯乙烯过程中,单体转化率最高可达100%,间规度(rrrr)最高可达100%,聚合物数均分子量随着苯乙烯与催化剂投料比的改变在8.4~93.5万之间可调,分子量分布最低可达1.21,熔点在266~273℃范围内。
附图说明
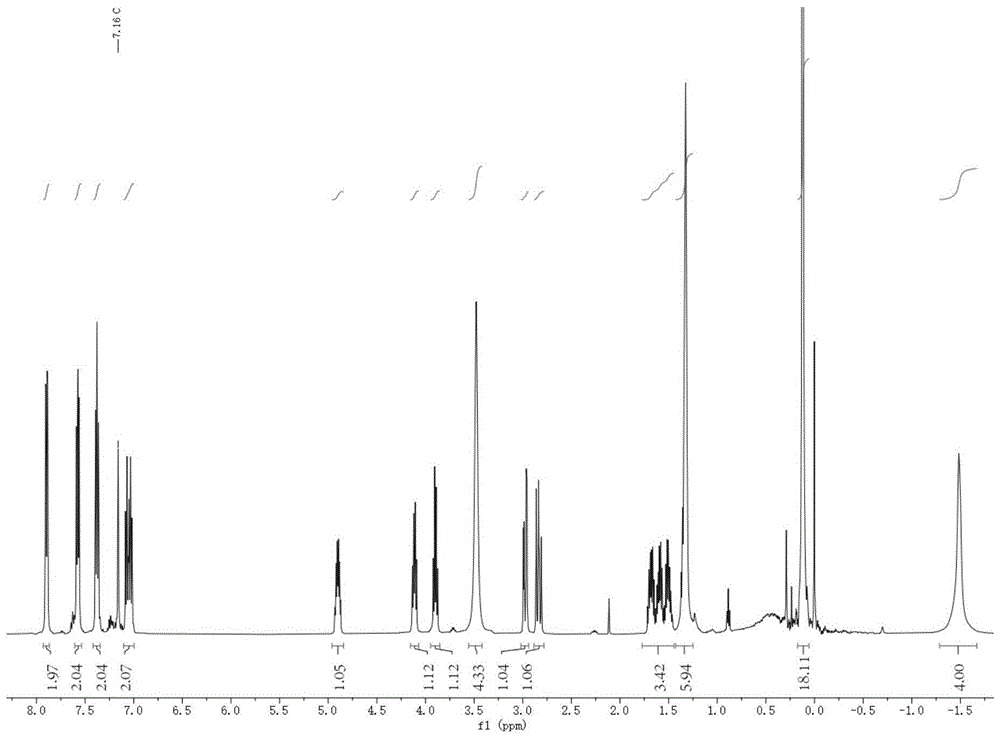
图1为本发明制备得到的具有式(I)结构的配合物的核磁共振氢谱图。
具体实施方式
以下通过具体实施例详细说明本发明的实施过程和产生的有益效果,旨在帮助阅读者更好地理解本发明的实质和特点,不作为对本案可实施范围的限定。
配合物制备实施例
各实施例的配合物结构如下表1所示:
表1
上表1各配合物的制备方法为:
配合物1所示稀土配合物的制备
在无水无氧的条件下,将式V(本发明对上述具有式V结构的配体的来源没有特别限制,以本领域技术人员熟知的合成方法制备即可,优选参照以下文献进行制备,(H.Miao,S.Wang,S.Zhou,Y.Wei,Z.Zhou,H.Zhu,S.Wu,H.Wang,Inorganica Chimica Acta,2010,363,1325-1331))所示R
配合物2所示稀土配合物的制备
在无水无氧的条件下,将式V所示R
配合物3所示稀土配合物的制备
在无水无氧的条件下,将式V所示R
配合物4所示稀土配合物的制备
在无水无氧的条件下,将式V所示R
配合物5所示稀土配合物的制备
在无水无氧的条件下,将式V所示R
对上述具有配合物5结构的稀土配合物进行核磁共振氢谱分析,如图1所示。
配合物6所示稀土配合物的制备
在无水无氧的条件下,将式V所示R
配合物7所示稀土配合物的制备
在无水无氧的条件下,将式V所示R
配合物8所示稀土配合物的制备
在无水无氧的条件下,将式V所示R
配合物9所示稀土配合物的制备
在无水无氧的条件下,将式V所示R
配合物10所示稀土配合物的制备
在无水无氧的条件下,将式V所示R
配合物11所示稀土配合物的制备
在无水无氧的条件下,将式V所示R
配合物12所示稀土配合物的制备
在无水无氧的条件下,将式V所示R
配合物13所示稀土配合物的制备
在无水无氧的条件下,将式V所示R
配合物14所示稀土配合物的制备
在无水无氧的条件下,将式V所示R
配合物15所示稀土配合物的制备
在无水无氧的条件下,将式V所示R
配合物16所示稀土配合物的制备
在无水无氧的条件下,将式V所示R
配合物17所示稀土配合物的制备
在无水无氧的条件下,将式V所示R
配合物18所示稀土配合物的制备
在无水无氧的条件下,将式V所示R
配合物19所示稀土配合物的制备
在无水无氧的条件下,将式V所示R
配合物20所示稀土配合物的制备
在无水无氧的条件下,将式V所示R
催化体系溶液实施例:
催化体系溶液1的制备:25℃下,向25ml经无水、无氧处理的聚合容器中加入10μmol配合物1所示的稀土配合物、10μmol[Ph
催化体系溶液2的制备:25℃下,向25ml经无水、无氧处理的聚合容器中加入10μmol配合物2所示的稀土配合物、10μmol[Ph
催化体系溶液3的制备:0℃下,向25ml经无水、无氧处理的聚合容器中加入10μmol配合物2所示的稀土配合物、10μmol[Ph
催化体系溶液4的制备:-60℃下,向25ml经无水、无氧处理的聚合容器中加入10μmol配合物2所示的稀土配合物、10μmol[Ph
催化体系溶液5的制备:40℃下,向25ml经无水、无氧处理的聚合容器中加入10μmol配合物2所示的稀土配合物、10μmol[Ph
催化体系溶液6的制备:80℃下,向25ml经无水、无氧处理的聚合容器中加入10μmol配合物2所示的稀土配合物、10μmol[Ph
