一种修复HBA2基因突变的方法、组合物及其应用
文献发布时间:2024-04-18 20:01:23
技术领域
本发明涉及基因编辑技术领域,特别是涉及一种修复HBA2基因突变的方法、组合物及其应用。
背景技术
地中海贫血症是一种遗传性溶血性的贫血,在我国长江以南广泛分布,其中广西地区最为严重。根据基因分型,临床上主要有α地中海贫血、β地中海贫血。引发α地中海贫血的突变分缺失型和非缺失型。非缺失型α地贫是由于α基因核苷酸的“点突变”导致。广东、广西及四川等地的α地贫中,非缺失型占35%~60%。我国较常见的非缺失型α-地贫有3种:Hb-CS(CD142)、Hb-QS(CD125)和Hb-WS(CD122)。其中Hb-CS型患者病情相对最严重,血红蛋白低,生存率不高。
血红蛋白CS(Hb-CS,Haemoglobin-Constant Spring,α142,Term→Gln,TAA>CAA(α2),α
造血干细胞(HSC,hematopoietic stem cell)移植是治愈严重地中海贫血的有效手段和唯一途径,而缺乏HLA匹配的健康供体、免疫并发症和病毒载体安全问题限制了其使用。
常规技术中,也有一些针对α地贫的基因治疗方法,如过表达α珠蛋白、ζ珠蛋白等基因,但此类方法存在显著的安全问题,例如过表达的珠蛋白基因拷贝随机整合到基因组数千个不同的位点,导致插入突变和恶性肿瘤的可能性。CN108165581B报导通过核酸酶切割靶位点,以及同源介导的基因修复方式对Hb-CS位点进行修复,但是编辑效率低,不足以达到治疗的水平。
发明内容
基于此,有必要针对上述问题,提供一种修复HBA2基因突变的方法、组合物及其应用,采用该方法或组合物,对HBA2基因的Hb-CS致病突变位点进行HDR修复,编辑效率高,能够达到治疗水平,并且,gRNA导向序列的靶区域为HBA2特异性靶位点,能够避免对HBA1进行切割;同时,修复的目的基因表达受天然的HBA2全部调控元件调控,具有较高的安全,也更能平衡α/β珠蛋白的表达。
本发明公开了一种修复HBA2基因突变的方法,包括以下步骤:将核酸酶、gRNA和供体修复模板与待编辑修复的HBA2基因序列接触,使突变的HBA2基因CD142密码子中的胞嘧啶碱基修复为胸腺嘧啶;
所述gRNA的导向序列与Chr16:173596-173624靶区域结合或反向互补。
本发明所述染色体定位对应于NCBI数据库GRch38版本。
上述修复HBA2基因突变的方法,将核酸酶、gRNA和供体修复模板与待编辑修复的HBA2基因序列接触,使HBA2基因CD142密码子中的胞嘧啶碱基修复为胸腺嘧啶,gRNA导向序列的靶区域为HBA2特异性靶位点,能够避免对HBA1进行切割,并且,编辑效率高,能够达到治疗水平,提供了一种更高效、更安全的修复Hb-CS突变的方法。
在一些实施例中,所述修复HBA2基因突变的方法用于非治疗目的,如科学研究等。
本发明还公开了上述的修复HBA2基因突变的方法在制备用于治疗α地中海贫血症的药物中的应用。
本发明还公开了一种H修复HBA2基因突变的组合物,包括:核酸酶、gRNA和供体修复模板,用于使突变的HBA2基因CD142密码子中的胞嘧啶碱基修复为胸腺嘧啶;
所述gRNA的导向序列与Chr16:173596-173624靶区域结合或反向互补。
对于组合物中的gRNA,其可以是单分子gRNA(single gRNA)、双分子gRNA(dgRNA,dual gRNA)或多分子gRNA。gRNA可替换为其它包含可与靶位点互补配对结合的导向序列、与核酸酶连接或结合的骨架序列的RNA分子。
