一种超级迷你型基因编辑器及其应用
文献发布时间:2024-04-18 19:58:53
技术领域
本发明涉及基因工程技术领域,尤其涉及一种超级迷你型基因编辑器及其应用。
背景技术
基因编辑技术是指对基因进行靶向修饰(敲除、插入、替换等)而获得新的特征或功能的技术,从第一代DNA核酸酶编辑系统ZFNs、第二代TALENs到第三代CRISPR/Cas9系统,以及以CRISPR/Cas9为基础的BE(Base editing)系统和PE(Prime ediing)系统,基因编辑效率不断提高、编辑类型不断丰富,成本逐渐降低,应用范围不断扩大,已经深入广泛地用于了医疗、农业、能源、材料与环境等领域,并已经有不少的相关基因编辑产品上市,而更多的产品正在临床试验或者上市前准备之中。特别重要的是,基因编辑为人体细胞或组织及其他生命体的遗传改造和基因治疗提供了前所未有的强大工具,这将为整个生物产业带来巨大的革命性发展机遇。
早期研究发现,CRISPR/Cas9系统来源于细菌和古细菌的天然获得性免疫系统,通过CRISPR RNA(crRNA)和trans-activating crRNA(tracrRNA)以及Cas9蛋白组成的复合体抵御外源性DNA的入侵。2012年CRISPR/Cas9的体外重构和2013年在人类细胞中证明了其基因编辑功能,标志着新一代基因编辑时代的开始,随后此技术成功的在不同类型的细胞及动植物多个物种中行使基因编辑功能。但CRISPR/Cas9域ZFNs和TALENs一样,都是在基因组靶标位点引起DNA双链断裂,进而激活细胞内部修复机制的基础上实现编辑的。细胞内DNA双链断裂的修复机制包括易引起随机插入、缺失的异源末端连接和需要同源模板存在才可以激活的同源重组修复。但是异源末端连接容易引起随机插入,进而影响靶基因的功能及基因组稳定性;同源重组(HDR)尽管精确性高于末端连接(NHEJ),但是其在细胞中的同源重组修复效率低,约为0.1%~5%,这些都大大限制了精确基因编辑技术的应用。随后针对上述缺陷,2016年4月,美国哈佛大学David Liu实验室开发了不需要依赖DNA双链断裂即可进行单碱基转换的基因编辑技术—BE技术。该技术基于无核酸酶活性的dCas9(Inactive,ordead Cas9)或有单链DNA切口酶活性的Cas9n(Cas9 nickase)、胞嘧啶脱氨酶、尿嘧啶糖基化酶抑制子(uracil DNA glycosylase inhibitor,UGI)以及sgRNA形成的复合体,在不引起双链DNA断裂的情况下,直接使靶向位点的胞嘧啶(Cytosine,C)脱氨基变成尿嘧啶(Uracil,U);由于尿嘧啶糖基化酶抑制子的存在,抑制了U的切除;随着DNA复制,U被胸腺嘧啶(Thymine,T)取代;同时,互补链上原来与C互补的鸟嘌呤(Guanine,G)将会替换为腺嘌呤(Adenine,A),最终实现了在一定的活性窗口内C-T和G-A的单碱基精准编辑。随后2017年,基于E.coli TadA(ecTadA)的新型单碱基编辑器——腺嘌呤碱基编辑器(adenine baseeditors,ABEs),实现了A.T碱基对向G.C碱基对的转换。随后国内外科学家又对BE技术进行扩展,陆续开发了GCBE及双BE等编辑器,并且上述各种BE编辑器在不同类型细胞及不同的动植物物种都成功应用。但是这些研究主要用于部分类型的单碱基突变,不能实现其他类型的精确突变,比如任意类型的点突变,精确的删除和插入等等。2019年,David R.Liu实验室开发了PE(Prime editing)编辑系统,该系统不仅可以引入插入和缺失(indels),还可以实现12种碱基之间的任意转换。