一种基于转录组数据的microRNA预测方法及系统
文献发布时间:2024-04-18 19:58:30
技术领域
本发明涉及生物技术领域,尤其是一种基于转录数据的microRNA预测方法及系统。
背景技术
随着生物学研究的不断深入和高通量测序技术的迅猛发展,转录组数据在揭示基因调控网络、生物学特性以及疾病机制方面发挥着愈发重要的作用。目前,在转录组数据分析中,通常都是针对蛋白质编码基因的研究,但大部分转录组数据无法被注释为蛋白质编码基因,然而这些非蛋白编码序列在生物的生命活动中发挥着同样重要的作用。由于大多物种缺少基因组数据,在公共数据库中的非编码RNA信息也较少,使得转录组数据中未能被注释为蛋白质编码基因的非蛋白编码序列往往被丢弃。
microRNA作为一类内源性非编码单链RNA分子,长度在18~25nt(核苷酸,Nucleotide),其在动植物中参与转录后基因表达调控。当前,microRNA的预测通常从基因组数据和小RNA文库中获得,目前针对从转录组数据中预测microRNA的流程研究甚少,并未有利用转录组数据进行有效地microRNA精准预测,因此,亟需一种基于转录组数据的microRNA预测方法及系统以准确预测microRNA。
发明内容
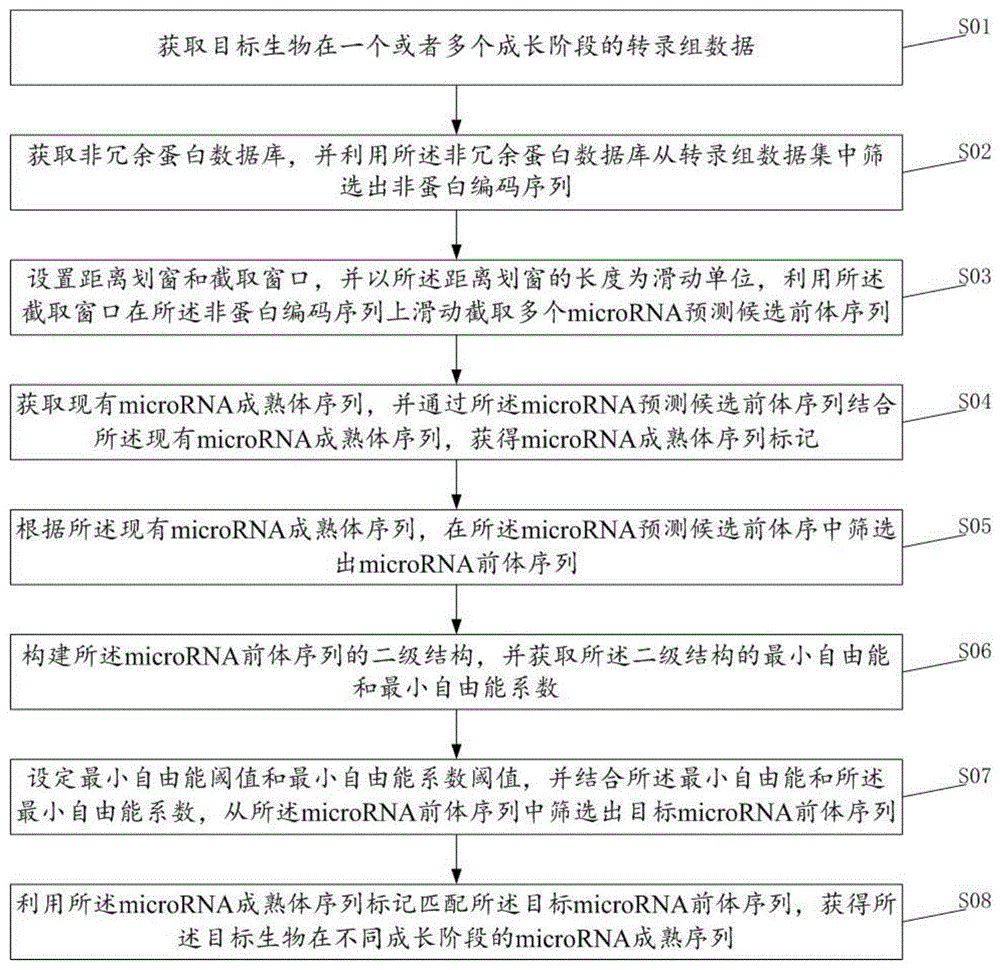
针对当前技术的不足以及实际应用的需求,第一方面,本发明提供了一种基于转录组数据的microRNA预测方法,旨在利用转录组数据精准预测目标生物的microRNA。本发明提供的基于转录组数据的microRNA预测方法,包括如下步骤:获取目标生物在一个或者多个成长阶段的转录组数据;获取非冗余蛋白数据库,并利用所述非冗余蛋白数据库从转录组数据集中筛选出非蛋白编码序列;设置距离划窗和截取窗口,并以所述距离划窗的长度为滑动单位,利用所述截取窗口在所述非蛋白编码序列上滑动截取多个microRNA预测候选前体序列;获取现有microRNA成熟体序列,并通过所述microRNA预测候选前体序列结合所述现有microRNA成熟体序列,获得microRNA成熟体序列标记;根据所述现有microRNA成熟体序列,在所述microRNA预测候选前体序中筛选出microRNA前体序列;构建所述microRNA前体序列的二级结构,并获取所述二级结构的最小自由能和最小自由能系数;设定最小自由能阈值和最小自由能系数阈值,并结合所述最小自由能和所述最小自由能系数,从所述microRNA前体序列中筛选出目标microRNA前体序列;利用所述microRNA成熟体序列标记匹配所述目标microRNA前体序列,获得所述目标生物在不同成长阶段的microRNA成熟序列。本发明利用转录组数据充分结合已知的蛋白编码序列和microRNA成熟体序列,实现了对目标生物在不同成长阶段的microRNA精准预测,为生物学研究和基因调控网络的探索提供了强大的工具。
可选地,所述目标生物包括小菜蛾。本可选项针对小菜蛾这种农业害虫,通过本发明方法精准预测其microRNA,可以为控制这些害虫带来新的思路和方法,有助于创造更环保和可持续的农业生产模式。
进一步可选地,所述成长阶段包括小菜蛾卵阶段、小菜蛾幼虫阶段、小菜蛾蛹阶段以及小菜蛾成虫阶段。本可选项结合小菜蛾不同成长阶段的转录组数据,精准预测microRNA,进一步为控制这些害虫带来新的思路和方法。
