镍掺杂氮碳纳米材料及其制备方法和应用
文献发布时间:2024-04-18 19:58:26
技术领域
本发明涉及纳米材料技术领域,具体地,涉及一种镍掺杂氮碳纳米材料及其制备方法和应用。
背景技术
随着工业化进程的推进,人类对化石能源的依赖导致了CO
在CO
金、银催化剂在CO
因此,设计出一种非贵金属催化剂代替贵金属催化剂来催化CO
发明内容
本发明的目的是提供一种镍掺杂氮碳纳米材料及其制备方法和应用,该镍掺杂氮碳纳米材料作为电催化剂对催化二氧化碳还原为一氧化碳表现出极高的活性和选择性,并且该镍掺杂氮碳纳米材料的制备方法操作简单,易于控制,耗时短,成本低,该镍掺杂氮碳纳米材料在二氧化碳的还原反应中具有广泛的pH适用性,为二氧化碳电化学还原体系提供了一种新途径。
为了实现上述目的,本发明第一方面提供了一种镍掺杂氮碳纳米材料,该镍掺杂氮碳纳米材料包括:节状碳氮纳米管以及负载在所述节状碳氮纳米管上的镍纳米颗粒;
所述镍掺杂氮碳纳米材料中镍元素、碳元素、氮元素和氧元素的质量比为1:2-5:0.1-0.3:0.5-1。
本发明第二方面提供了一种如第一方面所述的镍掺杂氮碳纳米材料的制备方法,该制备方法包括:
1)将三聚氰胺溶液和三聚氰酸溶液混合,得到三聚氰胺-三聚氰酸超分子;
2)将所述三聚氰胺-三聚氰酸超分子与镍源混合,得到前驱体材料;
3)在保护性气体的存在下,将所述前驱体材料进行煅烧。
本发明第三方面提供了一种上述的镍掺杂氮碳纳米材料或上述制备方法所制备的镍掺杂氮碳纳米材料在二氧化碳还原中的应用。
本发明第四方面提供了一种二氧化碳还原为一氧化碳的方法,该方法包括:采用三电极体系,将催化剂负载在碳纸上作为工作电极,Ag/AgCl电极作为参比电极,钛网电极作为对电极,在流动电解池中,通入流动的二氧化碳和电解液进行二氧化碳还原反应;
其中,所述催化剂为第一方面所述的镍掺杂氮碳纳米材料或第二方面所述的制备方法所制备的镍掺杂氮碳纳米材料。
在上述技术方案中,本发明的镍掺杂氮碳纳米材料包括节状碳氮纳米管和负载在节状碳氮纳米管上的镍纳米颗粒,这使得该材料对于CO
进一步,本发明通过一步煅烧法制备镍掺杂氮碳纳米材料,制备方法简单方便,易于控制,反应周期短,成本低。
本发明制备的镍掺杂氮碳纳米材料在二氧化碳还原领域应用,表现出对CO的高选择性,可达90%以上,同时,该材料还具有广泛的pH适用性,可以在宽的pH范围内保持优异的催化活性。
本发明的其他特征和优点将在随后的具体实施方式部分予以详细说明。
附图说明
附图是用来提供对本发明的进一步理解,并且构成说明书的一部分,与下面的具体实施方式一起用于解释本发明,但并不构成对本发明的限制。在附图中:
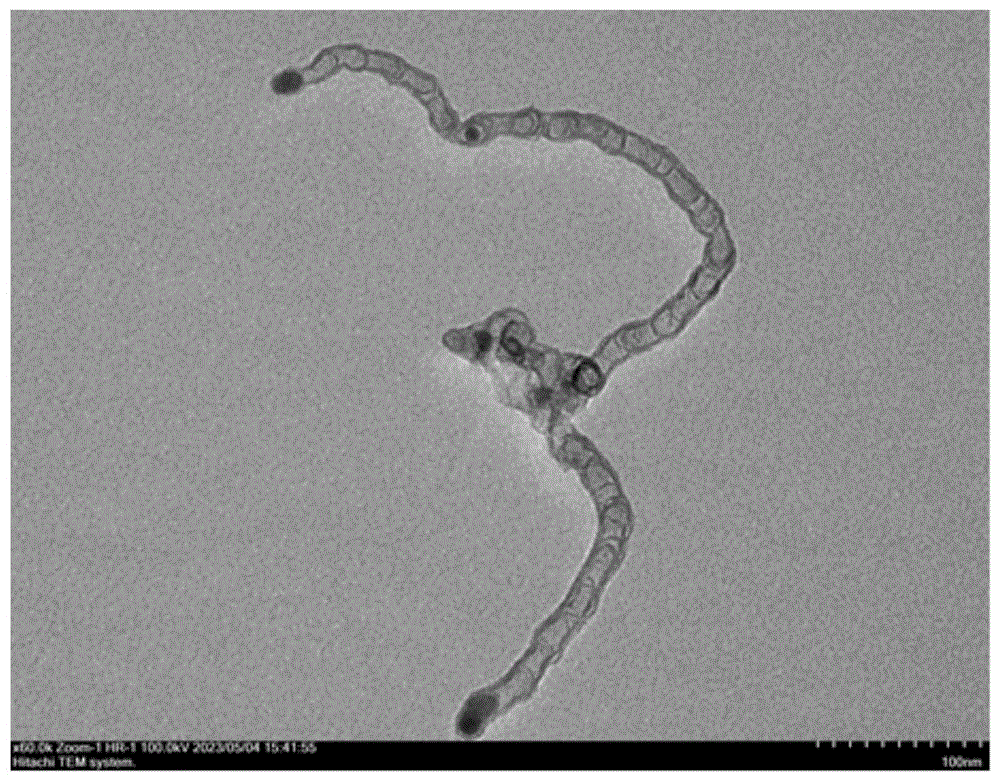
图1是实施例1制得的镍掺杂氮碳纳米材料B1放大60000倍的透射电子显微(TEM)图;
图2是实施例1制得的镍掺杂氮碳纳米材料B1放大30000倍的TEM图;
图3是对比例1制得的铁掺杂氮碳纳米材料D1放大40000倍的TEM图;