该系统的工作原理是:sgRNA被改造成pegRNA(primeediting guide RNA),pegRNA包含引物结合位点(primer binding site,PBS)区域;还携带着逆转录的模板序列,可以和逆转录酶(RT)相结合,而pegRNA序列引导将其带入到目标基因序列中实现基因编辑。随后国内外课题组对PE细胞进行各方面优化,比如提高效率,删除和插入的片段增大等等,而且此技术成功在各种常用的细胞,小鼠,斑马鱼等模型动物以及许多植物中成功应用,但目前在原代细胞中编辑效率很低,而且在大动物的细胞和个体水平都未有成功报道,更重要的是PE系统与BE系统类似,都因为其自身体量过大,从而限制其在AAV基因治疗系统中广泛应用,而这恰是人类基因治疗的核心关键所在。
针对上述Cas9,以及基于其发展的BE系统和PE系统,显著弱点是元件过大,造成细胞转染或者投递效率低,从而限制了其应用范围,特别重要的是由于编辑器分子量巨大,远远超过AAV基因治疗系统的承载量(小于4.7kb),因此限制了上述这些基因编辑作为药物或者治疗手段以及体内外细胞遗传修饰的应用。当前编辑器过大的原因主要是Cas9这个底盘核心工具分子量过大,达到了1368个氨基酸之多,以此为基础融合其他功能域(domain)构建的BE和PE系统进一步增大分子量,而这个问题不可能从底盘解决。因此国内外多个课题组,开始通过寻找新的底盘Cas系统的小型酶,以发展新的编辑工具。目前主要有,CasMINI(557AA),AsCas12f1(422AA),OgeuIscB(499AA)和SpCas12f(497AA),但是其效率远低于Cas9(平均只有2%左右),且很多研究仅于细胞水平,很少扩展到动植物研究及基因治疗研究。同时这些小型酶虽然比Cas9酶(1368AA)小,但还不足以小到可以融合不同domain,用于未来各种应用。显然,开发出超级小和效率高的底盘编辑器,仍然是当前基因编辑研发领域的核心关键任务。2021年nature报道发现了一个新的小型的TnpB编辑工具,认为是Cas12系统的祖先,而且仅有408个AA,但此报道仅仅在293T细胞中做了靶点的验证,截至目前,尚未见到此编辑工具在其他类型细胞和动物水平的研究报道。2023年王皓毅等人在nature上又报道了从64个注释的IS605成员中筛选获得的25个在大肠杆菌中活跃的TnpBs,通过进一步细胞研究发现仅仅有3个有活性。随后又通过生物信息学分析,基因组库筛选最终又获得369个氨基酸和382个氨基酸的编辑器ISAam1和ISYmu1编辑器,但是其仅在细胞水平验证,并没有进行基因编辑小鼠的制备研究,同时效率也低于传统的SaCas9。这类研究主要是通过大数据挖掘,生物信息分析及体外功能验证的方法,寻找不同类型的小型编辑器,但显而易见,这种方法工作量极大,筛选出的编辑工具也未必有效率,比如AsCas12f,OgeuIscB等,在细胞水平就几乎无编辑效率,更重要的是,现有的这些编辑器也没有在基因编辑动物制备中的应用报道。
发明内容
为解决上述技术难题,本发明提供了一种基因编辑器。
本发明首次通过对TnpB编辑器进行功能域精简的工程化操作,系统开发了新型的380-400个AA的超级迷你型编辑器,命名为SuperMini-(GE380-GE400)-SWL,并且其在不同类型的哺乳细胞中可以实现高效的基因编辑,效率在20%-60%中间,平均达到了32%,远远高于原始TnpB及其他小型基因编辑器,并且与经典的Cas9编辑效率相似,甚至稍高一些。同时,本发明首次将新型超级迷你编辑器应用于基因编辑小鼠,并获得了巨大的成功,证明了本发明的超级迷你新型编辑器,可以高效实现从哺乳类动物细胞到活体动物的精准编辑。基于此,本发明提出如下技术方案。