在一些实施例中,所述导向序列与Chr16:173596-173615、Chr16:173597-173616、Chr16:173598-173617、Chr16:173599-173618、Chr16:173600-173619、Chr16:173596-173616、Chr16:173596-173617、Chr16:173596-173618、Chr16:173596-173619、Chr16:173596-173620、Chr16:173597-173617、Chr16:173597-173618、Chr16:173597-173619、Chr16:173597-173620、Chr16:173597-173621、Chr16:173598-173618、Chr16:173598-173619、Chr16:173598-173620、Chr16:173598-173621、Chr16:173598-173622、Chr16:173599-173619、Chr16:173599-173620、Chr16:173599-173621、Chr16:173599-173622、Chr16:173599-173623、Chr16:173600-173620、Chr16:173600-173621、Chr16:173600-173622、Chr16:173600-173623、Chr16:173600-173624任一靶区域结合或反向互补。
在一些实施例中,所述导向序列选自SEQ ID NO.10-SEQ ID NO.39所示任一序列。
在一些实施例中,所述gRNA的导向序列的长度为20-25bp;和/或
所述gRNA为化学合成修饰的gRNA。
综合gRNA结构和上述导向序列匹配靶核酸区域设计而言,gRNA的导向序列的长度应至少为20bp,在对HBA2基因CD142密码子中的胞嘧啶碱基进行HDR修复的同时,避免对HBA1基因进行编辑,保证对HBA2靶位点的特异性。
在一些实施例中,所述核酸酶选自Cas9、CasX、CasY、Cpf1、C2c1、cas12b2、cas12h、cas12i、cas12g、C2c2、C2c3、C2c4、C2c5、C2c6、C2c7、c2c8、c2c9、及其变体中的至少一种。
可以理解的,所述核酸酶可以有最优PAM(识别位点的毗邻基,protospaceradjacent motif,PAM)位点,也可以无最优PAM位点或无PAM位点限制。
在一些实施例中,所述核酸酶没有PAM位点限制。
在一些实施例中,所述核酸酶具有最优PAM位点。例如,最优PAM位点为NGG、NG、NNNNCC、NAA、NAAN、NGGNG、NNNNGATT、NNNNCNDD、NNNNCAAA、NNNNCRAA、NNAGAAW、NNGRRN、TTN、TTTV/TTTN、ATTN或TTCN(N表示A/T/C/G,R表示A/G,W表示A/T,V表示A/C/G)等。
在一些实施例中,所述核酸酶选自SpRY Cas9变体、NG-Cas9变体、SpCas9中的至少一种。上述SpRY Cas9变体即为无PAM位点限制的SpRY核酸酶变体、NG-Cas9变体为最优PAM为NG的Cas9、SpCas9为最优PAM为NGG的Cas9。
在一些实施例中,所述核酸酶为SpRY Cas9,所述导向序列选自SEQ ID NO.13-SEQID NO.14、SEQ ID NO.30-SEQ ID NO.39所示任一序列。
在一些实施例中,所述核酸酶为NG-Cas9,所述导向序列选自SEQ ID NO.11-SEQID NO.12、SEQ ID NO.20-SEQ ID NO.29所示任一序列。
在一些实施例中,所述核酸酶为SpCas9,所述导向序列选自SEQ ID NO.10、SEQ IDNO.15-SEQ ID NO.19所示任一序列。
在一些实施例中,所述供体修复模板包含5'同源臂、替换序列和3'同源臂;
所述供体修复模板选自双链DNA或单链DNA。
在一些实施例中,所述供体修复模板为单链DNA;
