一种双-1,2,3-三唑-双烷基醇类衍生物及其制备方法与在核素萃取中的应用
文献发布时间:2024-04-18 19:58:30
技术领域:
本发明属于化学合成领域和应用化学领域,具体涉及一种双-1,2,3-三唑-双烷基醇类衍生物及其制备方法与在核素萃取中的应用。
背景技术:
人们开发核能的途径有两条:一是重元素的裂变,如铀的裂变;二是轻元素的聚变,如氘、氚、锂等。铀是目前最重要的核燃料,然而陆地上铀的储藏量并不丰富,且分布极不均匀。而在巨大的海水水体中,却含有丰富的铀矿资源。据估计,海水中溶解的铀的数量可达45亿吨,相当于陆地总储量的几千倍。如果能将海水中的铀全部提取出来,所含的裂变能可保证人类几万年的能源需要。不过,海水中含铀的浓度很低,1000吨海水只含有3克铀。只有先把铀从海水中提取出来,才能应用。而要从海水中提取铀,从技术上讲是件十分困难的事情,需要处理大量海水,技术工艺十分复杂。但是,人们已经试验了很多种海水提铀的办法,如吸附法、共沉法、气泡分离法以及藻类生物浓缩法等。核能源开发过程所产生的放射性废水的处理方式也越来越受到重视。在各种提纯方式中,萃取是有机化学实验室中用来提纯和纯化化合物的高效且操作简单的手段之一,且由于萃取是利用物质在两种互不相溶或微溶的溶剂中溶解度或分配系数的不同,使溶质物质从一种溶剂内转移到另外一种溶剂中的方法,在操作过程中并不造成被萃取物质化学成分的改变,因此能完全的保留所需物质的化学性质。
基于此,本发明的目的是通过研究用于核反应的放射性元素铀,发明了一种双-1,2,3-三唑-双烷基醇类衍生物,可用作核素萃取的有效溶剂,且提供了一种简单高效的制备方法。
发明内容:
针对现有化合物的欠缺,本发明的第一方面目的是提供一种双-1,2,3-三唑-双烷基醇类衍生物。
一种双-1,2,3-三唑-双烷基醇类衍生物,其结构式如下:
式中:R选自吡啶或菲咯啉;R’选自乙基、丙基、羟丙基、3-(2-氧代-2-((1,2,3-三羟基丙基-2-基)氨基)乙基)。
进一步地,本发明还提供以下具体化合物:
本发明的第二方面目的是提供一种上述化合物的制备方法,其特征在于,包括以下步骤:以含双端链炔烃的氮杂六元环和含叠氮基的醇为原料,以活泼铜为催化剂,在溶剂中进行叠氮-炔环加成反应,制备得双-1,2,3-三唑-双烷基醇类衍生物。
涉及的反应方程式如下:
式中:R选自吡啶或菲咯啉;R’选自乙基、丙基、羟丙基、3-(2-氧代-2-((1,2,3-三羟基丙基-2-基)氨基)乙基)。
进一步地,所述制备方法中:
所述含双端链炔烃的氮杂六元环,选自2,6-二乙炔基吡啶或2,9-二乙炔基-1,10-菲咯啉。
所述含叠氮基的醇选自2-叠氮基乙醇、3-叠氮基丙醇、3-叠氮基-1,2-丙二醇或2-叠氮基-N-(1,3-二羟基-2-(羟甲基)丙烷-2-基)乙酰胺。