本发明的基因编辑器的开发策略包括如下步骤:
(1)解析基因编辑器元件结构,确定基因编辑核心功能区及非核心功能区,对非核心功能区进行区域截短,构建不同截短体类型的基因编辑器突变体;
(2)对基因编辑器突变体进行细胞水平和/或个体水平基因编辑效率的验证。
第一方面,本发明提供了一种基因编辑器,其具有以下至少一种氨基酸序列:
SEQ ID NO.1所示氨基酸序列的第1~400位、第1~399位、第1~398位、第1~397位、第1~396位、第1~395位、第1~394位、第1~393位、第1~392位、第1~391位、第1~390位、第1~389位、第1~388位、第1~387位、第1~386位、第1~385位、第1~384位、第1~383位、第1~382位、第1~381位、第1~380位的氨基酸序列。
优选地,所述基因编辑器包括上述氨基酸序列以及功能蛋白;或包括上述氨基酸序列以及多肽。
所述功能蛋白或多肽与上述氨基酸序列连接后,不会影响上述氨基酸序列的活性(与导向RNA结合的活性、核酸内切酶活性、在导向RNA引导下与靶序列特定位点结合并切割的活性等)。
通过连接功能蛋白或多肽功能性元件后,能够赋予上述氨基酸序列更广泛的应用。如将其与核定位信号序列连接,可以提高本发明的蛋白进入细胞核的活性;与细胞修复机制关键调控因子连接,可以通过调控体内修复机制,提高特定修复所占比例。
优选地,所述功能蛋白或多肽选自以下至少一种:表位标签、报告基因序列、核定位信号序列、穿膜肽、靶向部分、转录激活结构域、转录抑制结构域、核酸酶结构域、具有下列活性的结构域:核苷酸脱氨酶、甲基化酶活性、去甲基化酶、转录激活活性、转录抑制活性、转录释放因子活性、组蛋白修饰活性、核酸酶活性、单链RNA切割活性、双链RNA切割活性、单链DNA切割活性、双链DNA切割活性、核酸结合活性。
在具体实施过程中,表位标签为本领域技术人员所熟知的,包括但不限于His、V5、FLAG、HA、Myc、VSV-G、Trx等。本领域技术人员根据期望目的(例如,纯化、检测或示踪)可以选择合适的表位标签。
在具体实施过程中,报告基因序列为本领域技术人员所熟知的,包括但不限于GST、HRP、CAT、GFP、HcRed、DsRed、CFP、YFP、BFP等。
在具体实施过程中,核定位信号序列(NLS)包含1个或多个NLS序列。所述核定位信号序列位于、靠近或接近本发明的基因编辑器的末端(例如,N端或C端)。
在具体实施过程中,所述穿膜肽包括但不限于TAT,CPP5等,以增加本发明基因编辑器穿透细胞膜的能力。
第二方面,本发明提供了编码上述基因编辑器的核酸。
在具体实施过程中,所述核酸可以经过密码子优化用于在原核细胞或真核细胞中的表达。
第三方面,本发明提供了一种复合物,包括蛋白组分和核酸组分;
所述蛋白组分为上述基因编辑器;
所述核酸组分中含有TTGAT/TTTAT特征以及能够与靶序列杂交的导向序列;
或所述核酸组分中含有导向RNA的核苷酸序列。
蛋白组分和核酸组分能够相互结合形成复合物。
蛋白组分和核酸组分可以是天然存在的、也可以是经过修饰的。
在一些实施方案中,靶序列是非天然存在的DNA或RNA序列。
在具体实施过程中,当靶序列为DNA时,所述靶序列位于原间隔序列临近基序(TAM)的5’端。
在一些实施方案中,靶序列存在于细胞内。
在一些实施方案中,靶序列存在于细胞核内或细胞质(如细胞器)内。
优选地,所述细胞为原核细胞。
第四方面,本发明提供了一种生物材料,其中含有上述基因编辑器、或核酸、或复合物,所述生物材料为表达盒、载体、宿主细胞、转基因细胞系或重组微生物。