所述5'同源臂或所述3'同源臂的序列长度任选自:30-100nt。
在其中一些实施例中,所述供体修复模板为单链供体修复模板(Single-strandeddonor oligonucleotides,ssODN),ssODN可为HBA2基因编辑序列的正义链或反义链。
在其中一些实施例中,所述5'同源臂或3'同源臂的序列长度任选自:30-100nt。例如可以为30-100nt、30-80nt、30nt-60nt、30nt-50nt、30nt-45nt、30nt-40nt。上述任选指5'同源臂或3'同源臂可具有不同的序列长度,如在其中一些实施例中,5′同源臂、3′同源臂选用长度分别为41nt和39nt。
在一些实施例中,所述5'同源臂和/或所述3'同源臂末端具有硫代磷酸(Phosphorothioate)修饰。上述修饰可增强ssODN的稳定性和提高其在基因编辑中的活性。
在一些实施例中,所述5'同源臂和/或所述3'同源臂末端的2个核苷酸具有硫代磷酸修饰。可以理解的,也可根据不同的实际情况,对供体修复模板5'同源臂或3'同源臂末端的1个,3个,4个,5个等核苷酸进行硫代磷酸修饰。
在一些实施例中,当所述供体修复模板序列对应于HBA2基因正义链,所述供体修复模板序列选自SEQ ID NO.1、SEQ ID NO.3和SEQ ID NO.5所示任一序列;
当所述供体修复模板序列对应于HBA2基因反义链,所述供体修复模板序列选自SEQ ID NO.2、SEQ ID NO.4和SEQ ID NO.6所示任一序列。
在其中一些实施例中,组合物可采用递送系统递送,可采用本领域的常规递送系统,如质粒、脂质体递送、脂质纳米颗粒递送、外囊泡EV、病毒颗粒或电穿孔等均可。
在其中一些实施例中,所述供体修复模板为ssODN,可将ssODN的cDNA序列装载在病毒载体中,递送入造血干/祖细胞或者体内表达,即可达到基因修复的治疗目的。
在其中一些实施例中,可以基因工程的方式对上述组合物进行制备、转化等程序。
本发明还公开了一种用于修复HBA2基因突变的gRNA,包含上述的gRNA。
本发明还公开了一种细胞,所述细胞包含上述的修复HBA2基因突变的组合物;和/或
所述细胞经上述的组合物进行HDR编辑修复获得。
在一些实施例中,所述细胞为细胞系和/或原代细胞。
在一些实施例中,所述细胞为造血干/祖细胞、诱导多能干细胞、红系祖细胞等具有诱导分化或转分化或重编程为成熟红细胞潜能的细胞。
在一些实施例中,可将含有Hb-CS突变的体细胞重编程为iPSC细胞,导入修复Hb-CS突变的gRNA和单碱基编辑器递后,修复Hb-CS突变,并诱导分化为造血干/祖细胞或红系祖细胞。
本发明还公开了一种修复HBA2基因突变的试剂,包括:
1)上述的核酸酶,和/或编码所述核酸酶的核苷酸序列;和/或
2)上述的gRNA,和/或编码所述gRNA的核苷酸序列,和/或包含编码所述gRNA的核苷酸序列的核苷酸组合物;和/或
3)上述的供体修复模板,和/或编码所述供体修复模板的核苷酸序列,和/或包含编码所述供体修复模板的核苷酸序列的核苷酸组合物。
本发明还公开了上述的组合物、上述的gRNA、上述的细胞、上述的试剂在制备用于治疗α地中海贫血症的药物中的应用。
与现有技术相比,本发明具有以下有益效果:
本发明的一种修复HBA2基因突变的方法,采用HDR编辑的方式,将核酸酶、gRNA和供体修复模板与待编辑修复的HBA2基因序列接触,使HBA2基因CD142密码子中的胞嘧啶碱基修复为胸腺嘧啶,gRNA的导向序列与Chr16:173596-173624靶区域结合或反向互补,编辑效率高,能够达到治疗水平;并且,gRNA导向序列的靶区域为HBA2特异性靶位点,能够避免对HBA1进行切割;同时,修复的目的基因表达受天然的HBA2全部调控元件调控,具有较高的安全,也更能平衡α/β珠蛋白的表达。
