用于检测宫颈癌的试剂盒及其应用
文献发布时间:2024-04-18 20:01:23
技术领域
本发明涉及生物检测领域,特别涉及一种于检测宫颈癌的试剂盒及其应用。
背景技术
DNA甲基化为DNA化学修饰的一种形式,能够在不改变DNA序列的前提下,改变遗传表现。DNA甲基化是指在DNA甲基化转移酶的作用下,在基因组CpG二核苷酸的胞嘧啶5'碳位共价键结合一个甲基基团。大量研究表明,DNA甲基化能引起染色质结构、DNA构象、DNA稳定性及DNA与蛋白质相互作用方式的改变,从而控制基因表达,在肿瘤的发生和发展中起着重要的调控作用。因此,差异表达的甲基化DNA是肿瘤早期诊断和预后监测的重要生物标记物。亚硫酸盐依赖的检测方法(BS)是临床上核酸基因甲基化检测的金标准。这种方法是DNA经亚硫酸盐处理后,未甲基化的胞嘧啶(C)被转化成尿嘧啶(U),甲基化的胞嘧啶保持不变。处理后的DNA通过测序或使用特异性引物PCR得到非常准确的DNA序列甲基化位点信息。但是其操作步骤繁琐、耗时时间长、实验条件严苛,加上靶基因易降解、转化不完全且回收率低,这些固有的缺点导致结果基因甲基化(尤其是丰度极低的基因)检测结果分析不准确和结果重复性较差,严重限制了其在临床检测效果。
而甲基化敏感限制性内切酶(MSRE)刚好可以克服BS的缺点,这种方法是利用甲基化敏感性限制性内切酶对甲基化区域不切割的特性,将DNA消化为不同大小的片段后再进行后续分析。由于其反应条件较为温和,可以极大的避免了DNA的降解。此外,与BS需要产物回收和纯化相比,MSRE反应的酶切产物通常不需要纯化即可进行后续的检测反应。但是此检测方法通常检测灵敏度比较低,需要产物的进一步扩增,即需要昂贵的PCR仪。PCR是使用最为广泛的核酸扩增技术,以其灵敏性、特异性得到广泛应用,然而PCR需要反复的热变性,无法摆脱依赖仪器设备的局限,从而限制了其在临床现场检测中的应用。
自20世纪90年代初,很多实验室开始发展无需热变性的恒温扩增技术,现已开发出重组聚合酶扩增技术(RPA)、环介导等温扩增技术(LAMP)、链替代等温扩增技术(SDA)、滚环等温扩增技术(RCA)等温扩增技术等技术。其中,如SDA、RCA和RPA已经成功的用于甲基化基因的检测。SDA和RCA是一个耗时较长的实验,需要数小时才能使目的产物达到峰值,导致其在甲基化检测中灵敏度较低。然而RPA能够在半小时内使目的产物扩增量达到峰值且具有极高的准确度,因此被广泛用于疾病检测中。
近年来,基于规律间隔成簇短回文重复序列(CRISPR)技术的核酸检测如SHERLOCK、DETECTR,因其快速又灵敏的特性备受关注。在先前的研究中,CRISPR/Cas12b结合BS技术(称为HOLMESv2)已经被用于DNA甲基化检测中。然而,HOLMESv2检测过程复杂限制了其在临床上的应用。因此,开发快速、灵敏、简单易用的甲基化基因检测技术,对于临床上疾病检测显得非常重要。
发明内容
为了解决上述问题,本发明提供一种用于检测宫颈癌的试剂盒,所述试剂盒包括检测SEPT9基因启动子SEPT9:77373475-77373595的甲基化试剂。