在具体实施过程中,所述载体为克隆载体或表达载体。所述载体能够表达上述基因编辑器、或编码基因编辑器的核酸、或上述复合物。所述载体包括但不限于质粒、噬菌体、柯斯质粒。
在具体实施过程中,宿主细胞包括但不限于原核细胞(例如大肠杆菌细胞)、真核细胞(例如酵母细胞)、昆虫细胞、植物细胞或动物细胞(例如哺乳动物细胞,例如小鼠细胞或人细胞)。宿主细胞还可以是细胞系,例如293T细胞系。
第五方面,本发明提供了一种递送组合物,其中含有递送载体以及以下至少一种组分:上述基因编辑器、核酸、复合物、生物材料。
优选地,所述递送载体为以下至少一种:脂质颗粒、糖颗粒、金属颗粒、蛋白颗粒、脂质体、外泌体、微泡、基因枪载体、病毒载体(例如复制缺陷型逆转录病毒、慢病毒、腺病毒或腺相关病毒)。
本发明的递送组合物可以通过本领域已知的任何方法进行递送,包括但不限于电穿孔、脂转染、核转染、显微注射、声孔效应、基因枪、磷酸钙介导的转染、阳离子转染、脂质体转染、树枝状转染、热激转染、核转染、磁转染、脂转染、穿刺转染、光学转染、试剂增强性核酸摄取、经脂质体、免疫脂质体、病毒颗粒、人工病毒体等载体递送。
第六方面,本发明提供了一种药品,其中含有以下至少一种组分:上述基因编辑器、核酸、复合物、生物材料、递送组合物。
所述药品包括但不限于用于基因编辑。
第七方面,本发明提供了一种试剂或试剂盒,其中含有以下至少一种组分:上述基因编辑器、核酸、复合物、生物材料、递送组合物。
本发明的试剂或试剂盒中包含的组分可以被提供于任何适合的容器中。
在一些实施方案中,所述试剂或试剂盒中还包含一种或多种缓冲液。缓冲液可以是任何缓冲液,包括但不限于碳酸钠缓冲液、碳酸氢钠缓冲液、硼酸盐缓冲液、Tris缓冲液、MOPS缓冲液、HEPES缓冲液及其组合。
在一些实施方案中,所述试剂或试剂盒中还包含一个或多个寡核苷酸,该一个或多个寡核苷酸对应于一个用于插入进载体中的导向序列,以便可操作地连接该导向序列和调节元件。
在一些实施方案中,所述试剂或试剂盒包括同源重组模板多核苷酸。
第八方面,本发明提供了上述基因编辑器、核酸、复合物、生物材料、递送组合物、药品、试剂或试剂盒在基因编辑中的应用。
优选地,所述基因编辑为真核生物的基因编辑。
所述真核生物包括但不限于人、猴子、小鼠、猪、牛、羊、兔子等。
第九方面,本发明提供了一种靶基因的修饰方法,包括采用上述基因编辑器、核酸、复合物、生物材料、递送组合物、药品、试剂或试剂盒对靶基因进行修饰。
在具体实施过程中,将上述基因编辑器、核酸、复合物、生物材料、递送组合物、药品、试剂或试剂盒与靶基因接触进行修饰,或者递送至含有靶基因的细胞或胚胎中,靶序列存在于所述靶基因中。
在一些实施方案中,靶基因存在于胚胎或细胞中。
优选地,靶基因存在于真核细胞中,更优选存在于哺乳动物细胞中。所述哺乳动物细胞包括但不限于人类细胞,猴子细胞,非人灵长类动物、牛、猪或啮齿类动物细胞。
在一些实施方案中,所述细胞是非哺乳动物真核细胞,例如家禽或鱼等。
在一些实施方案中,所述细胞是植物细胞,例如栽培植物(如玉米、高粱、小麦或水稻)、藻类、树或蔬菜细胞。
在一些实施方案中,所述靶基因存在于体外的核酸分子(如质粒)中。
在一些实施方案中,所述方法导致靶基因中的靶序列断裂(如使DNA双链断裂或RNA单链断裂)。
在一些实施方案中,所述断裂导致靶基因的转录降低。
优选地,所述修饰方法还包括利用编辑模板(例如外源核酸)对靶基因进行编辑。
在具体实施过程中,将编辑模板与靶基因接触进行编辑,或者递送至含有靶基因的细胞或胚胎中进行编辑。
在一些实施方案中,所述方法通过与编辑模板(例如外源核酸)同源重组修复断裂的靶基因,其中修复导致一种突变,包括所述靶基因的一个或多个核苷酸的插入、缺失或取代。