所述活泼铜选择一价铜盐,优选为碘化亚铜。
所述溶剂选择N,N-二甲基甲酰胺,优选为N,N-二甲基甲酰胺与三乙胺的混合物。
所述含双端链炔烃的氮杂六元环、含叠氮基的醇、催化剂的投料摩尔比为:1:1~3:0.08~0.09。
所述反应的反应温度为50~80℃,反应时间12~48小时。优选在60℃温度下反应20小时。
所述反应结束后,对产物进行后处理,后处理包括萃取、洗涤。
进一步地:
本发明提供一种双-1,2,3-三唑-双烷基醇类衍生物的制备方法,其特征在于,包括如下步骤:
(1)在50ml反应瓶中,将叠氮化钠溶于N,N-二甲基甲酰胺中,开启搅拌,在氩气条件下,向反应瓶中缓慢加入2-溴乙醇,在60℃下反应48小时后,冷却至室温得到2-叠氮基乙醇;其中:叠氮化钠与2-溴乙醇的摩尔比为1.5:1,N,N-二甲基甲酰胺为反应溶剂,投入量5~10eq,将反应物完全溶解即可;
(2)将步骤(1)得到的2-叠氮基乙醇无需后处理,直接用于第(2)步反应,向2-叠氮基乙醇中加入2,6-二乙炔基吡啶,再加入三乙胺和碘化亚铜作催化剂,开启搅拌,在氩气条件下,将反应置于60℃下20小时;其中:2-叠氮基乙醇与2,6-二乙炔基吡啶的摩尔比为2:1,仍以N,N-二甲基甲酰胺作为反应溶剂;
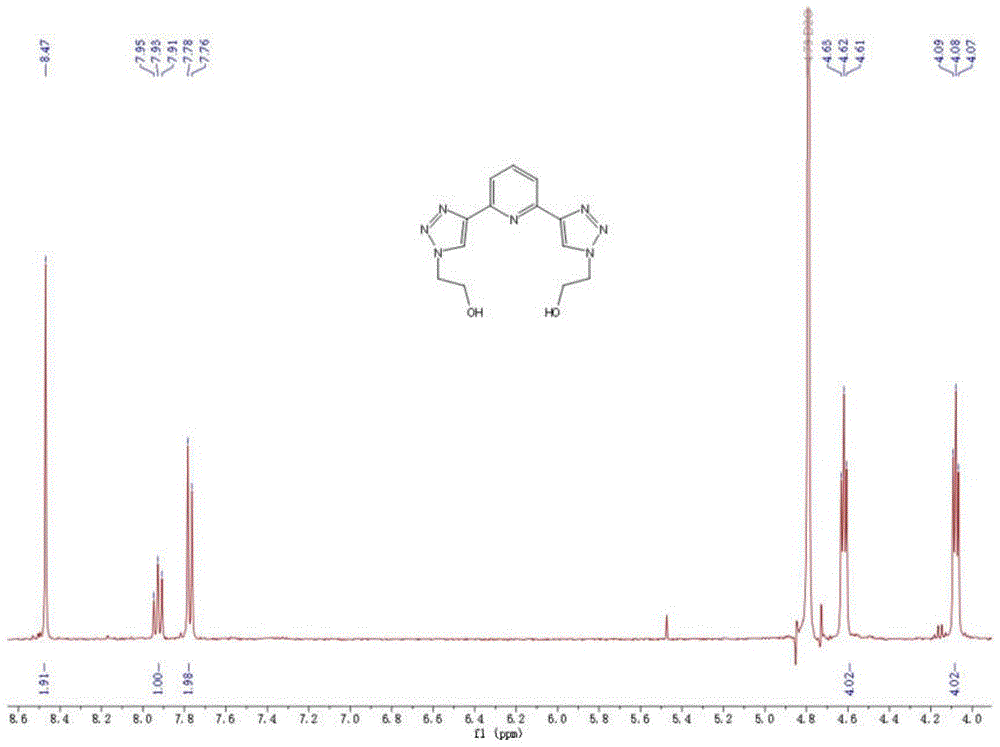
(3)向步骤(2)反应后的溶液中,加入浓盐酸充分混合后将产物质子化,再用饱和食盐水与乙酸乙酯进行萃取,留取水相,向水相中加入氢氧化钠,充分溶解后用乙酸乙酯进行萃取,留取有机相,用无水硫酸钠干燥后,减压去除有机溶剂,得到2,2’-(吡啶-2,6-二基双(1H-1,2,3-三唑-4,1-二基))双(乙烷-1-醇)。
本发明的第三方面目的是提供一种双-1,2,3-三唑-双烷基醇类衍生物在核素萃取方面的应用,具体包括以下步骤:
(1)采用丁二酸酰基过氧化物与氧化石墨烯发生反应,制得羧基化氧化石墨烯;
(2)通过酯化反应将双-1,2,3-三唑-双烷基醇类衍生物固载在上述制得的羧基化氧化石墨烯上,利用石墨烯极好的稳定性可以使化合物长久保存,且由于石墨烯有极好的吸脱附性与表面延展性,在萃取过程中可有效增大接触面积与萃取效率;
(3)将固载了双-1,2,3-三唑-双烷基醇类衍生物的羧基化氧化石墨烯溶于乙酸乙酯,加入少量的氢氧化钠,再与含铀量稍高的海水进行萃取,分离出有机相与水相;加入氢氧化钠能使酯化反应向逆方向进行,将固载于氧化石墨烯上的双-1,2,3-三唑-双烷基醇类衍生物分离出来溶于有机溶液;
(4)萃取后分离出的水相与有机相,将水相通过过滤孔径为22μm的膜过滤器进一步过滤出石墨烯固体后留取分离液,通过分光光度计测量分离液的吸光度,检测结果表明分离液中含铀量已明显降低。