图4是对比例2制得的钴掺杂氮碳纳米材料D2放大30000倍的TEM图;
图5是实施例1制得的镍掺杂氮碳纳米材料B1的EDS能谱图;
图6是实施例1制得的镍掺杂氮碳纳米材料B1、对比例1制得的铁掺杂氮碳纳米材料D1和对比例2制得的钴掺杂氮碳纳米材料的X射线衍射图谱;
图7是实施例1制得的镍掺杂氮碳纳米材料进行电化学测试,在碱性条件下得到的二氧化碳还原反应性能图;
图8是实施例1制得的镍掺杂氮碳纳米材料进行电化学测试,在中性条件下得到的二氧化碳还原反应性能图;
图9是实施例1制得的镍掺杂氮碳纳米材料进行电化学测试,在pH为2酸性条件下得到的二氧化碳还原反应性能图;
图10是对比例1制得的铁掺杂氮碳纳米材料进行电化学测试,在碱性条件下得到的二氧化碳还原反应性能图;
图11是对比例2制得的掺杂氮碳纳米材料进行电化学测试,在碱性条件下得到的二氧化碳还原反应性能图。
具体实施方式
以下对本发明的具体实施方式进行详细说明。应当理解的是,此处所描述的具体实施方式仅用于说明和解释本发明,并不用于限制本发明。
在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。
本发明第一方面提供了一种镍掺杂氮碳纳米材料,该镍掺杂氮碳纳米材料包括:节状碳氮纳米管以及负载在节状碳氮纳米管上的镍纳米颗粒;
所述镍掺杂氮碳纳米材料中镍元素、碳元素、氮元素和氧元素的质量比为1:2-5:0.1-0.3:0.5-1。
本发明的镍掺杂氮碳纳米材料制备的工作电极对二氧化碳还原为一氧化碳提供了极大的促进作用,表现出很高的转化效率和选择性。
在本发明一种优选的实施方式中,所述镍纳米颗粒的尺寸为15-30nm。
在本发明一种优选的实施方式中,所述节状碳氮纳米管的表面粗糙。
本发明第二方面提供了一种如上述的镍掺杂氮碳纳米材料的制备方法,该制备方法包括:
1)将三聚氰胺溶液和三聚氰酸溶液混合,得到三聚氰胺-三聚氰酸超分子;
2)将所述三聚氰胺-三聚氰酸超分子与镍源混合,得到前驱体材料;
3)在保护性气体的存在下,将所述前驱体材料进行煅烧。
本发明通过简单的制备方法得到镍掺杂氮碳纳米材料,该制备方法对环境友好,易于控制,反应周期短,产率高,成本低,产品质量稳定。
在本发明一种优选的实施方式中,为了使制得的三聚氰胺-三聚氰酸超分子更好的提供氮化碳分子,在步骤1)中,所述三聚氰胺溶液中的三聚氰胺与三聚氰酸溶液中的三聚氰酸摩尔比为1:1。
在本发明一种优选的实施方式中,为了使三聚氰胺溶液和三聚氰酸溶液快速稳定的发生反应,在步骤1)中,所述三聚氰胺溶液的浓度为0.02-0.1mol/L。
在本发明一种优选的实施方式中,为了使三聚氰胺溶液和三聚氰酸溶液快速稳定的发生反应,在步骤1)中,所述三聚氰胺溶液的浓度为0.04-0.08mol/L。
优选地,所述三聚氰胺溶液的浓度为0.02mol/L、0.04mol/L、0.06mol/L、0.08mol/L、0.1mol/L。