在一些实施方案中,所述突变导致在从包含该靶序列的基因表达的蛋白质中的一个或多个氨基酸的改变。
在一些实施方案中,所述修饰还包括将编辑模板(例如外源核酸)插入断裂的靶基因中。
在一些实施方案中,所述修饰方法用于改变靶基因或编码靶基因产物的核酸分子中的一个或多个靶序列来修饰细胞、细胞系或生物体。
与现有技术相比,本发明的有益效果在于:
本发明首次通过对TnpB编辑器进行功能域精简的工程化操作,系统开发了新型的380-400个AA的超级迷你型编辑器,本发明的超级迷你新型编辑器,可以高效实现从哺乳类动物细胞到活体动物的精准、高效编辑,具有极大的推广应用价值。
附图说明
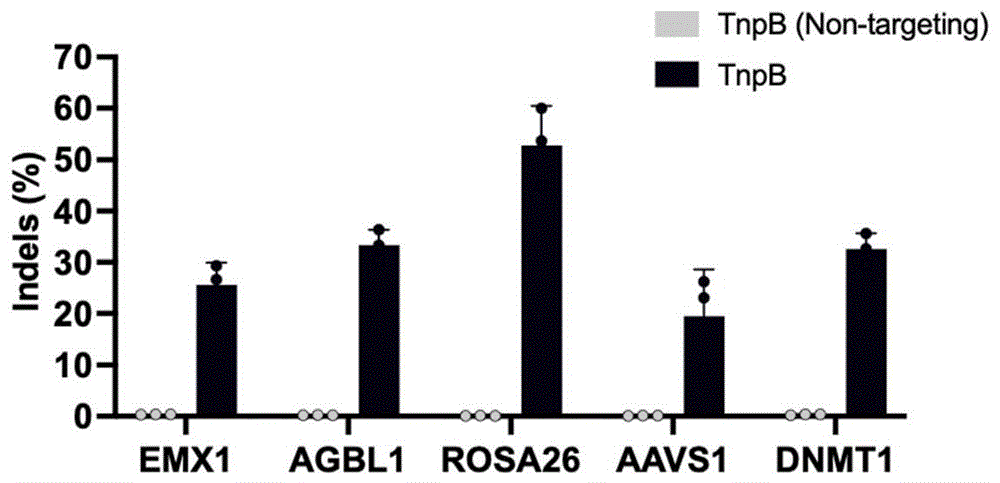
图1为原始TnpB在HEK293T细胞中介导基因编辑效率验证图。
图2为基因编辑器SuperMini-(GE380-GE400)-SWL的开发原理图。
图3为基因编辑器SuperMini-(GE380-GE400)-SWL的开发技术路线图。
图4为基因编辑器SuperMini-(GE380-GE400)-SWL细胞水平介导基因编辑效率验证图。
图5为基因编辑器SuperMini-(GE380-GE400)-SWL体外囊胚率及小鼠出生率效率统计图。
图6为基因编辑器SuperMini-(GE380-GE400)-SWL小鼠囊胚水平介导基因编辑效率验证图。
图7为基因编辑器SuperMini-(GE380-GE400)-SWL小鼠个体水平介导基因编辑效率验证图。
具体实施方式
为使本发明的目的、技术方案和优点更加清楚,下面将结合本发明中的附图,对本发明中的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
下述实施例中所使用的实验方法如无特殊说明,均为常规方法。
下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
本发明提供的可编程核酸酶的基因编辑原理如图2所示,具体为:在SuperMini-(GE380-GE400)-SWL识别TAM序列存在时,reRNA和SuperMini-(GE380-GE400)-SWL蛋白形成的复合物,靶向与reRNA碱基互补配对的DNA序列,DNA双链打开,reRNA与其中一条单链结合,即目标链(TS),此时SuperMini-(GE380-GE400)-SWL蛋白酶的切割活性就会被激活,目标链(TS)和非目标链(NTS)就会被SuperMini-(GE380-GE400)-SWL蛋白酶切割。在合适的TAM存在下,通过设计不同的reRNA序列,可以靶向不同的靶标DNA片段。
实施例1 SuperMini-(GE380-GE400)-SWL的克隆
1.原始TnpB表达载体的构建
实验中所用原始TnpB的氨基酸序列如SEQ ID NO.1所示,基因序列如SEQ ID NO.2所示。基因合成片段由上海生物工程有限公司进行合成。基因片段,pST1374(Addgene,#13426)载体经EcoRI(购自NEB)酶切,然后在重组酶(购自诺唯赞)的作用下重组基因片段和载体。测序鉴定重组质粒载体,然后提取质粒。
2.reRNA的构建
在HEK293T细胞中候选5个内源基因,设计5个reRNA。位点序列如下表1所示。
表1 reRNA识别序列表
根据靶向位点序列设计20个碱基互补配对的上下游引物,加灭菌水溶解至10uM。经过退火分别连接至pGL3-U6-sgRNA(Addgene,#51133)载体上,经过测序验证,构建出靶向特异性reRNA。