附图说明
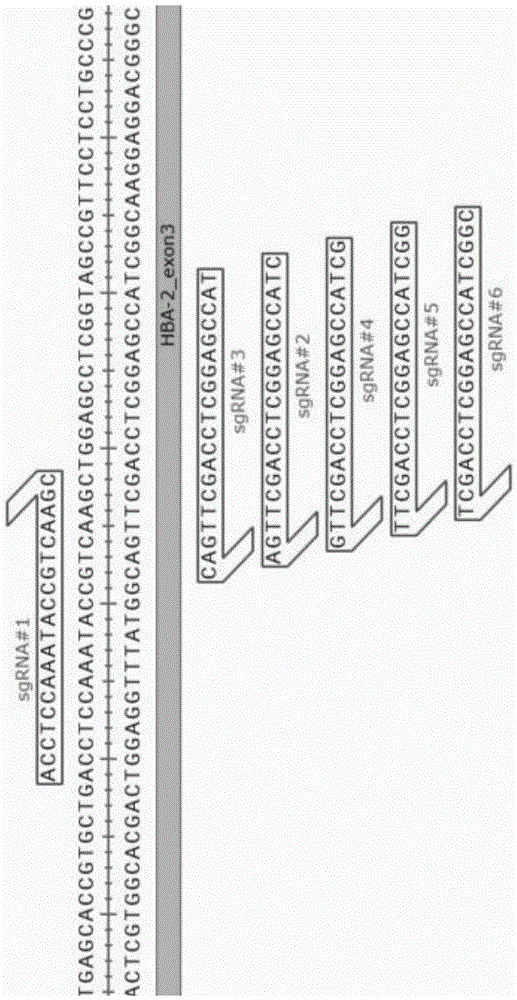
图1为实施例2中gRNA导向序列靶向区域示意图;
图2为实施例3中HL60(--
图3为实施例3中HL60(--
图4为实施例4HL60(--
图5为实施例4HL60(--
图6为实施例4各组别HL60(--
图7为实施例6hCD34(--
图8为实施例6hCD34(--
图9为实施例6采用不同sgRNA修复hCD34(--
图10为实施例7hCD34(--
图11为实施例7hCD34(--
图12为实施例8hCD34(--
图13为实施例8hCD34(--
图14为实施例9hCD34(--
图15为实施例10hCD34(--
具体实施方式
术语定义
本文使用的术语“HDR”,即同源重组修复(Homology directed repair),指使用同源核酸(例如,内源同源序列(例如姐妹染色单体)或外源核酸(例如模板核酸))修复DNA损伤的过程。
本文使用的术语“变体”指一组高度相似的蛋白质成员,这些成员同源于单个蛋白或蛋白家族,具有相同或相似的生物学作用。例如,包含核酸酶或其片段的蛋白质可称为“核酸酶变体”,如Cas9变体。核酸酶变体与核酸酶或其片段共享同源性。例如,核酸酶与野生型核酸酶至少约70%相同、至少约80%相同、至少约90%相同、至少约95%相同、至少约96%相同、至少约97%相同、至少约98%相同、至少约99%相同、至少约99.5%相同或至少约99.9%相同。
在一些实施方案中,核酸酶变体与野生型核酸酶相比,可以具有1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、21、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50个或更多个氨基酸变化,但其依然具有与可编程核苷酸如gRNA等结合并靶向核酸靶点的功能。
本文使用的术语“化学合成修饰的gRNA”指可采用本领域技术人员熟知的化学修饰方法进行修饰,如在一些实施例中,以2’-O-甲基(2’-O-Me)修饰、2’-氟(2'-F)修饰。在一些实施例中,所述gRNA核苷酸之间的修饰方式包含硫代磷酸酯(PS)键。在一些实施例中,5’末端处的前三个核苷酸,及3’末端处的最后三个核苷酸经修饰。在一些实施方案中,5’末端处的前四个核苷酸,及3’末端处的最后四个核苷酸与硫代磷酸酯(PS)键连接。
在一些实施例中,gRNA也可使用修饰的核糖核苷酸、脱氧核糖核苷酸、其他合成碱基和合成骨架连接(诸如肽核酸(PNA)、锁核酸(LNA)等)。在一些实施例中,构成引导核苷酸序列的核酸包含天然核苷(例如腺苷、胸苷、鸟苷、胞苷、尿苷、脱氧腺苷、脱氧胸苷、脱氧鸟苷和脱氧胞苷);核苷类似物(例如2-氨基腺苷、2-硫代胸苷、肌苷、吡咯并嘧啶、3-甲基腺苷、5-甲基胞苷、2-氨基腺苷、C5-溴尿苷、C5-氟尿苷、C5-碘尿苷、C5-丙炔基-尿苷、C5-丙炔基-胞苷、C5-甲基胞苷、2-氨基腺苷、7-脱氮腺苷、7-脱氮鸟苷、8-氧代腺苷、8-氧代鸟苷、O(6)-甲基鸟嘌呤和2-硫代胞苷);化学修饰的碱基;生物修饰的碱基(例如甲基化碱基);插入的碱基;经修饰的糖(例如2’-氟核糖、核糖、2’-脱氧核糖、阿拉伯糖和己糖);和/或经修饰的磷酸基团(例如硫代磷酸酯和5’-N-亚磷酰胺连接)。
为了便于理解本发明,下面将参照相关附图对本发明进行更全面的描述。附图中给出了本发明的较佳实施例。但是,本发明可以以许多不同的形式来实现,并不限于本文所描述的实施例。相反地,提供这些实施例的目的是使对本发明的公开内容的理解更加透彻全面。
除非另有定义,本文所使用的所有的技术和科学术语与属于本发明的技术领域的技术人员通常理解的含义相同。本文中在本发明的说明书中所使用的术语只是为了描述具体的实施例的目的,不是旨在于限制本发明。本文所使用的术语“和/或”包括一个或多个相关的所列项目的任意的和所有的组合。