进一步优选地,所述三聚氰胺溶液的浓度为0.04mol/L、0.06mol/L、0.08mol/L。
在步骤1)中,所述三聚氰酸溶液的溶剂可以是本领域使用的常规溶剂,例如可以为水、乙醇和乙二醇中的一种或两种以上。
在本发明一种优选的实施方式中,所述三聚氰胺溶液的配制条件包括:将溶剂与三聚氰胺混合在80-90℃下加热搅拌形成澄清透明溶液。
在本发明一种优选的实施方式中,所述三聚氰胺溶液的配制条件包括:将溶剂与三聚氰胺混合在83-86℃下加热搅拌形成澄清透明溶液。
在本发明一种优选的实施方式中,为了使三聚氰胺溶液和三聚氰酸溶液快速稳定的发生反应,以得到所需三聚氰胺-三聚氰酸超分子,在步骤1)中,所述三聚氰酸溶液的浓度为0.02-0.1mol/L。
在本发明一种优选的实施方式中,为了使三聚氰胺溶液和三聚氰酸溶液快速稳定的发生反应,在步骤1)中,所述三聚氰酸溶液的浓度为0.04-0.08mol/L。
优选地,所述三聚氰酸溶液的浓度为0.02mol/L、0.04mol/L、0.06mol/L、0.08mol/L、0.1mol/L。
进一步优选地,所述三聚氰酸溶液的浓度为0.04mol/L、0.06mol/L、0.08mol/L。
在步骤1)中,所述三聚氰酸溶液的溶剂可以是本领域使用的常规溶剂,例如可以为水、乙醇和乙二醇中的一种或两种以上。
在本发明一种优选的实施方式中,所述三聚氰酸溶液的配制条件包括:将溶剂与三聚氰酸混合在80-90℃下加热搅拌形成澄清透明溶液。
在本发明一种优选的实施方式中,所述三聚氰酸溶液的配制条件包括:将溶剂与三聚氰酸混合在83-86℃下加热搅拌形成澄清透明溶液。
在本发明一种优选的实施方式中,为了使三聚氰胺溶液和三聚氰酸溶液可以自组装形成所需超分子,在步骤1)中,所述混合的条件包括:转速为300-1000rpm,温度为80-100℃,时间为3-8h。
在本发明一种优选的实施方式中,为了使三聚氰胺溶液和三聚氰酸溶液可以自组装形成所需超分子,在步骤1)中,所述混合的条件包括:转速为500-800rpm,温度为82-90℃,时间为4-6h。
在本发明一种优选的实施方式中,所述在步骤1)还包括,将所述混合后的反应物冷却至25-40℃,去除溶液接着将固体产物进行清洗、真空干燥。
进一步优选地,所述清洗分别采用超纯水和乙醇离心、抽滤、洗涤3-5次。
在本发明一种优选的实施方式中,所述真空干燥的条件可以在宽的范围内选择,例如,所述真空干燥的温度为60-100℃,时间为10-15h。
在本发明一种优选的实施方式中,为了使制得的镍掺杂氮碳纳米材料上镍纳米颗粒更好的附着,在步骤2)中,所述镍源选自乙酰丙酮镍、硝酸镍、氯化镍和硫酸镍中的一种或两种以上。
在本发明一种优选的实施方式中,为了使制得的镍掺杂氮碳纳米材料拥有更为优异的催化活性,所述三聚氰胺-三聚氰酸超分子和镍源的质量比为2:0.010-0.025,其中,镍源以镍离子计。
优选地,所述三聚氰胺-三聚氰酸超分子和镍源的质量比为2:0.011、2:0.015、2:0.018、2:0.020和2:0.023。