可选地,所述利用所述非冗余蛋白数据库从转录组数据集中筛选出非蛋白编码序列,包括如下步骤:组装转录组数据集中的转录数据,获得多条非重复连续序列;比对所述非冗余蛋白数据库中已知蛋白编码序列和所述非重复连续序列,获得所述非重复连续序列中与所述已知蛋白编码序列相似的编码区域,并计算所述编码区域中的序列与已知蛋白编码序列的相似度;设置相似度阈值,并通过所述相似度和所述相似度阈值的比较,判断所述非重复连续序列为非蛋白编码序列或者蛋白编码序列。本可选项借助非冗余蛋白数据库,通过序列比对与相似度计算,从转录组数据中辨识非蛋白编码序列,确保有效获取潜在microRNA前体序列,优化预测方法的信准度与效率。
可选地,所述距离划窗的长度范围包括18nt至25nt,所述截取窗口的长度至少为120nt。本可选项中对距离划窗和截取窗口的长度设计,有助于捕获潜在microRNA前体序列,提升预测方法的准确性和全面性。
进一步可选地,设置所述距离划窗的长度为25nt,设置所述截取窗口的长度为120nt;以所述距离划窗的长度为滑动单位,利用所述截取窗口在所述非蛋白编码序列上滑动截取多个microRNA预测候选前体序列,任一个microRNA预测候选前体序列,满足如下计数模型:L
可选地,所述通过所述microRNA预测候选前体序列结合所述现有microRNA成熟体序列,获得microRNA成熟体序列标记,包括如下步骤:比对所述现有microRNA成熟体序列和所述microRNA预测候选前体序列,获得具有比对位点的microRNA预测候选前体序列;根据所述具有比对位点的microRNA预测候选前体序列,标记与比对位点序列相似的microRNA成熟体序列作为microRNA成熟体序列标记。本可选项通过将现有的microRNA成熟体序列与microRNA预测候选前体序列进行比对,识别具有比对位点的候选前体序列,并将与比对位点相似的部分标记为microRNA成熟体序列,从而为预测结果添加成熟体序列标记,提高预测的可靠性和准确性。本可选项通过与现有microRNA成熟体序列比对,标记与比对位点序列相似的microRNA成熟体序列,为microRNA成熟体序列标记提供了准确依据,提升了预测结果的可靠性。
可选地,所述获取所述二级结构的最小自由能和最小自由能系数,包括如下步骤:分别搭建最小自由能模型和最小自由能系数模型;利用所述最小自由能模型和所述最小自由能系数模型,分别获得所述二级结构对应的最小自由能和最小自由能系数。本可选项采用最小自由能模型和最小自由能系数模型,计算并获取microRNA前体序列的二级结构的最小自由能和最小自由能系数,为精准microRNA预测提供了稳固的理论支持。
可选地,所述利用所述microRNA成熟体序列标记匹配所述目标microRNA前体序列,获得所述目标生物在不同成长阶段的microRNA成熟序列,包括如下步骤:利用所述microRNA成熟体序列标记所对应的microRNA成熟体序列,匹配所述目标microRNA前体序列的二级结构中stem区域的序列;根据匹配结果,获得所述目标生物在不同成长阶段的microRNA成熟序列。本可选项通过匹配microRNA成熟体序列标记和目标microRNA前体序列的二级结构中的stem区域,以获取不同成长阶段的目标生物的microRNA成熟序列,进一步加强了microRNA预测的可信度与准确性。
第二方面,为更好地实施上述基于转录组数据的microRNA预测方法,本发明还提供了一种基于转录组数据的microRNA预测系统。本发明所提出的基于转录组数据的microRNA预测系统包括输入设备、处理器、存储器和输出设备,所述输入设备、所述处理器、所述存储器和所述输出设备相互连接,其中,所述存储器用于存储计算机程序,所述计算机程序包括程序指令,所述处理器被配置用于调用所述程序指令,执行本发明第一方面所述的基于转录组数据的microRNA预测方法。本发明所提供的基于转录组数据的microRNA预测系统,通过输入设备、处理器、存储器和输出设备的互联,存储并执行计算机程序,有效地实现了前述的基于转录组数据的microRNA预测方法,为生物学研究和农业害虫管理提供了便捷且高效的工具。