以下实施例所用试剂,如非特别说明,均为市售可得;以下实施例所用方法,如非特别说明,均为常规方法可实现。
实施例1
用于α地贫基因治疗的供体修复模板ssODN的设计和构建。
ssODNs(单链供体寡核苷酸)结构上包含5′同源臂、替换序列、3′同源臂。同源臂是与HBA2基因组Hb-CS突变位点两侧的DNA区域同源的序列,单侧同源臂序列长度可约为30nt-100nt,本实施例中5′同源臂、3′同源臂选用长度分别为60nt和60nt。所涉及的ssODN序列如下表所示。
表1ssODN序列信息
注:下划线碱基表示相对于HBA2基因突变序列的替换。编号中的s表示HBA2基因正义链(sense),as表示HBA2基因反义链(antisense)。
实施例中ssODN均为5'同源臂和3'同源臂末端的2个核苷酸具有硫代磷酸修饰的序列,即序列中以s表示。ssODN两侧末端的2个核苷酸使用硫代磷酸(Phosphorothioate)修饰以增强ssODN的稳定性和提高其在基因编辑中的活性。
设计完成后,ssODN交给序列合成公司进行合成,并将商业化合成的ssODN粉末(Invitrogen公司)离心,用dd H
实施例2
gRNA的设计和构建
sgRNA包含导向序列、骨架序列,导向序列与HBA2基因的靶向序列互补。本实施例选择长度为100nt的sgRNA,其5′端为20nt的导向序列,后面80nt为通用的sgRNA骨架序列,具体序列为:5’-GTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGT CCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCTTTT-3’(SEQ ID NO.7),如sgRNA#1的完整序列为5’-
表2各sgRNA信息
上表中sgRNA#1-sgRNA#6的靶向序列分布如图1所示。
依据上表2的gRNA导向序列信息,通过化学方式合成和修饰获得甲基和硫代磷酸酯键修饰的gRNA,备用。
如合成修饰的单分子的sgRNA#2合成序列和修饰为:5'-
实施例3
HBA2基因CS突变的人源类红细胞HL60(--
1、RNP电穿孔1
将商业化的spCas9蛋白(恺佧生物,货号#EE109)和化学合成的sgRNA(sgRNA指导序列为:5’-GGTCGTCCCCACTGTCGTCG-3’,SEQ ID NO.40和5’-GATGGCTACTCGGAGATATA-3’,SEQ ID NO.41),按照100pmol:300pmol比例混合,在体外孵育包装成RNP。通过lonza-4D电转仪将与SpCas9/sgRNARNP导入人原髓系白血病细胞HL60(αα
2、HL60(--
电转72小时后,取少量细胞悬液计数,将细胞悬液移至刻度离心管中连续倍数稀释至10个细胞/ml。将稀释好的细胞悬液接种到96孔板中,每孔加液0.1ml。然后放入培养箱内培养。96小时后,在倒置显微镜下观察培养板各孔细胞数,挑选只含单个克隆细胞团的孔,补加0.1ml培养液继续培养。扩增传代至细胞数量达到5×10
3、RNP电穿孔2
将商业化的spCas9蛋白和化学合成的sgRNA(sgRNA指导序列为:5’-ACCTCCAAATACCGTTAAGC-3’,SEQ ID NO.46和5’-CTACCGAGGCTCCAGCTTAA-3’,SEQ IDNO.47),按照100pmol:300pmol比例混合,在体外孵育包装成RNP。与ssODN(C-s-A-s-CGCCTCCCTGGACAAGTTCCTGGCTTCTGTGAGCACCGTGCTGACCTCCAAATACCGT
4、HL60(--