3.HEK293FT细胞的培养与转染
复苏HEK293FT细胞(购自ATCC),并培养在10cm培养皿(Corning,430167)中,培养基为含有10%体积比的胎牛血清(HyClone,SV30087)的DMEM(HyClone,SH30243.01)。培养温度为37℃,二氧化碳浓度为5%。传代后当细胞密度为80%时,细胞分盘至12孔板。
(1)细胞分盘12-14h后,细胞浓度约为80%时,使用lipo3000细胞转染试剂(购自Thermo)进行转染;
(2)每孔转染的质粒的总量为1ug,其中TnpB表达质粒和reRNA表达质粒以1:1混合。将混合质粒混匀在75ul的Opti-MEM(Gibco,11058021)培养基中,同时加入2.5μlLipo3000细胞转染试剂,静置5分钟;
(3)另外,将2.75μl的lipo3000细胞转染试剂Lipo3000混入75μl的Opti-MEM培养基,静置5分钟;
(4)将(2)和(3)慢速吹打混匀,静置20分钟;
(5)将上述混和静置好的转染液分别加入培养的细胞中;
(6)转染48-72小时后,去培基,用PBS清洗一次细胞,然后用TE(Thermo Fisher,R001100)将细胞消化下来,再用含有10%FBS的DMEM终止消化,并离心收集细胞,最后用培养基重悬。
4.切割效率验证
取上述收集的细胞的1/6进行直接裂解,并PCR扩增靶向位点片段,引物设计如表2所示。用诺唯赞高保真酶试剂盒(Vazyme,p501-d2)PCR扩增各基因组靶向位点片段。
表2不同靶位点PCR扩增引物
PCR反应体系如表3所示:
表3 PCR反应体系
PCR反应程序参照试剂盒。PCR扩增产物经过AxyPrep PCR Clean-up试剂盒(Axygen,AP-PCR-500G)纯化回收,进行二代测序分析(上海生物工程有限公司)。通过分析发现在检测的5个位点中均有编辑效率,效率为20%-60%(图1)。
实施例2新型超级迷你型编辑器SuperMini-GE380-GE400-SWL的开发及细胞中介导基因编辑活性的验证
1.TnpB功能域截短及效率检测
基于原始TnpB高效的基因编辑,本发明对其结构进行解析,并进行非核心功能域的截短(图2)。本发明中主要针对C端进行功能域的截短,具体不同大小的截短突变体及验证的过程详见图3所示。以实施例1中构建的pST1374-TnpB作为骨架载体,利用FseI和BsaI(均购自NEB)进行酶切,并利用AxyPrep PCR Clean-up试剂盒(Axygen,AP-PCR-500G)纯化回收。随后设计不同截短体,以原始TnpB(SEQ ID 2)作为模板,设计上下游引物(引物如表4所示),利用Forward/Reverse-1,Forward/Reverse-2,Forward/Reverse-3,Forward/Reverse-4,Forward/Reverse-5,Forward/Reverse-6,Forward/Reverse-7分别PCR扩增884bp,911bp,812bp,842bp,902bp,932bp,962bp片段,获得不同截短体,其主要不同在于TnpB-N端存在34aa,25aa,58aa,48aa,28aa,18aa,8aa的氨基酸缺失。利用同源重组方法,将骨架载体与不同截短体扩增产物进行连接,获得不同截短类型的新型基因编辑器。
表4截短体PCR扩增引物设计
随后,对6个截短体进行功能活性的验证,验证方法同实施例1中2-3所示。将截短体TnpB与靶向内源基因ROSA26,EMX1的reRNA分别共转HEK293T细胞,72h后裂解基因组,经过区域PCR扩增及片段回收,首先利用T7E1进行效率验证。