附图说明
图1为本发明实施例所提供的基于转录组数据的microRNA预测方法流程图;
图2为本发明实施例所提供的基于转录组数据的microRNA预测系统结构图。
具体实施方式
下面将详细描述本发明的具体实施例,应当注意,这里描述的实施例只用于举例说明,并不用于限制本发明。在以下描述中,为了提供对本发明的透彻理解,阐述了大量特定细节。然而,对于本领域普通技术人员显而易见的是:不必采用这些特定细节来实行本发明。在其他实例中,为了避免混淆本发明,未具体描述公知的电路,软件或方法。
在整个说明书中,对“一个实施例”、“实施例”、“一个示例”或“示例”的提及意味着:结合该实施例或示例描述的特定特征、结构或特性被包含在本发明至少一个实施例中。因此,在整个说明书的各个地方出现的短语“在一个实施例中”、“在实施例中”、“一个示例”或“示例”不一定都指同一实施例或示例。此外,可以以任何适当的组合和、或子组合将特定的特征、结构或特性组合在一个或多个实施例或示例中。此外,本领域普通技术人员应当理解,在此提供的示图都是为了说明的目的,并且示图不一定是按比例绘制的。
在一个可选的实施例中,请参见图1,图1为本发明实施例所提供的基于转录组数据的microRNA预测方法流程图。如图1所示,所述基于转录组数据的microRNA预测方法,包括如下步骤:
S01、获取目标生物在一个或者多个成长阶段的转录组数据。
本发明所述的目标生物是指本发明所关注的生物体,可以是动物、植物或其他有机体,可以理解的是,通过本发明所述基于转录组数据的microRNA预测方法,可基于特定的目标生物,了解其在不同成长阶段的microRNA的表达情况。
所述的成长阶段是指目标生物在生命周期中特定的时期,即从胚胎发育到成熟的不同发育时期。可以理解的是,生物的不同成长阶段往往伴随着基因表达的变化,包括microRNA的表达情况。
所述的转录数据是指在特定成长阶段,目标生物体所有基因的RNA转录产物的集合。转录组数据可以通过高通量测序技术(如RNA-Seq)获得,转录组数据展示了在特定生长阶段内,每个基因的转录水平。
为预测小菜蛾在不同生长阶段microRNA的表达偏好,以有效防治和持续控制小菜蛾的种群暴发提供新思路,在一个可选的实施例中,所关注的目标生物为小菜蛾。
进一步地,在本实施例中,所关注的生长阶段包括小菜蛾卵阶段(Egg)、小菜蛾幼虫阶段(Larva)、小菜蛾蛹阶段(Pupa)以及小菜蛾成虫阶段(Adult)。
在本实施例中,步骤S01所述的获取目标生物在一个或者多个成长阶段的转录组数据,包括如下步骤:
S011、收集目标生物在一个或者多个成长阶段的样本,并基于不同成长阶段的样本提取所述目标生物在不同成长阶段的RNA信息。
步骤S011分别收集了小菜蛾在Egg、Larva、Pupa以及Adult四个成长阶段的样本;再分别将四个阶段的样本利用液氮研磨;然后,利用RNA提取试剂Trizol从不同成长阶段的研磨样本中提取小菜蛾的总RNA。
S012、根据所述RNA信息构建总RNA文库,并利用高通量测序技术结合所述总RNA文库获得转录组数据集。
由于分析和实验过程中,RNA容易降解,而DNA更稳定且易于操作,因此,将RNA经过连接反应和逆转录,生成对应的cDNA(互补DNA)以供后续分析。
可以理解的是,所述cDNA是一种通过逆转录过程合成的DNA分子,它的序列与RNA分子的相应部分互补。进一步地,所述高通量测序技术为RNA测序(RNA-Seq)技术。