催化体系溶液7的制备:25℃下,向25ml经无水、无氧处理的聚合容器中加入10μmol配合物2所示的稀土配合物、20μmol干燥的甲基铝氧烷、100μmol三甲基铝以及二甲苯溶剂,催化体系溶液中稀土配合物的浓度为1.0mmol·L
催化体系溶液8的制备:25℃下,向25ml经无水、无氧处理的聚合容器中加入10μmol配合物3所示的稀土配合物、10μmol[PhNHMe
催化体系溶液9的制备:-40℃下,向50ml经无水、无氧处理的聚合容器中加入10μmol配合物4所示的稀土配合物、10μmol[Ph
催化体系溶液10的制备:25℃下,向250ml经无水、无氧处理的聚合容器中加入10μmol配合物5所示的稀土配合物、100μmol甲基铝氧烷、100μmol三乙基铝以及甲苯溶剂,催化体系溶液中稀土配合物的浓度为1.0mmol·L
催化体系溶液11的制备:25℃下,向50ml经无水、无氧处理的聚合容器中加入10μmol配合物5所示的稀土配合物、10μmol[Ph
催化体系溶液12的制备:60℃下,向25ml经无水、无氧处理的聚合容器中加入10μmol配合物6所示的稀土配合物、10μmol[PhNHMe
催化体系溶液13的制备:0℃下,向100ml经无水、无氧处理的聚合容器中加入10μmol配合物7所示的稀土配合物、20μmol的甲基铝氧烷、以及戊烷溶剂,催化体系溶液中稀土配合物的浓度为0.2mmol·L
催化体系溶液14的制备:25℃下,向50ml经无水、无氧处理的聚合容器中加入10μmol配合物8所示的稀土配合物、1000μmol的甲基铝氧烷、300μmol三甲基铝以及己烷溶剂,催化体系溶液中稀土配合物的浓度为0.2mmol·L
催化体系溶液15的制备:40℃下,向50ml经无水、无氧处理的聚合容器中加入10μmol配合物9所示的稀土配合物、50μmol三甲基铝氧烷、10mmol的三异丁基铝以及己烷溶剂,催化体系溶液中稀土配合物的浓度为0.25mmol·L
催化体系溶液16的制备:25℃下,向50ml经无水、无氧处理的聚合容器中加入10μmol配合物10所示的稀土配合物、10μmol[Ph
催化体系溶液17的制备:0℃下,向50ml经无水、无氧处理的聚合容器中加入10μmol配合物11所示的稀土配合物、20μmol[PhNHMe
催化体系溶液18的制备:80℃下,向50ml经无水、无氧处理的聚合容器中加入10μmol配合物12所示的稀土配合物、10μmol B(C
催化体系溶液19的制备:25℃下,向50ml经无水、无氧处理的聚合容器中加入10μmol配合物13所示的稀土配合物、10mmol甲基铝氧烷、500μmol三乙基铝以及二甲苯溶剂,催化体系溶液中稀土配合物的浓度为0.33mmol·L
催化体系溶液20的制备:0℃下,向50ml经无水、无氧处理的聚合容器中加入10μmol配合物14所示的稀土配合物、10μmol[Ph
催化体系溶液21的制备:40℃下,向50ml经无水、无氧处理的聚合容器中加入10μmol配合物15所示的稀土配合物、10μmol[PhNHMe
催化体系溶液22的制备:25℃下,向25ml经无水、无氧处理的聚合容器中加入10μmol配合物16所示的稀土配合物、10μmol B(C
催化体系溶液23的制备:–60℃下,向50ml经无水、无氧处理的聚合容器中加入10μmol配合物17所示的稀土配合物、10μmol[Ph
催化体系溶液24的制备:80℃下,向100ml经无水、无氧处理的聚合容器中加入10μmol配合物17所示的稀土配合物、10μmol[Ph
催化体系溶液25的制备:0℃下,向50ml经无水、无氧处理的聚合容器中加入10μmol配合物18所示的稀土配合物、100μmol修饰的甲基铝氧烷、5mmol三甲基铝以及戊烷溶剂,催化体系溶液中稀土配合物的浓度为0.2mmol·L