本发明提供一种基于等温扩增方法和CRISPR/Cas方法结合直接检测宫颈癌的POCT试剂盒,所述试剂盒是检测SEPT9基因启动子SEPT9:77373475-77373595片段的甲基化试剂盒,所述试剂盒包括:组分1:其包括甲基化敏感限制性内切酶,所述限制性内切酶消化人基因组DNA,此时甲基化修饰的目的序列片段会保持完整而非甲基化的序列片段会被特异性的切割;组分2:其包括等温扩增试剂,所述等温扩增试剂对于所述甲基化修饰的目的序列片段进行扩增,得到双链DNA,而所述非甲基化的序列片段则不进行等温扩增;和组分3:CRISPR/Cas12a试剂,所述试剂对所述双链DNA进行特异性识别,进而激活Cas12a酶反式切割活性切割具有荧光基团的单链报告DNA,产生荧光信号以直接检测所述甲基化修饰的目的序列片段。
在一种实施方式中,所述甲基化敏感限制性内切酶是核酸内切酶BstUI,所述等温扩增试剂是RPA等温扩增试剂。
在一种实施方式中,所述CRISPR/Cas12a试剂包括crRNA、Cas12a酶和具有荧光基团的单链报告DNA,所述crRNA引导Cas12a酶识别所述双链DNA,对所述甲基化修饰的目的序列片段进行特异性切割,所述双链DNA中包括与crRNA特异性结合的核酸片段,该核酸片段起始序列是TTTN序列,N为A、C、G三者中任意一种碱基;所述具有荧光基团的单链报告DNA被激活的Cas12a酶切割后产生荧光信号以进行特异性的检测。
在一种实施方式中,所述基因组DNA浓度为不高于2.5ng/μL。
在一种实施方式中,所述核酸内切酶BstUI浓度为不低于0.75U/μL,优选地为1U/μL。
在一种实施方式中,所述RPA等温扩增试剂中镁离子浓度为10-25mM,优选地为10mM。
在一种实施方式中,所述RPA等温扩增的扩增时间为5-30分钟,优选为15分钟。
在一种实施方式中,所述RPA等温扩增试剂中引物浓度为100-600nM,优选地为400nM。
在一种实施方式中,所述RPA等温扩增试剂中温度为38-43℃,优选地为42℃。
在一种实施方式中,所述Cas12a反应温度为35-45℃,优选地为45℃。
当前已有的甲基化基因检测方法,如金标准亚硫酸盐依赖方法需要强酸和高温等严苛条件导致的DNA降解和检测失败,MSRE虽然反应条件温和可以避免DNA降解,但消化不完全导致检测准确度和灵敏度有待提高。
与现有技术的DNA甲基化检测方法相比,当前其他技术用的都是线性的DNA检测难度较低,没有用具有超螺旋结构的基因组DNA,因此本发明所述方法用的是基因组DNA更贴近临床应用,且检出限较低,检测范围较宽。此外,整个反应可以在1h内完成,因此非常适合即时检测(POCT)。
具体地,本发明提供了一种基于甲基化敏感限制性内切酶(MSRE)技术的等温扩增(RPA)与CRISPR/Cas12a(MeCRISPR)系统结合DNA甲基化检测系统,与传统的甲基化特异性PCR相比,基于核酸内切酶BstUI的RPA反应条件温和,检测速度快(整个反应低于1h)。在DNA甲基化检测中表现出良好的性能,能够对极低低浓度(1copy/μL)和含量(0.01%)的甲基化水平进行准确检测。此外,MeCRISPR在宫颈癌临床检测中的灵敏度和特异性可高达100%和92.3%,具有极好的应用价值和前景。