1)PCR产物进行梯度退火
将上述得到的回收PCR产物进行浓度测定,对PCR产物进行梯度退火,得到梯度退火的PCR产物。
上述梯度退火的体系如下表5所示。
表5为梯度退火的体系
上述PCR产物梯度退火程序如表6所示。
表6为PCR产物梯度退火程序
2)对上述1)得到的梯度退火的PCR产物进行T7E1酶切,酶切体系如下表7所示。
表7酶切体系
反应程序:将表7所示的酶切反应在37℃恒温培养箱中进行,酶切反应时间1h,得到酶切产物。
将上述酶切产物进行聚丙烯酰胺凝胶(PAGE)电泳检测,采用8%的聚丙烯酰胺凝胶,电泳结束后,EB对PAGE胶进行染色。T7E1切割效率检测结果如图4A所示,WT为HEK293T未经任何转染的细胞,ROSA26代表原始TnpB在内源基因ROSA26上的切割活性,T1-T7代表不同截短后的TnpB(即超级新型迷你型TnpB,也就是本发明命名的SuperMini-(GE380-GE400)-SWL)在内源基因ROSA26上的切割活性,Cas代表在该区域设计的CRISPR/Cas9对照靶点的切割活性;结果表明,在HEK293T细胞中,靶向内源基因ROSA26位点,截短至380个aa时,其仍然具有同原始TnpB相近的切割活性。当对其进一步进行截短时,其切割活性失活。与设计的Cas9靶点切割效率相比,未见明显显著性差异。
靶向内源基因EMX1,AGBL1的T7E1结果进一步证实上述截短体的结果,即当TnpB截短至380个aa时,其仍然具有同原始TnpB相近的切割活性。当对其进一步进行截短时,其切割活性失活(图4C,E)。值得注意的是,与该位点的Cas9靶点切割效率相比,TnpB及各截短体的切割活性显著高于Cas9。
随后对上述PCR产物进行扩增子测序,测序结果利用在线软件CRISPResso2进行效率统计分析。二代测序分析结果进一步证实了T7E1实验结果(图4B,D,F)。以上结果充分说明,本发明获得的新型超级迷你型基因编辑器SuperMini-(GE380-GE400)-SWL可以有效的介导细胞水平的基因编辑。
实施例3新型超级迷你型基因编辑器SuperMini-(GE380-GE400)-SWL介导小鼠囊胚及个体水平基因编辑活性验证
本实施例以380AA,390AA,400AA截短型SuperMini-GE380-SWL/SuperMini-GE390-SWL/SuperMini-GE400-SWL以及原始TnpB作为底盘,对其介导囊胚及个体水平的基因编辑活性进行验证。为了便于观察小鼠表型,我们候选tyr基因作为靶目标,该基因的纯合突变可以导致小鼠的体毛由黑色变为白色,流程图如图5A所示。
1.靶向Tyr基因SuperMini-(GE380-GE400)-SWL表达载体的构建
本研究选择Tyr基因外显子1区域作为打靶位点,设计reRNA(图5B所示)。随后设计靶向位点序列设计20个碱基互补配对的上下游引物,Forward如SEQ ID NO.26所示,Reverse如SEQ ID NO.27所示,加灭菌水溶解至10uM。经过退火分别连接至pGL3-U6-sgRNA(Addgene,#51133)载体上,经过测序验证,构建出靶向特异性reRNA。
2.显微注射制备tyr基因编辑囊胚及小鼠
1)对适龄小鼠进行超数排卵,具体为给每只小鼠腹腔注射10IU的PMSF(购自宁波激素第二制药厂),48h后,再腹腔注射10IU的HCG(购自宁波激素第二制药厂);
2)注射HCG后,将母鼠与种公鼠1:1进行交配,同时将适龄的受体母鼠与结扎公鼠1:1进行交配;
3)交配的第二天,挑选见栓的母鼠和受体母鼠;