在本发明一种优选的实施方式中,步骤2)中,所述第二混合的条件可以在宽的范围内选择,例如,所述混合的方式为研磨。
优选地,所述研磨的时间为0.5-2h,研磨的温度为15-30℃。
在本发明一种优选的实施方式中,为了在煅烧阶段不引入新的杂质,且保护产品不被氧化,在步骤3)中,所述保护性气体为氩气或氮气。
在本发明一种优选的实施方式中,为了使镍元素以镍纳米颗粒的形式附着在节状碳氮纳米管上,使镍掺杂氮碳纳米材料拥有更为优异的催化活性,在步骤3)中,所述煅烧的条件包括:升温速率为3-7℃/min,温度为800-950℃,时间为1-5h。
在本发明一种优选的实施方式中,为了使制得的镍掺杂氮碳纳米材料拥有更为优异的催化活性,在步骤3)中,所述煅烧的条件还包括:将煅烧产物进行研磨10-30min。
本发明第三方面提供了一种上述的镍掺杂氮碳纳米材料或上述制备方法所制备的镍掺杂氮碳纳米材料在二氧化碳还原中的应用。
本发明第四方面提供了一种二氧化碳还原为一氧化碳的方法,该方法包括:采用三电极体系,将催化剂负载在碳纸上作为工作电极,Ag/AgCl电极作为参比电极,钛网电极作为对电极,在流动电解池中,通入流动的二氧化碳和电解液进行二氧化碳还原反应;
其中,所述催化剂为上述的镍掺杂氮碳纳米材料或上述的制备方法所制备的镍掺杂氮碳纳米材料。
在本发明一种优选的实施方式中,为了保证催化剂制备的工作电极可以正常工作,提供更好的催化性能,所述电解液的pH为2-14。
在本发明一种优选的实施方式中,为了保证电解质中电子的传输效率,所述电解液选自氢氧化钾、硫酸钾、氯化钾和碳酸氢钾中的一种或两种以上。
本发明利用镍掺杂氮碳纳米材料作为电催化剂促进二氧化碳电化学还原,在流动态电解池中,由镍掺杂氮碳纳米材料所制备的工作电极对二氧化碳还原为一氧化碳提供了极大的促进作用,表现出很高的转化效率和选择性。
以下将通过实施例对本发明进行详细描述,但本发明的保护范围并不局限于此。以下实例中,药品和药剂均为常规市售品。
实施例1
(1)分别称取0.03mol的三聚氰胺和0.03mol的三聚氰酸于干净的烧杯中,分别加入500ml去离子水,均在85℃下搅拌形成澄清透明溶液,然后将二者混合,在85℃条件下,搅拌反应5h;
降至室温(25℃)后,将得到的反应液离心,分离出固体物,将固体物先用水洗2次,再用乙醇洗2次,最后在70℃的条件下干燥12h,得到三聚氰胺-三聚氰酸超分子,记作MA-CA超分子;
(2)取2gMA-CA超分子和0.08g乙酰丙酮镍于研钵中(其中,MA-CA超分子:镍元素的质量比为2:0.018),顺时针研磨1h,得到均匀的金属镍氮碳材料前驱体;
(3)将上述前驱体转移至磁舟中,将磁舟放置在在管式炉中,在氩气氛围下,升温速度4℃/min,升温至920℃下煅烧2h,待冷却至室温(25℃)后,将得到的粉末研磨20min,得到镍掺杂氮碳纳米材料,记作B1。
实施例2