电转72小时后,取少量HL60(--
实施例4
在人源类红细胞HL60(--
将商业化的SpCas9/NG-Cas9/SpRY Cas9 mRNA和各自对应的化学合成的sgRNA#1-sgRNA#6(单分子gRNA,两端末三个核苷酸进行硫代磷酸化和甲基化修饰),按照2μg:2μg比例混合。其中,SpCas9对应sgRNA#1-sgRNA#2,NG-Cas9对应sgRNA#3-sgRNA#4,SpRY Cas9对应sgRNA#5-sgRNA#6,利用Lonza4D电转仪将SpCas9/NG-Cas9/SpRY Cas9mRNA与sgRNA、ssODN(1μL)导入人类红细胞HL60(--
经过电穿孔处理后72小时,收集细胞提取基因组DNA,PCR扩增HBA2基因编辑片段,以实施例3中的测序引物组进行测序分析。
该实验重复三次,测序结果经EditR软件分析,实验结果见图4-6所示。图4为sgRNA#3+ssODN1-as组HL60(--
图6显示,仅电转sgRNA#2,人类红细胞HL60(--
实施例5
不同长度gRNA介导HDR编辑修复Hb-CS突变实验。
本实施例参照实施例4方法,与sgRNA#3+ssODN1-as组的处理区别仅在于:采用不同长度gRNA,即将sgRNA#3替换为sgRNA#12、sgRNA#13、sgRNA#14、sgRNA#15或sgRNA#16,在人源类红细胞HL60(--
表3不同长度gRNA介导HDR编辑修复Hb-CS突变的编辑效率结果
以上结果表明,靶区域相同条件下,21nt-25nt导向序列的gRNA与20nt导向序列的gRNA相比,介导HDR编辑修复Hb-CS突变的编辑效率无明显区别。
实施例6
在病人来源的造血干祖细胞中HDR修复Hb-CS突变实验。
通过广州市某三甲医院获取含有HBA2基因Hb-CS纯合突变(--
将商业化的SpCas9/NG-Cas9/SpRY Cas9 mRNA和各自对应的化学合成的sgRNA#1-sgRNA#6(单分子gRNA,两端末三个核苷酸进行硫代磷酸化和甲基化修饰),按照2μg:2μg比例混合,加入1μL ssODN,利用Lonza4D电转仪将SpCas9/NG-Cas9/SpRY Cas9 mRNA与sgRNA、ssODN导入CD34阳性细胞,电转程序为EO-100。其中,SpCas9对应sgRNA#1-sgRNA#2,NG-Cas9对应sgRNA#3-sgRNA#4,SpRY Cas9对应sgRNA#5-sgRNA#6。
经过电穿孔处理后12小时,按照1μM终浓度加入SCR7(MCE,货号#HY-12742)继续培养。经过电穿孔处理后72小时,收集细胞提取基因组DNA,PCR扩增HBA2基因编辑片段,以实施例3中的测序引物组进行测序分析。另PCR扩增HBA2基因编辑片段进行NGS(二代测序)分析(PCR正向引物为CGCCTCCCTGGACAAGTTC,SEQ ID NO.52;反向引物为TTGGCACATTCCGGGATAGA,SEQ ID NO.53)。
该实验重复3次,实验结果见表4和图7-9所示。图7为组合4(ssODN#1-as和sgRNA#3)效率Sanger测序分析结果图,图8为组合4(ssODN#1和sgRNA#3)效率NGS分析结果,表4和图9为实施例6采用不同sgRNA修复hCD34(--
表4 ssODN和sgRNA组合及其目的修复效率
基因编辑效率显示,尽管ssODN#1-as联合gRNA#1编辑效率较低,而ssODN#1-as联合gRNA2-6,编辑效率显著上升,表明较靶向Chr16:173583-173602区域相比,采用HDR进行CS位点修复,靶向Chr16:173596-173615、Chr16:173597-173616、Chr16:173598-173617、Chr16:173599-173618、Chr16:173600-173619能够获得更高的编辑效率。