应当理解,通过步骤S011至步骤S012,可实现小菜蛾在Egg、Larva、Pupa以及Adult四个成长阶段转录组数据的获取,任一成长阶段的转录组数据中包括蛋白质编码序列信息和非蛋白质编码序列信息。
在其他的一个或者一些实施例中,步骤S01所述的获取目标生物在一个或者多个成长阶段的转录组数据,可以通过现有数据库完成。
进一步地,在一个具体的实施例中,从NCBI(National Center forBiotechnology Information,美国国立生物技术信息中心)的SRA数据库(Sequence ReadArchive)中下载了小菜蛾在Egg、Larva、Pupa以及Adult四个成长阶段的转录组数据,其编号分别为SRR179062、SRR179508、SRR179509、SRR179510。
S02、获取非冗余蛋白数据库,并利用所述非冗余蛋白数据库从转录组数据集中筛选出非蛋白编码序列。
本发明所述的非冗余蛋白数据库是指存储来自各种生物学研究的已知蛋白质编码序列的数据库,例如,NCBI的nr数据库(Non-Redundant Protein Database,非冗余蛋白数据库)。
进一步地,基于上述实施例中从NCBI的SRA数据库(Sequence Read Archive)中小菜蛾在Egg、Larva、Pupa以及Adult四个成长阶段的转录组数据,步骤S02中所述利用所述非冗余蛋白数据库从转录组数据集中筛选出非蛋白编码序列,包括如下步骤:
S021、组装转录组数据集中的转录数据,获得多条非重复连续序列。
步骤S021可选用Trinity软件进行转录数据中的序列组装,其中,Trinity是一个用于转录组组装的开源软件,用于从RNA-Seq(转录组测序)数据中重建多个基因的转录本。
在本实施例中,利用所述Trinity软件(默认参数)分别对上述四个成长阶段的转录组数据进行组装,获取了多条非重复连续序列。
S022、比对所述非冗余蛋白数据库中已知蛋白编码序列和所述非重复连续序列,获得所述非重复连续序列中与所述已知蛋白编码序列相似的编码区域,并计算所述编码区域中的序列与已知蛋白编码序列的相似度。
步骤S022可选用Blastx软件,将组装得到的多条非重复序列分别与非冗余蛋白数据库(nr数据库)进行比对;其中,Blastx是一种基于比对的序列相似性搜索工具,用于将核酸序列与蛋白质数据库进行比对,以确定序列之间的相似性和匹配度。
在本实施例中,除了利用Blastx软件将组装得到的非重复序列与非冗余蛋白数据库(nr数据库)进行比对,还利用Blastx软件识别出非重复连续序列中与所述已知蛋白编码序列相似的编码区域,并利用Blastx软件计算所述编码区域中序列与已知蛋白编码序列的相似度。
S023、设置相似度阈值,并通过所述相似度和所述相似度阈值的比较,判断所述非重复连续序列为非蛋白编码序列或者蛋白编码序列。
在本实施例中,基于上述Blastx软件所获得的比对结果,设置相似度阈值为0.00001,以判断所述非重复连续序列为非蛋白编码序列或者蛋白编码序列:
当编码区域中序列与已知蛋白编码序列的相似度大于等于0.00001时,该编码区域所对应的非重复连续序列为蛋白编码序列。
当编码区域中序列与已知蛋白编码序列的相似度小于0.00001时,该编码区域所对应的非重复连续序列为非蛋白编码序列。
在本实施例中,经过步骤S021至步骤S023,本实施例在各个阶段所对应的序列数量如下表所示:
其中,所述原始序列对应的数据是指在不同成长阶段,小菜蛾转录组数据集中的序列数量;所述非重复序列对应的数据是指在不同成长阶段,原始序列经过组装后获得的序列数量;所述注释的序列对应的数据是指在不同成长阶段,非冗余蛋白数据库中已知蛋白编码序列数量;所述非编码序列对应的数据是指在不同成长阶段,非蛋白编码序列的数量;所述最长编码序列是指在不同成长阶段,最长非编码序列中的核苷酸数量;所述最短编码序列是指在不同成长阶段,非编码序列中最短非蛋白编码序列中的核苷酸数量;所述非编码序列比例(%)对应的数据是指在不同成长阶段,非编码序列占非重复序列的比例。
S03、设置距离划窗和截取窗口,并以所述距离划窗的长度为滑动单位,利用所述截取窗口在所述非蛋白编码序列上滑动截取多个microRNA预测候选前体序列。
由于microRNA的长度在18~25nt内,为确保在扫描非蛋白编码序列的过程中涵盖了典型的microRNA长度范围,因此,所述距离划窗的长度范围包括18nt至25nt,所述截取窗口的长度至少为120nt。
在一个具体的实施例中,步骤S03中设置所述距离划窗的长度为25nt,设置所述截取窗口的长度为120nt。即以25nt为滑动单位,利用所述截取窗口在所述非蛋白编码序列上滑动截取多个microRNA预测候选前体序列,任一个microRNA预测候选前体序列,满足如下计数模型:L
进一步地,任一长度为N的非蛋白编码序列对应的microRNA预测候选前体序列,包括:L
L
S04、获取现有microRNA成熟体序列,并通过所述microRNA预测候选前体序列结合所述现有microRNA成熟体序列,获得microRNA成熟体序列标记。
本发明所述的现有microRNA成熟体序列是指已经在数据库中或者文献中确认并公开的microRNA成熟体序列。进一步地,所述现有microRNA成熟体序列可通过相关数据库获得,例如,miRBase数据库。
在一个可选的实施例中,步骤S04所述的通过所述microRNA预测候选前体序列结合所述现有microRNA成熟体序列,获得microRNA成熟体序列标记,包括如下步骤:
S041、比对所述现有microRNA成熟体序列和所述microRNA预测候选前体序列,获得具有比对位点的microRNA预测候选前体序列。
在本实施例中,可选用Seqmap将microRNA成熟体序列与microRNA预测候选前体序列比对。其中,所述Seqmap(Sequence Mapping and Assembly Program)是一个用于序列比对和组装的计算工具,旨在从高通量测序数据中快速准确地映射和组装序列。
S042、根据所述具有比对位点的microRNA预测候选前体序列,标记与比对位点序列相似的microRNA成熟体序列作为microRNA成熟体序列标记。
在本实施例中,使用1.0.13版本的Seqmap将microRNA成熟体序列与microRNA预测候选前体序列比对,分别按2_1_1和3_1_1(错配_插入_缺失)的标准得到具有比对位点的序列,其中,microRNA成熟体序列来自与miRBase数据库。
S05、根据所述现有microRNA成熟体序列,在所述microRNA预测候选前体序中筛选出microRNA前体序列。
在一个可选的实施例中,步骤S05可选用triplet-SVM算法相关软件识别具有比对位点的microRNA预测候选前体序列中的特征序列,以确定具有比对位点microRNA预测候选前体序列是否为microRNA。
进一步地,所述triplet-SVM算法相关软件是一种基于机器学习算法的软件,它是基于支持向量机(SVM)的一种扩展形式,主要用于处理三元组数据的排序和排名问题。
可以理解的是,triplet-SVM算法相关软件是用已知的microRNA成熟序列和非microRNA序列数据作为训练集(和验证)构建一个学习模型,用于对预测的序列进行分类,可初步判断具有比对位点microRNA预测候选前体序列是否为microRNA。