本发明利用BstUI的特异性,能够对甲基化和非甲基化DNA进行有效区分,同时,RPA对靶DNA的高效扩增可以产生大量的dsDNA(双链DNA),随后dsDNA被Cas12a特异性的识别,识别过程中需要PAM位点,从而避免了RPA技术非特异性扩增的缺陷,最后激活CRISPR/Cas12a的反式切割活性对荧光基团修饰的单链报告基因进行非特异性切割产生荧光信号,从而使本发明所述检测方法具有较高的灵敏度和选择性,良好的抗干扰能力和重复性。
由于产生的dsDNA与crRNA部分互补,从而激活Cas12a的反式切割活性。Cas12a可以任意切割附近的荧光报告探针(诸如FAM-DNA-BHQ1)来释放绿色荧光信号。此外,本发明所述方法还可以通过改变检测方式,使用紫外灯分析以实验可视化即时检测。当SEPT9存在甲基化修饰时,可以激活RPA反应短时间扩增出大量dsDNA,进而激活Cas12a的反式切割活性,从而非特异性切割单链的FAM-DNA-BHQ1报告基因。含有FAM集团修饰的报告探针切割后,在480nm激发光波长下,在525nm波长下有吸收峰,且在紫外灯下能够检测到绿色荧光信号。相反,未甲基化的SEPT9,由于无法激活Cas12a反式切割活性,因此在480nm激发波长或紫外灯下,无明显的绿色荧光信号。基于传感器的检测原理,可以成功区分SEPT9的甲基化修饰情况,并可应用于即时检测或高通量检测平台。
在本发明中,SEPT9基因启动子SEPT9:77373475-77373595(121bp)含有3个5’-CGCG-3’的酶切位点。甲基化DNA和未甲基化DNA首先经过BstUI甲基化敏感限制性内切酶处理,该酶可以识别和消化“5’-CGCG-3’”位点。因此,未甲基化的DNA会被BstUI切断。非甲基化修饰的SEPT9完全被BstUI消化。相反,甲基化SEPT9没有影响,结构保持完整。结构完整的甲基化SEPT9被用来激活RPA扩增反应。在RPA系统中,重组酶与引物结合定位双链中的同源序列,并以指数方式扩增靶区。因此,通过RPA扩增实验可以获得大量的dsDNA。同时,获得的dsDNA,因PAM序列位点的存在,使其可以被Cas12a酶特异性识别,增加了其检测的特异性。而CRISPR/Cas12a系统的高特异性和自放大能力进一步提高了本检测系统的检测性能。
在本发明中我们所选的MSRE(BstUI)在该片段中能够识别并切割3个含有5'-CGCG-3’的序列,进而保证非甲基化的DNA被完全消化。此外,RPA反应扩增出的大量dsDNA,在PAM位点的辅助下被CRISPR/Cas12a特异性识别,激活其反式切割活性,用于荧光检测(可视化和高通量)。MeCRISPR系统由于在真实临床样本检测中具有较高的灵敏度和特异性,可用于实际临床诊断中宫颈癌的检测。
附图说明
为了更清楚地说明本申请实施例中的技术方案,下面将对实施例中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本申请中记载的一些实施例,对于本领域普通技术人员来说,在不付出创造性劳动的前提下,还可以根据这些附图获得其它的附图。