本发明的有益效果如下:
(1)本发明创造性生成了一种双-1,2,3-三唑-双烷基醇类衍生物。
(2)本发明提供了一种简单、高效、成本低的上述化合物的合成方法。
(3)本发明提供了一种双-1,2,3-三唑-双烷基醇类衍生物在核素萃取方面的应用,经实验发现,上述化合物在核素萃取方面具有很好的效果,可以有效分离出海水中的放射性元素铀。
附图说明:
图1为实施例1制备的2,2’-(吡啶-2,6-二基双(1H-1,2,3-三唑-4,1-二基))双(乙烷-1-醇)的
图2为实施例1制备的2,2’-(吡啶-2,6-二基双(1H-1,2,3-三唑-4,1-二基))双(乙烷-1-醇)的
图3a是铀含量分别为0、5ppm、10ppm、50ppm的模拟海水萃取前的紫外光谱图;
图3b是铀含量分别为5ppm、10ppm、50ppm的模拟海水萃取后的分离液的紫外光谱图。
具体实施方式:
下面结合具体实施例和附图对本发明做进一步说明:
实施例1:制备2,2’-(吡啶-2,6-二基双(1H-1,2,3-三唑-4,1-二基))双(乙烷-1-醇)
(1)2-叠氮基乙醇的合成
在50ml反应瓶中,将2.35g(36mmol)叠氮化钠溶于20ml的N,N-二甲基甲酰胺中,开启搅拌,在氩气条件下,向反应瓶中缓慢加入2-溴乙醇1.7ml(24mmol,ρ=1.763g/ml),在60℃下反应48小时后,冷却至室温得到2-叠氮基乙醇,产物无需后处理;
(2)2,2’-(吡啶-2,6-二基双(1H-1,2,3-三唑-4,1-二基))双(乙烷-1-醇)的合成
向(1)所得的2-叠氮基乙醇的反应瓶中加入2,6-二乙炔基吡啶1.5g(11.8mmol),再加入1ml三乙胺和200mg碘化亚铜,开启搅拌,在氩气条件下,将反应置于60℃中搅拌反应20小时,再冷却至室温。向反应后的溶液中加入2ml浓盐酸(12mol/L),用饱和食盐水和乙酸乙酯进行第一步萃取,留取水相,再向所得水相中加入2g氢氧化钠,充分溶解后用乙酸乙酯进行第二步萃取,将所得有机相用无水硫酸钠干燥,减压除去有机溶剂,得到2,2’-(吡啶-2,6-二基双(1H-1,2,3-三唑-4,1-二基))双(乙烷-1-醇)粗产物3.42g,将粗产物用10ml溶液(正戊烷:二氯甲烷=1:1)洗涤,减压除去有机溶剂,得到纯净的2,2’-(吡啶-2,6-二基双(1H-1,2,3-三唑-4,1-二基))双(乙烷-1-醇)产物3.39g,产率95.5%。
实施例2:制备4,4’-(2,6-吡啶二基)双[1H-1,2,3-三唑-1-丙醇]