S06、构构建所述microRNA前体序列的二级结构,并获取所述二级结构的最小自由能和最小自由能系数。
在一个可选的实施例中,步骤S06可选用RNAfold预测二级结构,并利用RNAfold软件得到二级结构的最小自由能(Minimum Free Energy,MFE)以及对应的计算最小自由能指数(Minimum Free Energy Index,MFEI)。其中,所述RNAfold是一种用于预测RNA分子二级结构的计算工具。
进一步地,在本实施例中,步骤S06所构建的microRNA前体序列的任一二级结构,其最小自由能和最小自由能系数可利用上述RNAfold软件计算。
在其他的一个或者一些实施例中,基于所搭建的二级结构,所述获取所述二级结构的最小自由能和最小自由能系数,包括如下步骤:
S061、分别搭建最小自由能模型和最小自由能系数模型。
在本实施例中,最小自由能和最小自由能系数分别满足如下模型:
其中,MFE表示二级结构的最小自由能,MFEI表示二级结构的最小自由能系数,i和j均表示microRNA前体序列中碱基位置,w(i,j)表示microRNA前体序列中第i个位置处的碱基与第j个位置处的碱基之间的能量,δ(i,j)表示microRNA前体序列中第i个位置处的碱基与第j个位置处的碱基之间的配对指示函数,w(i)表示microRNA前体序列中第i个位置处碱基能量,δ(i)表示microRNA前体序列中第i个位置处碱基的稳定指示函数,R表示理想气体常数,T表示绝对温度,l表示预测二级结构中的碱基数目,m(G&C)表示microRNA前体序列中碱基G和碱基C的数目,m(G&C&A&U),表示microRNA前体序列中碱基G、碱基C、碱基A和碱基U的数目。
进一步地,针对配对指示函数δ(i,j):当microRNA前体序列中第i个位置处的碱基与第j个位置上处的碱基能配对,δ(i,j)=1,当microRNA前体序列中第i个位置处的碱基与第j个位置处的碱基不能配对,δ(i,j)=0。针对稳定指示函数δ(i):当microRNA前体序列中第i个位置处碱基处于稳定状态,δ(i)=1,当microRNA前体序列中第i个位置处碱基处于非稳定状态,δ(i)=0。
S062、利用所述最小自由能模型和所述最小自由能系数模型,分别获得所述二级结构对应的最小自由能和最小自由能系数可通过如下最小自由能模型和最小自由能系数模型计算。
S07、设定最小自由能阈值和最小自由能系数阈值,并结合所述最小自由能和所述最小自由能系数,从所述microRNA前体序列中筛选出目标microRNA前体序列。
在一个可选的实施例中,基于上述RNAfold软件所预测的二级结构,以及RNAfold软件所计算的最小自由能和所述最小自由能系数,设置有对应的最小自由能阈值和最小自由能系数阈值:最小自由能阈值MFEMFE≤
-25Kcal/mol,最小自由能系数阈值MFEI≥0.85。
进一步地,当RNAfold软件所构建二级结构,其最小自由能和所述最小自由能系数满足上述对应阈值,则该二级结构对应的microRNA前体序列即为目标microRNA前体序列;反之,则该二级结构对应的microRNA前体序列不是目标microRNA前体序列。
S08、利用所述microRNA成熟体序列标记匹配所述目标microRNA前体序列,获得所述目标生物在不同成长阶段的microRNA成熟序列。
在一个可选的实施例,步骤S07所述的利用所述microRNA成熟体序列标记匹配所述目标microRNA前体序列,获得所述目标生物在不同成长阶段的microRNA成熟序列,包括如下步骤:
S081、利用所述microRNA成熟体序列标记所对应的microRNA成熟体序列,匹配所述目标microRNA前体序列的二级结构中stem区域的序列。
在本实施例中,所述stem区域指的是microRNA候选前体序列中形成stem-loop结构的那部分序列。具体来说,stem区域是由两段互补的序列组成,它们通过互补配对形成了稳定的双螺旋结构。
S082、根据匹配结果,获得所述目标生物在不同成长阶段的microRNA成熟序列。
具体地,若所述microRNA成熟体序列标记所对应的microRNA成熟体序列能完全匹配所述目标microRNA前体序列的二级结构中stem区域的序列,则microRNA成熟体序列标记所对应的microRNA成熟体序列为目标生物的microRNA成熟序列。进一步地,在预测结果中去除位置重复或前体序列一致的冗余序列。
在一个具体的实施例中,基于上述实施例中从NCBI的SRA数据库(Sequence ReadArchive)中小菜蛾在Egg、Larva、Pupa以及Adult四个成长阶段的转录组数据,通过上述步骤S02至步骤S08,从小菜蛾转录组数据中预测得到的microRNA在Egg、Larva、Pupa、Adult中的分布数量分别为62、35、69、76。
在一个可选的实施例中,为更好地实施上述基于转录组数据的microRNA预测方法,本发明还提供了一种基于转录组数据的microRNA预测系统,请参见图2,图2为本发明实施例所提供的基于转录组数据的microRNA预测系统结构图。
如图2所示,本发明所提出的基于转录组数据的microRNA预测系统包括输入设备、处理器、存储器和输出设备,所述输入设备、所述处理器、所述存储器和所述输出设备相互连接,其中,所述存储器用于存储计算机程序,所述计算机程序包括程序指令,所述处理器被配置用于调用所述程序指令,执行本发明所提供的基于转录组数据的microRNA预测方法。
进一步地,所述输入设备可以是键盘、鼠标、触摸屏等,用于用户与系统进行交互。例如,研究人员可以通过输入设备提供待分析的转录组数据,以便系统进行后续处理。
进一步地,所述处理器是系统的核心部件,用于执行计算机程序并处理数据。应当理解,处理器负责调用存储在存储器中的计算机程序指令,执行基于转录组数据的microRNA预测方法。
进一步地,所述存储器用于存储计算机程序、数据和中间结果。在本发明中,存储器存储了执行microRNA预测方法所需的计算机程序指令。这可能包括转录组数据、非冗余蛋白数据库、现有microRNA成熟体序列等。
进一步地,所述输出设备用于将系统处理的结果显示给用户。例如,系统可以将预测的microRNA成熟序列结果通过输出设备展示给研究人员,以便他们分析和研究。
在本实施例中,本发明的基于转录组数据的microRNA预测系统利用输入设备获取转录组数据,经由处理器执行计算机程序,利用存储器存储所需的程序和数据,最终将预测结果通过输出设备呈现给用户。本发明所提供的基于转录组数据的microRNA预测系统可以高效且有效地实现前述的基于转录组数据的microRNA预测方法,为生物学研究和农业害虫管理提供了便捷且高效的工具。
最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围,其均应涵盖在本发明的权利要求和说明书的范围当中。
- 一种基于星天牛转录组数据开发SSR引物的方法
- 一种基于Illumina的转录组测序数据和PeakCalling方法的细菌ncRNA预测方法
- 一种基于转录组测序预测分泌蛋白序列的方法或系统