催化体系溶液26的制备:25℃下,向50ml经无水、无氧处理的聚合容器中加入10μmol配合物18所示的稀土配合物、100μmol修饰的甲基铝氧烷、5mmol三甲基铝以及己烷溶剂,催化体系溶液中稀土配合物的浓度为0.2mmol·L
催化体系溶液27的制备:80℃下,向25ml经无水、无氧处理的聚合容器中加入10μmol配合物18所示的稀土配合物、10μmol[Ph
催化体系溶液28的制备:60℃下,向25ml经无水、无氧处理的聚合容器中加入10μmol配合物18所示的稀土配合物、10μmol BPh
催化体系溶液29的制备:25℃下,向25ml经无水、无氧处理的聚合容器中加入10μmol配合物19所示的稀土配合物、20μmol[Ph
催化体系溶液30的制备:40℃下,向25ml经无水、无氧处理的聚合容器中加入10μmol配合物20所示的稀土配合物、100μmol干燥的甲基铝氧烷、500μmol氢化二异丁基铝以及甲苯溶剂,催化体系溶液中稀土配合物的浓度为0.5mmol·L
催化体系溶液31的制备:0℃下,向50ml经无水、无氧处理的聚合容器中加入10μmol配合物20所示的稀土配合物、10μmol[Ph
催化体系溶液32的制备:-60℃下,向50ml经无水、无氧处理的聚合容器中加入10μmol配合物20所示的稀土配合物、10μmol[PhNHMe
苯乙烯聚合实施例:
聚合实施例1
取催化体系溶液1 5ml,置于经过无水、无氧处理的聚合瓶中,加入20mmol苯乙烯单体,聚合反应在25℃下进行5分钟,加入2ml体积浓度为10%盐酸的乙醇溶液终止聚合反应,将反应溶液倒入100ml甲醇中沉降,得到间规聚苯乙烯;再将得到的该聚合物置于真空干燥箱中干燥48小时,得到干燥恒重的间规聚苯乙烯,净重2.08g。总转化率1000%。计算聚合活性为2496·(mol
聚合实施例2–32
为所述的本发明提供的配位催化体系在间规苯乙烯聚合中的实施例。其步骤同聚合实施例1,具体的条件和所得的结果如表2所示:
表2配位聚合方法合成间规苯乙烯
/>
从聚合实施例1–32的聚合数据,可以得出:通过配位聚合方法,由本发明提供催化体系催化苯乙烯聚合反应可以实现苯乙烯的高活性、高间规聚合,活性范围在520kgmol
本发明提供的一种具有式I结构的配合物,该配合物合成方法简单,收率高达50~95%。本发明还提供一种聚合用催化体系及其制备方法,该催化体系由上述稀土配合物、有机硼盐和烷基化试剂按摩尔比1∶(1~2)∶(2~1000)组成,与现有技术相比,该聚合用催化体系催化苯乙烯间规聚合反应具有可控聚合的特征。另外,本发明还提供一种间规苯乙烯的制备方法。制备间规聚苯乙烯时采用本发明提供的包括eta5配位的稀土配合物和一种或一种以上助催化剂的催化剂组合,稀土配合物由于存在茂环(环戊二烯基及其衍生物、茚基及其衍生物和芴基及其衍生物)的eta5配位方式以及含氮芳香环的吸电子作用,并且还存在氧原子配位到中心金属,对中心金属空间具有限制作用。因此,在催化间规苯乙烯聚合时可以使苯乙烯单体选择性的插入,并且通过改变加料量,来获得具有不同分子量的间规聚苯乙烯。实验结果表明,本发明提供的间规苯乙烯聚合物制备方法,在制备间规聚苯乙烯过程中,单体转化率最高可达100%,活性最高可达19980kgmol
- 一种卟啉萘菁双层金属配合物及其制备方法和应用
- 一种稳定的多孔钴配合物及其制备方法和在吸附、分离气体小分子中的应用
- 一种氨基亚胺铱配合物及其制备方法和应用
- 一种两性离子型半三明治铱配合物及其制备方法和应用
- 一种锆基金属有机配合物UiO-66/泡沫炭复合材料的制备方法和应用
- 一类取代高哌嗪氨荒酸铋配合物、配合物的可药用盐以及配合物的制备方法和抗肿瘤应用
- 一类取代高哌嗪氨荒酸铋配合物、配合物的可药用盐以及配合物的制备方法和抗肿瘤应用