在100ml反应瓶中,将2,6-二乙炔基吡啶1.5g(11.8mmol)溶于40ml的N,N-二甲基甲酰胺中,再加入1ml三乙胺和200mg碘化亚铜,开启搅拌,在氩气条件下,缓慢加入3-叠氮基丙醇2.4g(23.6mmol),在60℃下反应20小时后,再冷却至室温。向反应后的溶液中加入2ml浓盐酸(12mol/L),用饱和食盐水和乙酸乙酯进行第一步萃取,留取水相,再向所得水相中加入2g氢氧化钠,充分溶解后用乙酸乙酯进行第二步萃取,将所得有机相用无水硫酸钠干燥,减压除去有机溶剂,得到化合物2:4,4’-(2,6-吡啶二基)双[1H-1,2,3-三唑-1-丙醇]粗产物3.69g,将粗产物用16ml溶液(正戊烷:二氯甲烷=1:1)洗涤,减压除去有机溶剂,得到纯产物3.65g,产率93.8%。
实施例3:制备3,3-[2,6-吡啶二基双(1H-1,2,3-三唑-4,1-二基)]双[1,2-丙二醇]
在50ml反应瓶中,将2,6-二乙炔基吡啶1.5g(11.8mmol)溶于25ml的N,N-二甲基甲酰胺中,再加入1ml三乙胺和200mg碘化亚铜,开启搅拌,在氩气条件下,缓慢加入3-叠氮基-1,2-丙二醇2.76g(23.6mmol),在60℃下反应20小时后,再冷却至室温。向反应后的溶液中加入2ml浓盐酸(12mol/L),用饱和食盐水和乙酸乙酯进行第一步萃取,留取水相,再向所得水相中加入2g氢氧化钠,充分溶解后用乙酸乙酯进行第二步萃取,将所得有机相用无水硫酸钠干燥,减压除去有机溶剂,得到化合物3:3,3-[2,6-吡啶二基双(1H-1,2,3-三唑-4,1-二基)]双[1,2-丙二醇]粗产物4.19g,将粗产物用16ml溶液(正戊烷:二氯甲烷=1:1)洗涤,减压除去有机溶剂,得到化合物3纯产物4.13g,产率97.0%。
实施例4:制备4,4’-(2,6-吡啶二基)双[N-[2-羟基-1,1-双羟甲基)乙基]-1H-1,2,3-三氮唑-1-乙酰胺]
在100ml反应瓶中,将2,6-二乙炔基吡啶1.5g(11.8mmol)溶于40ml的N,N-二甲基甲酰胺中,再加入1ml三乙胺和200mg碘化亚铜,开启搅拌,在氩气条件下,缓慢加入2-叠氮基-N-(1,3-二羟基-2-(羟甲基)丙烷-2-基)乙酰胺4.8g(23.6mmol),在60℃下反应20小时后,再冷却至室温。向反应后的溶液中加入2ml浓盐酸(12mol/L),用饱和食盐水和乙酸乙酯进行第一步萃取,留取水相,再向所得水相中加入2g氢氧化钠,充分溶解后用乙酸乙酯进行第二步萃取,将所得有机相用无水硫酸钠干燥,减压除去有机溶剂,得到化合物4:4,4’-(2,6-吡啶二基)双[N-[2-羟基-1,1-双羟甲基)乙基]-1H-1,2,3-三氮唑-1-乙酰胺]粗产物6.27g,将粗产物用20ml溶液(正戊烷:二氯甲烷=1:1)洗涤,减压除去有机溶剂,得到化合物4纯产物6.20g,产率98.3%。