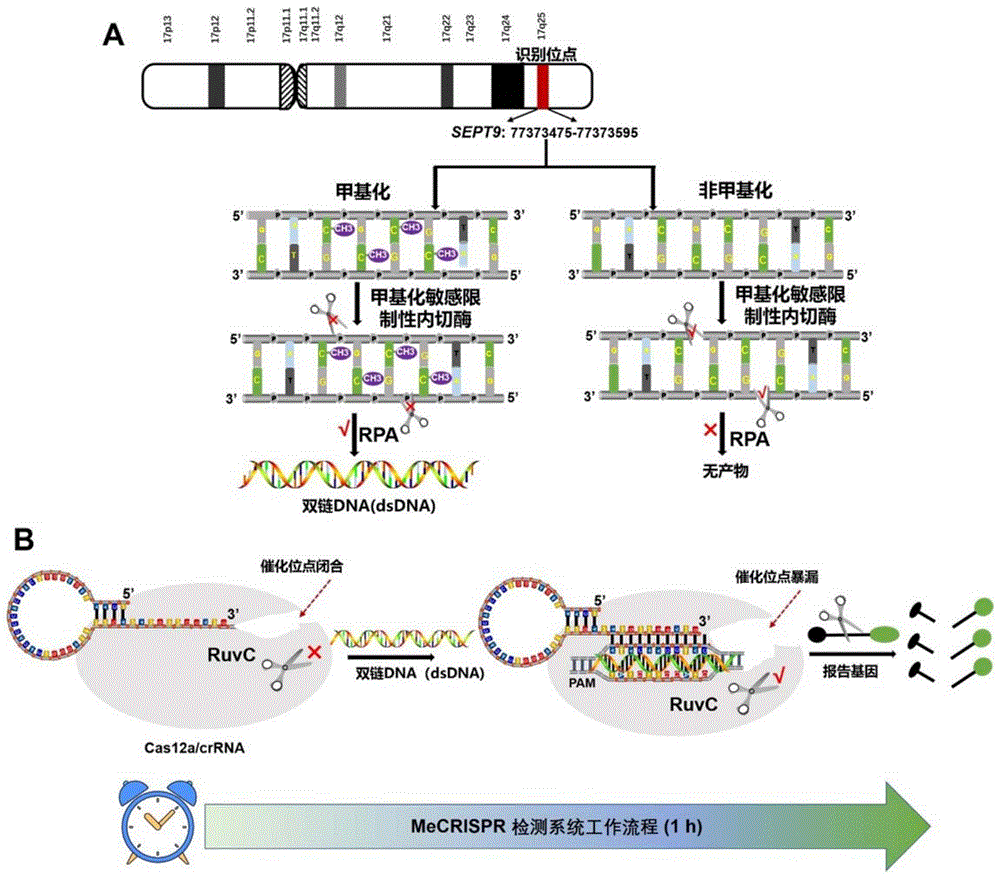
图1是本发明的基本原理图,其中图1A是甲基化DNA和未甲基化DNA经过BstUI甲基化敏感限制性内切酶处理及其后续等温扩增反应的激活示意图,和图1B是产生的dsDNA激活CRISPR/Cas12a系统切割报告基因产生荧光信号的反应示意图;
图2是靶基因片段经RPA扩增的dsDNA电泳结果图;
图3是CRISPR/Cas12a检测结果图,其中图3A是荧光光谱检测结果图,图3B是可视化检测结果图;
图4是本发明不同实验参数优化结果图,其中图4A是不同底物浓度的扩增产物电泳结果图,图4B是不同浓度BstUI的扩增产物电泳结果图,图4C是不同浓度Mg
图5是本发明甲基化检测检测系统的性能结果图,其中图5A是不同浓度的mSEPT9测试结果图,图5B是不同含量的mSEPT9测试结果图;
图6是本发明甲基化检测检测系统的可视化结果图,其中图6A是本发明甲基化检测检测系统可视化的检测原理示意图,图6B是不同浓度的mSEPT9可视化结果图,和图6C是不同含量的mSEPT9可视化结果图;
图7是本发明甲基化检测检测系统的检测mSEPT9在宫颈癌临床诊断中的效果图,图7A和7B分别是在相同量的基因组DNA条件下,宫颈癌患者的脱落细胞样本的荧光信号值明显与非宫颈癌患者样本的信号值结果图(图7A)和可视化结果图(图7B)。
具体实施方式
为了使本领域技术领域人员更好地理解本申请中的技术方案,下面将结合实施例对本发明作进一步说明,显然,所描述的实施例仅仅是本申请一部分实施例,而不是全部的实施例。基于本申请中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其它实施例,都应当属于本申请保护的范围。
实施例一本发明方法检测宫颈癌样品
SEPT9启动子区域的甲基化被认为是宫颈癌诊断的有效生物标志物,通过如下方法对其进行检测。本实施例所述检测方法的原理见附图1。从附图1A可以看出,甲基化DNA和未甲基化DNA首先经过BstUI甲基化敏感限制性内切酶处理,该酶可以识别和消化“5’-CGCG-3’”位点。因此,未甲基化的DNA会被BstUI切断。
在本发明中,SEPT9基因启动子SEPT9:77373475-77373595(121bp)含有3个5’-CGCG-3’的酶切位点。因此,非甲基化修饰的SEPT9完全被BstUI消化。相反,甲基化SEPT9没有影响,结构保持完整。结构完整的甲基化SEPT9被用来激活RPA扩增反应。在RPA系统中,重组酶与引物结合定位双链中的同源序列,并以指数方式扩增靶区。因此,通过RPA扩增实验可以获得大量的dsDNA。