(1)分别称取0.02mol的三聚氰胺和0.02mol的三聚氰酸于干净的烧杯中,分别加入500ml去离子水,均在82℃下搅拌形成澄清透明溶液,然后将二者混合,在82℃条件下,搅拌反应4h;
降至室温后,将得到的反应液离心,分离出固体物,将固体物先用水洗2次,再用乙醇洗2次,最后在70℃的条件下干燥15h,得到三聚氰胺-三聚氰酸超分子,记作MA-CA超分子;
(2)分别取2gMA-CA超分子和0.05g乙酰丙酮镍于研钵中(其中,MA-CA超分子:镍元素的质量比为2:0.011),顺时针研磨1h,得到均匀的金属镍氮碳材料前驱体;
(3)将上述前驱体转移至磁舟中,将磁舟放置在管式炉中,在氩气氛围下,升温速度5℃/min,升温至800℃下煅烧4h,待冷却至室温后,将得到的粉末研磨10min,得到镍掺杂氮碳纳米材料,记作B2。
实施例3
(1)分别称取0.04mol的三聚氰胺和0.04mol的三聚氰酸于干净的烧杯中,分别加入500ml去离子水,均在90℃下搅拌形成澄清透明溶液,然后将二者混合,在90℃条件下,搅拌反应4h;
降至室温后,将得到的反应液离心,分离出固体物,将固体物先用水洗2次,再用乙醇洗2次,最后在70℃的条件下干燥10h,得到三聚氰胺-三聚氰酸超分子,记作MA-CA超分子;
(2)分别取2gMA-CA超分子和0.1g乙酰丙酮镍于研钵中(其中,MA-CA超分子:镍元素的质量比为2:0.023),顺时针研磨1h,得到均匀的金属镍氮碳材料前驱体;
(3)将上述前驱体转移至磁舟中,将磁舟放置在管式炉中,在氩气氛围下,升温速度6℃/min,升温至950℃下煅烧1.5h,待冷却至室温后,将得到的粉末研磨30min,得到镍掺杂氮碳纳米材料,记作B3。
实施例4
按照实施例1的方法进行,所不同的是,将0.08g乙酰丙酮镍换成0.04g氯化镍(其中,MA-CA超分子:镍元素的质量比为2:0.018),得到的镍掺杂氮碳纳米材料,记作B4。
实施例5
按照实施例1的方法进行,所不同的是,将0.08g乙酰丙酮镍换成0.057g硝酸镍(其中,MA-CA超分子:镍元素的质量比为2:0.018),得到的镍掺杂氮碳纳米材料,记作B5。
实施例6
按照实施例1的方法进行,所不同的是,将0.08g乙酰丙酮镍换成0.048g硫酸镍(其中,MA-CA超分子:镍元素的质量比为2:0.018),得到的镍掺杂氮碳纳米材料,记作B6。
实施例7
按照实施例1的方法进行,所不同的是,步骤(1)中,三聚氰胺溶液和三聚氰酸溶液的浓度均为0.02mol/L,得到的镍掺杂氮碳纳米材料,记作B7。
实施例8
按照实施例1的方法进行,所不同的是,步骤(1)中,三聚氰胺溶液和三聚氰酸溶液的浓度均为0.1mol/L,得到的镍掺杂氮碳纳米材料,记作B8。
对比例1
按照实施例1的方法进行,所不同的是,步骤(2)中,乙酰丙酮镍换成乙酰丙酮铁,得到的铁掺杂氮碳纳米材料,记作D1。
具体的制备方法包括以下步骤:
(1)分别称取0.03mol的三聚氰胺和0.03mol的三聚氰酸于干净的烧杯中,分别加入500ml去离子水,均在85℃下搅拌形成澄清透明溶液,然后将二者混合,在85℃条件下,搅拌反应5h;