4)对见栓的母鼠进行脱臼处死,剪开皮肤,用眼科剪刀和镊子取出小鼠左右两侧卵巢,找到输卵管的膨大部;
5)用胚胎操作针将受精卵从输卵管的膨大部剥离出,用透明质酸酶(购自Sigma)处理后,收集于胚胎操作液中(购自Sigma);
6)将SuperMini-GE380-SWL,SuperMini-GE390-SWL,SuperMini-GE400-SWL,TnpB及reRNA进行体外转录(购自Thermo),将体外转录物以1:1混合,共计200ng/ul,然后利用显微操作系统将混合物注入动物胚胎中,半小时后,将状态较好的注射后的受精卵或置于小鼠胚胎培养液中(购自Sigma)进行体外培养或移植至代孕母鼠体内。对于体外培养的受精卵,3.5d后可收集囊胚进行检测;对于移植的受精卵,19天后,小鼠出生,对其表型进行观察并对其基因型进行分析。对其囊胚率进行统计分析发现,SuperMini-GE380-SWL,SuperMini-GE390-SWL,SuperMini-GE400-SWL,TnpB注射后,囊胚率分别为73.3%,75%,78.7%,73.8%,无显著性差异,同时这一数据与野生型对照组体外培养囊胚率基本一致(图5C所示)。进一步对小鼠出生率进行统计分析SuperMini-GE380-SWL,SuperMini-GE390-SWL,SuperMini-GE400-SWL,TnpB注射后,小鼠出生率分别为37.5%,34%,40.4%,39.1%。与文献中报道的基因编辑个体出生率比较一致(图5D所示)。以上结果证实注射组分对囊胚形成及个体出生无毒副作用。
3.小鼠单囊胚检测分析
随后,收集小鼠的单囊胚,对单囊胚进行效率统计分析。具体操作如下:
1)将小鼠囊胚置于PBS中(购自Gibco),润洗3遍;
2)将10ul胚胎裂解液(购自Thermo)分装至200ul PCR操作管中,用显微注射针吸取单囊胚至裂解液中,上述体系放置于PCR仪中,运行以下程序:55度2h;95度10min;
3)处理好的单囊胚进行巢式PCR扩增,其中Tyr-F/R用于第一轮扩增;Tyr-F1/R1用于第二轮扩增;巢式扩增引物设计如下:
Tyr-F:TTAACCTATTGGTGCAG(SEQ ID NO.28);
Tyr-R:TTAACCTATTGGTGCAG(SEQ ID NO.29);
Tyr-F1:GTGGATGACCGTGAGTCCT(SEQ ID NO.30);
Tyr-R1:CCCAGTTAGTTCTCGAATTTC(SEQ ID NO.31);
PCR扩增体系同实施例1中1.4中所述。对单囊胚扩增获得的PCR扩增产物进行T7E1实验(方法见实施例2)及二代测序分析。结果如图6所示,SuperMini-GE380-SWL,SuperMini-GE390-SWL,SuperMini-GE400-SWL均可以有效介导小鼠囊胚水平的基因编辑。
4.小鼠个体水平基因型分析
待小鼠出生2周后,对小鼠表型进行观察。表型分析结果发现,注射后出生的小鼠表现出不同程度的白化病表型(图7A所示)。通过提取小鼠尾部基因组进行PCR扩增(扩增体系同上所述)及二代测序分析,小鼠在Tyr基因出现不同程度的基因编辑(图7B-C所示)。
综上所述,本发明有效克服了现有基因编辑工具的缺点,发现的新型超级迷你型基因编辑器可以高效介导细胞/个体水平的基因编辑,具有高度产业利用价值。
最后应说明的是:以上实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的精神和范围。
- 一种基因编辑融合蛋白、其构建的腺嘌呤碱基编辑器及其应用
- 应用转基因技术提高超级水稻产量的一种方法