同时,如附图1B所示,由于产生的dsDNA与crRNA部分互补,从而激活Cas12a的反式切割活性。Cas12a可以任意切割附近的荧光报告探针(FAM-DNA-BHQ1)来释放绿色荧光信号。此外,本发明所述方法还可以通过改变检测方式,使用紫外灯分析以实验可视化即时检测。当SEPT9存在甲基化修饰时,可以激活RPA反应短时间扩增出大量dsDNA,进而激活Cas12a的反式切割活性,从而非特异性切割单链的FAM-DNA-BHQ1报告基因。含有FAM集团修饰的报告探针切割后,在480nm激发光波长下,在525nm波长下有吸收峰,且在紫外灯下能够检测到绿色荧光信号。相反,未甲基化的SEPT9,由于无法激活Cas12a反式切割活性,因此在480nm激发波长或紫外灯下,无明显的绿色荧光信号。基于传感器的检测原理,可以成功区分SEPT9的甲基化修饰情况,并可应用于即时检测或高通量检测平台。
一.材料与试剂
HeLa基因组DNA购买于赛默飞世尔科技(中国)有限公司。RPA引物、PAGE相关试剂(DNA marker(25-500bp)、GelRed核酸染料、TE缓冲液和上样缓冲液)购于上海生物工程技术有限公司,crRNA和荧光基团标记的单链DNA(ssDNA-FQ reporter)购于广州莱博生物科技有限公司。
表1本发明实施例中所使用的DNA和RNA序列
注:红色序列为BstUI酶切识别位点;黄色序列为CRISPR/Cas12a识别位点;蓝色序列为crRNA上的间隔区(spacer)负责与双链DNA上名为原间隔序列临近基序(protospaceradjacent motif,PAM,通常为TTTN序列,N为A、C、G三者中任意一种碱基)下游的21个碱基互补。
二.宫颈脱落细胞基因组DNA提取
宫颈脱落细胞的基因组DNA采用商业化试剂盒进行提取,例如,敬善生物科技(江苏)有限公司的核酸提取试剂盒(磁珠法)。提取后的基因组DNA用Nanodrop检测浓度和质量,将质量合格的DNA样本(OD260/280=1.6~2.0;OD260/230≥3.0;DNA浓度≥20ng/μL,推荐为100ng/μL)浓度调整到20ng/μL,调好浓度的核酸样本备用。然后,将获得的基因组DNA作为目标DNA,验证该方法的实际应用能力。
三.BstUI甲基化敏感限制性内切酶对靶DNA的消化
用BstUI内切酶消化靶DNA(mSEPT9和unSEPT9),反应总体积为20μL。具体步骤如下,加入目标DNA溶液2μL,缓冲液2μL(10×),BstUI 2μL(20U)和14μL无DNA/RNA酶水,60℃反应30分钟,得到靶DNA的酶切产物。
四.RPA扩增
使用
五.电泳分析
用非变性3%聚丙烯酰胺凝胶验证上述扩增步骤得到的扩增产物。由于直接合成的双链靶DNA片段或含靶基因片段的质粒样本均为线性DNA,无法模拟真实的检测环境。因为真实临床样本提取的基因组DNA为超螺旋结构,远比线性DNA复杂的多,因此我们使用HeLa和Jurkat作为甲基化阳性和阴性的样本,将靶基因片段经RPA扩增的dsDNA作为电泳分析目标。因RPA反应中的物质会干扰琼脂糖电泳,如果不进行产物回收,跑出来的带会出现拖尾,所以我们使用20μL苯酚/氯仿(1:1)溶液与RPA产物充分混匀后,离心,取上清。将上清液与6×的DNA上样缓冲液混匀后,然后在1×TAE缓冲液中进行PAGE电泳实验,电压为130V、时间为40分钟。最后,经电泳后的凝胶,用Tanon UV image拍照,进行图像的采集。