实施例5:制备2,2’-((1,10-菲咯啉基-2,9-二基)双(1H-1,2,3-三唑-4,1-二基))双(乙烷-1-醇)
在50ml反应瓶中,将2,9-二乙炔基-1,10-菲咯啉2.7g(11.8mmol)溶于20ml的N,N-二甲基甲酰胺中,再加入1ml三乙胺和200mg碘化亚铜,开启搅拌,在氩气条件下,缓慢加入2-叠氮基乙醇2.1g(23.6mmol),在60℃下反应20小时后,再冷却至室温。向反应后的溶液中加入2ml浓盐酸(12mol/L),用饱和食盐水和乙酸乙酯进行第一步萃取,留取水相,再向所得水相中加入2g氢氧化钠,充分溶解后用乙酸乙酯进行第二步萃取,将所得有机相用无水硫酸钠干燥,减压除去有机溶剂,得到化合物5:2,2’-((1,10-菲咯啉基-2,9-二基)双(1H-1,2,3-三唑-4,1-二基))双(乙烷-1-醇)粗产物4.57g,将粗产物用12ml溶液(正戊烷:二氯甲烷=1:1)洗涤,减压除去有机溶剂,得到化合物5纯产物4.49g,产率94.3%。δH(400MHz;DMSO-d6)9.02(s,2H),8.59(d,J=8.2Hz,2H),8.43(d,J=8.2Hz,2H),8.00(s,2H),5.05(s,2H),4.31-4.35(m,4H),3.32-3.36(m,4H).
实施例6:制备3,3’-((1,10-邻菲罗基-2,9-二基)双(1H-1,2,3-三唑-4,1-二基))双(丙烷-1,2-二醇)
在100ml反应瓶中,将2,9-二乙炔基-1,10-菲咯啉2.7g(11.8mmol)溶于40ml的N,N-二甲基甲酰胺中,再加入1ml三乙胺和200mg碘化亚铜,开启搅拌,在氩气条件下,缓慢加入3-叠氮基-1,2-丙二醇2.76g(23.6mmol),在60℃下反应20小时后,再冷却至室温。向反应后的溶液中加入2ml浓盐酸(12mol/L),用饱和食盐水和乙酸乙酯进行第一步萃取,留取水相,再向所得水相中加入2g氢氧化钠,充分溶解后用乙酸乙酯进行第二步萃取,将所得有机相用无水硫酸钠干燥,减压除去有机溶剂,得到化合物6:3,3’-((1,10-邻菲罗基-2,9-二基)双(1H-1,2,3-三唑-4,1-二基))双(丙烷-1,2-二醇)粗产物5.23g,将粗产物用16ml溶液(正戊烷:二氯甲烷=1:1)洗涤,减压除去有机溶剂,得到化合物6的纯产物5.17g,产率94.5%。δH(400MHz;DMSO-d6)8.90(s,2H),8.57(d,J=8.2Hz,2H),8.43(d,J=8.0Hz,2H),7.99(s,2H),5.26(s,2H),4.93(s,2H),4.56(d,J=11.2Hz,2H),4.36(d,J=11.2Hz,2H),3.89-3.93(m,2H),3.38-3.42(m,4H).