降至室温后,将得到的反应液离心,分离出固体物,将固体物先用水洗2次,再用乙醇洗2次,最后在70℃的条件下干燥12h,得到三聚氰胺-三聚氰酸超分子,记作MA-CA超分子;
(2)分别取2gMA-CA超分子和0.08g乙酰丙酮铁于研钵中(其中,MA-CA超分子:铁元素的质量比为2:0.013),顺时针研磨1h,得到均匀的金属铁氮碳材料前驱体;
(3)将上述前驱体转移至磁舟中,将磁舟放置在管式炉中,在氩气氛围下,升温速度4℃/min,升温至920℃下煅烧2h,待冷却至室温后,将得到的粉末研磨20min,得到铁掺杂氮碳纳米材料,记作D1。
对比例2
照实施例1的方法进行,所不同的是,步骤(2)中,乙酰丙酮镍换成乙酰丙酮钴,得到的钴掺杂氮碳纳米材料,记作D2。
具体的制备方法包括以下步骤:
(1)分别称取0.03mol的三聚氰胺和0.03mol的三聚氰酸于干净的烧杯中,分别加入500ml去离子水,均在85℃下搅拌形成澄清溶液,然后将二者混合,在85℃条件下,搅拌反应5h;
降至室温后,将得到的反应液离心,分离出固体物,将固体物先用水洗2次,再用乙醇洗2次,最后在70℃的条件下干燥12h,得到三聚氰胺-三聚氰酸超分子,记作MA-CA超分子;
(2)分别取2gMA-CA超分子和0.08g乙酰丙酮钴于研钵中(其中,MA-CA超分子:钴元素的质量比为2:0.018),顺时针研磨1h,得到均匀的金属钴氮碳材料前驱体;
(3)将上述前驱体转移至磁舟中,将磁舟放置在管式炉中,在氩气氛围下,升温速度4℃/min,升温至920℃下煅烧2h,待冷却至室温后,将得到的粉末研磨20min,得到金属钴掺杂氮碳纳米材料,记作D2。
检测例1
(1)将实施例1的镍掺杂氮碳纳米材料B1、对比例1的铁掺杂氮碳纳米材料D1和对比例2的钴掺杂氮碳纳米材料D2进行透射电子显微镜检测,具体结果见图1-4。
(2)将实施例1和对比例1-2制备的材料进行X射线衍射(XRD)分析,其结果如图6所示。
(3)将实施例1的镍掺杂氮碳纳米材料B1进行EDS能谱分析,结果如图5所示。
图1和图2分别为实施例1的镍掺杂氮碳纳米材料B1放大60000倍和30000倍的透射电子显微(TEM)图,由图1-2可知实施例1制得的镍掺杂氮碳纳米材料B1是有镍纳米颗粒负载的节状碳氮纳米管结构,镍纳米颗粒的大小为15-30nm,纳米管表面粗糙;
图3和图4分别为对比例1的铁掺杂氮碳纳米材料D1放大40000倍的透射电子显微(TEM)图和对比例2的钴掺杂氮碳纳米材料D2放大30000倍的透射电子显微(TEM)图,可以看到对比例1的铁掺杂氮碳纳米材料D1和对比例2的钴掺杂氮碳纳米材料D2形貌同为节状碳氮纳米管结构,表面分别有铁颗粒和钴颗粒负载。
由图5的EDS能谱图可以看出,实施例1制得的镍掺杂氮碳纳米材料B1含有镍元素、碳元素、氮元素和氧元素,其质量比为Ni:C:N:O=1:3.23:0.15:0.71。