实施例7
对病人来源的造血干祖细胞HDR修复Hb-CS突变后分化RBC,并进行珠蛋白检测。
通过广州市某三甲医院获取含有HBA2基因Hb-CS纯合突变的地中海贫血患者动员外周血来源的人CD34阳性细胞(批次编号:HBA-R-202305-001),细胞复苏后,在富含人类细胞因子(SCF、TPO、Flt3L,各100ng/ml)的StemSpan无血清扩增培养基(StemCell公司,StemSpan
将商业化的NG-Cas9 mRNA和sgRNA#3/sgRNA#4(单分子gRNA,两端末三个核苷酸进行硫代磷酸化和甲基化修饰),按照2μg:2μg比例混合,利用Lonza4D电转仪将NG-Cas9mRNA/sgRNA混合物和ssODN1-as(1μL)导入CD34阳性细胞,电转程序为EO-100。其中,编辑-1组CD34阳性细胞导入了NG-Cas9 mRNA、sgRNA#3和ssODN1-as,编辑-2组CD34阳性细胞导入了NG-Cas9 mRNA、sgRNA#4和ssODN1-as,NC组未经电穿孔处理。
经过电穿孔处理后12小时,按照1μM终浓度加入SCR7(MCE,货号#HY-12742)继续培养。电转24h后,使用100X Erythroid Expansion Supplement试剂(StemSpan,货号#02692)诱导电转后细胞进行红系分化。红系诱导分化流程:在室温解冻或2-8℃过夜解冻SFEMII培养基(StemSpan,货号#09605)。分装,-20℃保存。将100X Erythroid ExpansionSupplement试剂和SFEMII培养基按照1:99比例混合配置分化完全培养基。诱导分化的第0天,使用分化完全培养基培养电转细胞,细胞密度为1x10
诱导分化的第18天,取出细胞,分成2份,一份利用商业化RIPA裂解液(碧云天,货号#P0013B)及PMSF(碧云天,货号#ST507)冰上裂解30min,加入SDS-PAGE蛋白染色及上样缓冲液(碧云天,货号#P0280)煮沸5min,冰上静置2min后用于进行Western Blot检测,结果如图10所示;另一份细胞直接冻沉淀于负八十,送样进行MALID-TOF-MS检测,蛋白检测结果如图11所示,统计结果如表5所示。
表4 MALID-TOF-MS检测结果统计
Western Blot检测结果显示,与NC组相比,编辑-1组和编辑-2组样本β珠蛋白表达水平无变化,而CS珠蛋白表达水平显著下降,提示CS基因得到修复,但是由于Western Blot过程中存在信号放大,正常α珠蛋白本底较高,编辑-1组和编辑-2组样本α珠蛋白并未观察到显著变化。同时将部分样本进行了MALID-TOF-MS检测,结果显示与NC样本相比,编辑-1组和编辑-2组正常α珠蛋白峰显著提升,表3统计结果显示编辑-1组和编辑-2组α/β样珠蛋白水平显著上升。综合以上结果可知CS突变修复后的患者CD34阳性细胞可有效表达正常α珠蛋白。
实施例8
动物实验研究患者来源细胞编辑后的体内维持水平。
本实施例将实施例7采用NG-Cas9 mRNA、sgRNA#3和ssODN1-as组合进行HDR编辑修复后的患者CD34阳性细胞通过尾静脉注射到高度免疫缺陷小鼠(小鼠品系:NCG-X,集萃药康)体内,在移植后第16周取骨髓细胞,收集细胞提取基因组DNA,PCR扩增HBA2基因编辑片段,以实施例6中的NGS测序引物组进行测序分析。检测结果示意图如图12所示,统计结果如图13所示。
图12结果显示,未编辑的细胞回输后小鼠检测编辑效率几乎为0,而编辑组小鼠可检测到有效编辑。图13统计结果显示编辑细胞回输小鼠体内16周后,编辑效率均有少量下降,编辑效率的个体差异较小,表明编辑后CS位点修复的CD34阳性细胞可有效在小鼠体内长期维持,表明其有潜在的植入能力。