应用实施例1:
一种实施例1制备的2,2’-(吡啶-2,6-二基双(1H-1,2,3-三唑-4,1-二基))双(乙烷-1-醇)在核素萃取提纯方面的应用,包括以下步骤:
(1)取双氧水、丁二酸酐、去离子水适量,并按m(H
(2)将0.1g氧化石墨烯(Graphene Oxide,以下简称GO)和20ml DMF加入到50ml三口烧瓶中,常温下超声分散2小时,即制得GO悬浮液;升温至85℃,反应72小时,在此过程中每24小时加人0.1g丁二酸酰基过氧化物;待反应完毕后冷却至室温,减压过滤制得固体产物,并在垫有微孔滤膜的抽滤瓶上用DMF反复洗涤产物去除杂质,于80℃真空干燥12小时,即得到羧基化氧化石墨烯,记为GO—COOH。羧基广泛分布在该羧基化氧化石墨烯中的石墨烯分子的表面,且都为活泼羧基。
(3)称取0.1g实施例1制备的化合物1:2,2’-(吡啶-2,6-二基双(1H-1,2,3-三唑-4,1-二基))双(乙烷-1-醇),在稀盐酸条件下发生酯化反应,将2,2’-(吡啶-2,6-二基双(1H-1,2,3-三唑-4,1-二基))双(乙烷-1-醇)通过共价结合固载到石墨烯分子上,由于羧基化氧化石墨烯中羧基的广泛分布,所以酯化反应过程中能基本完全将2,2’-(吡啶-2,6-二基双(1H-1,2,3-三唑-4,1-二基))双(乙烷-1-醇)固载在氧化石墨烯表面而不使平衡向左移动发生分解。
(4)将0.1g固载了2,2’-(吡啶-2,6-二基双(1H-1,2,3-三唑-4,1-二基))双(乙烷-1-醇)的氧化石墨烯溶于200ml乙酸乙酯,加入0.2g氢氧化钠,超声辅助溶解后倒入1L分液漏斗;
(5)取200ml含铀量稍高的模拟海水(铀含量:30ppm),倒入分液漏斗后充分摇晃进行萃取,静置分液后分别留取水相与有机相,将水相与有机相分别通过高速离心沉降,分离出未溶解的氧化石墨烯粉末固体,再将分离出的液体通过过滤孔径为22μm的膜过滤器,进一步过滤出未沉降的石墨烯固体,分别留取各分离液用于液体中铀含量的检测。
(6)将0.07g偶氮胂-III粉末溶于100mL浓度为3mol/L的高氯酸溶液中,超声辅助溶解,静置一周后可得显色剂溶液。将水相分离液与显色剂按体积比1﹕3的比例混和,通过分光光度计分别测量分离液的吸光度,检测结果表明水相的分离液中铀酰离子在特征吸收波长为651nm处已无吸收峰,证明溶液中残留的铀酰离子已经低于分光光度计的检测限,说明铀酰离子含量已明显降低。
(7)根据铀酰离子的浓度变化率的计算公式:
C
计算得知2,2’-(吡啶-2,6-二基双(1H-1,2,3-三唑-4,1-二基))双(乙烷-1-醇)对铀酰离子的吸附萃取率达到了96.7%。
应用实施例2:
方法同应用实施例1,区别在于:步骤(3)中所称取的化合物类型不同,分别采用实施例2-6制备的化合物2-6,并与氧化石墨烯载体进行对照,测试其对铀酰离子的萃取效率,如表1所示:
表1
。
从表1中数据可以看出:取代基的位次和羟基的数目对提取效率都有较大的影响,实施例3制备的化合物3:3,3-[2,6-吡啶二基双(1H-1,2,3-三唑-4,1-二基)]双[1,2-丙二醇]的提取效率最佳,达到了97.2%。可在该结构基础上进行进一步改进提高。
分析:
本发明提供了一种双-1,2,3-三唑-双烷基醇类衍生物,可用作核素萃取的有效溶剂,经上述实施例可以看出:采用本发明化合物固载并用于核素萃取,具有合成方法简单、原料易得、反应安全可行,对铀酰离子的萃取吸附率极高等优点。
- 一种取代1,2,3三氮唑类二芳基嘧啶衍生物及其制备方法与应用
- 一种双-氟喹诺酮噁二唑脲类N-乙酰基环丙沙星衍生物及其制备方法和应用
- 一种含N-甲基加替沙星的双-氟喹诺酮基噁二唑脲类衍生物及其制备方法和应用
- 一种含芦氟沙星的双-氟喹诺酮基噁二唑脲类衍生物及其制备方法和应用
- 一种含N-甲基莫西沙星的双-氟喹诺酮基噁二唑脲类衍生物及其制备方法和应用
- 双官能团的1,2,3-三氮唑衍生物中间体及制备方法与应用
- 双官能团的1,2,3-三氮唑衍生物中间体及制备方法与应用