由图6可知,实施例1制得的镍掺杂氮碳纳米材料B1、对比例1制得的铁掺杂氮碳纳米材料D1和对比例2制得的钴掺杂氮碳纳米材料D2均存在对应的金属颗粒。
检测例2
电化学测试
电化学测试采用三电极体系测试催化性能,对电极为钛网,参比电极为Ag/AgCl电极,将实施例1制得的镍掺杂氮碳纳米材料B1作为工作电极,在Flow Cell流动电解池中进行电化学CO
电解液为碱性,具体为阴极电解液均为1mol/L的氢氧化钾溶液,阳极电解液均为1mol/L的氢氧化钾溶液,测试结果如图7所示。
检测例3
按照检测例2的方法进行,所不同的是将电解液改为中性溶液,具体为阴极电解液为1mol/L氯化钾溶液,阳极为1mol/L氢氧化钾溶液,测试结果如图8所示。
检测例4
按照检测例2的方法进行,所不同的是将电解液改为酸性溶液,具体为阴极电解液为pH为2的0.5mol/L硫酸钾溶液,阳极为0.5mol/L硫酸溶液,测试结果如图9所示。
由图7-9可知实施例1制得的镍掺杂氮碳纳米材料B1作为电催化剂在碱性、中性及酸性条件下,电流密度在100mA·cm
应用例1
电化学测试采用三电极体系测试催化性能,对电极为钛网,参比电极为Ag/AgCl电极,分别将实施例1制得的镍掺杂氮碳纳米材料B1、对比例1的铁掺杂氮碳纳米材料D1和对比例2的钴掺杂氮碳纳米材料D2作为工作电极,在Flow Cell流动电解池中进行电化学CO
电解液为碱性,具体为阴极电解液均为1mol/L的氢氧化钾溶液,阳极电解液均为1mol/L的氢氧化钾溶液,结果如图7、10-11所示。
由图7可以看到,实施例1制得的镍掺杂氮碳纳米材料B1在碱性电解液中的电催化CO
由图10可以看到对比例1的铁掺杂氮碳纳米材料D1在碱性电解液中电催化CO
由图11可以看到对比例2的钴掺杂氮碳纳米材料D2在碱性电解液中电催化CO
可见实施例1制得镍掺杂氮碳纳米材料B1对CO
以上详细描述了本发明的优选实施方式,但是,本发明并不限于上述实施方式中的具体细节,在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,这些简单变型均属于本发明的保护范围。
另外需要说明的是,在上述具体实施方式中所描述的各个具体技术特征,在不矛盾的情况下,可以通过任何合适的方式进行组合,为了避免不必要的重复,本发明对各种可能的组合方式不再另行说明。
此外,本发明的各种不同的实施方式之间也可以进行任意组合,只要其不违背本发明的思想,其同样应当视为本发明所公开的内容。
- 中空锰掺杂氧化钴镍包覆的氮掺杂碳纳米复合材料及制备
- 中空铁掺杂氧化钴镍包覆的氮掺杂碳纳米复合材料及制备
- 钴镍双金属氢氧化物纳米片/氮化碳包覆氮掺杂中空石墨烯球复合材料及其制备方法和应用
- 一种中空氧化钴镍包覆氮掺杂碳纳米复合材料及制备
- 一种钴氮共掺杂的氮碳材料载体负载的纳米氮化镍铁复合材料及其制备方法和应用
- 一种钴氮共掺杂的氮碳材料载体负载的纳米氮化镍铁复合材料及其制备方法和应用