实施例9
动物实验研究患者来源细胞编辑后的生物活性。
本实施例将实施例7采用NG-Cas9 mRNA、sgRNA#3和ssODN1-as组合进行HDR编辑修复后的患者CD34阳性细胞通过尾静脉注射到高度免疫缺陷小鼠(小鼠品系:NCG-X,集萃药康)体内,在移植后第16周取骨髓细胞。利用hCD34磁珠(美天旎,货号#130-046-702)及hCD235(美天旎,货号#130-050-501)磁珠对骨髓中人源造血干细胞及人源红细胞前体细胞进行分选。分选后,使用100X Erythroid Expansion Supplement试剂(StemSpan,货号#02692)诱导细胞进行红系分化。
红系诱导分化流程:在室温解冻或2-8℃过夜解冻SFEMII培养基(StemSpan,货号#09605)。分装,-20℃保存。将100X Erythroid Expansion Supplement试剂和SFEMII培养基按照1:99比例混合配置分化完全培养基。诱导分化的第0天,使用分化完全培养基培养电转细胞,细胞密度为1x 10
诱导分化的第18天,取出细胞,利用商业化RIPA裂解液(碧云天,货号#P0013B)及PMSF(碧云天,货号#ST507)冰上裂解30min,加入SDS-PAGE蛋白染色及上样缓冲液(碧云天,货号#P0280)煮沸5min,冰上静置2min后,进行Western Blot检测,结果如图14所示。
体内来源的细胞分化后Western Blot结果显示,与NC相比,编辑组细胞的CS珠蛋白表达水平显著下降,该结果与体外结果一致,说明体内植入细胞可有效维持其生物活性。
实施例10
病人来源的造血干/祖细胞中通过HDR修复Hb-CS突变的同时不影响HBA1的正常功能。
通过广州市某三甲医院获取含有HBA2基因Hb-CS纯合突变(--
将NG-Cas9 mRNA和化学合成的sgRNA#3(单分子gRNA,两端末三个核苷酸进行硫代磷酸化和甲基化修饰),按照2μg:2μg比例混合,加入1μL ssODN,利用Lonza4D电转仪将SpRY/sgRNA/ssODN导入人源细胞hCD34(--
经过电穿孔处理后12小时,按照1μM终浓度加入SCR7(MCE,货号#HY-12742)继续培养。经过电穿孔处理后72小时,收集细胞提取基因组DNA,PCR扩增HBA1基因编辑片段进行NGS测序分析(PCR正向引物为CGGGTTGCGGGAGGTGTAGCGCAGG,SEQ ID NO.54;反向引物为AGGCCCAAGGGGCAAGAAGCATGGCC,SEQ ID NO.55)。分析结果如图15所示,结果显示,NC与编辑组细胞在HBA1位点均未出现插入缺失突变,图15中编辑组出现的效率低于1%的点突变片段在NC中也低频出现,猜测为PCR引入的点突变序列。该结果表明,采用sgRNA#3对Hb-CS位点进行HDR编辑不会诱发HBA2复等位基因HBA1的插入缺失突变,保证对HBA2靶位点的特异性,安全性高。而sgRNA#1除3’端第五位C碱基外,其余核苷酸序列与HBA1序列完全一致,且切割位点距离终止密码子较近,可能在HBA1位点引起脱靶,发生Indel,损伤病人原本正常的HBA1基因。
综上,本发明筛选出了修复Hb-CS突变的HDR编辑修复组合物,兼顾安全性的同时,基因修复效率高,同时,修复后的造血干组细胞血红蛋白表达得到很大提升,能够达到治疗水平,有望运用于临床实践中,以有效缓解患者的病情并持续摆脱输血的状况。
以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
- 一种基于改进1D-Lenet网络的海面小目标检测方法
- 一种基于改进相位特征的海面慢速小目标检测方法