甲基化的SEPT9(mSEPT9)和未甲基化的SEPT9(unSEPT9)碱基数均为121bp,unSEPT9含有3个BstUI的“5’-CGCG-3’”位点,因此unSEPT9与BstUI内切酶反应后,会被在酶切位点处切段。从图2中可以发现,在泳道3(mSEPT9组)中可以观察到经RPA反应特异性扩增出的条带,而泳道2(unSEPT9组)与泳道1(阴性对照组)均未发现明显的靶基因片段。因为mSEPT9因此甲基修饰无法被限制性内切酶消化保持完整,所以可以此为RPA扩增模板得到大量的dsDNA。这些结果表明mSEPT9和unSEPT9均可以被BstUI特异性识别。
六.CRISPR/Cas12a检测
基于荧光光谱的高通量检测和基于紫外灯的可视化检测是基于Cas12a的反式切割活性,因此Cas12a反应过程如下,反应总体积为20μL:8μL无DNA/RNA酶水,2μL Cas12酶反应缓冲液,4μL的crRNA(1μM),2μL的Cas12a(1μM),2μL的荧光探针(5μM FAM-DNA-BHQ1)和2μL反应产物,45℃孵育15分钟。对于高通量检测来说,加入80μL的无DNA/RNA酶水,在480nm激发下用多功能酶标仪(SpectraMax M5)测定其在525nm下荧光信号强度。对于可视化检测来说,将CRISPR/Cas12a切割产物(无需稀释)置于紫外灯下,用智能手机或数码相机拍照。
如图3所示,荧光光谱和可视化检测结果分别如图3A和图3B所示。结果发现,只有mSEPT9才会产生较强的荧光,而unSEPT9和阴性对照组的荧光可以忽略不计。上述结果表明,mSEPT9可以通过5mC修饰避免BstUI位点特异性的消化,从而触发RPA反应生成大量的dsDNA,进而特异性的激活Cas12a的反式切割活性切割荧光标记的单链报告基因,释放荧光信号。与此同时,unSEPT9由于可以被BstUI识别被消化,因此不能激活后续的RPA反应和CRISPR/Cas12a反应。在高通量检测中(图3A),只有mSEPT9组在525nm发射波长下出现明显的信号强度,而unSEPT9和NC信号值相当,这是由于在mSEPT9组,Cas12a的非特异性切割属性被激活,导致FAM-DNA-BHQ1被切割,FAM基团与猝灭基团分离,而FAM基团在525nm激发波长下有特意的吸收峰所致。在可视化检测中(图3B),只有mSEPT9组在紫外灯下出现明显的绿色荧光,这是由于FAM-DNA-BHQ1被切割,FAM基团与猝灭基团分离释放荧光所致。上述结果表明,MeCRISPR系统能够成功检测DNA甲基化,且背景干扰较低,灵敏度和准确性高。
实施例二本发明方法的优化实验
为了提高该方法的灵敏度,分别对MSRE实验(BstUI酶切底物浓度、BstUI浓度)、RPA实验(Mg
(一)MSRE实验参数优化
1.BstUI酶切底物浓度优化:为了实现MeCRISPR技术的POCT(即时检测),我们首先将酶切反应时间设定为30min,以优化酶切底物的浓度。MSRE(甲基化敏感限制性内切酶)实验条件优化:将2μL不同浓度的Jurkat gDNA(0/25/50/100/250/500ng,即0、1.25、2.5、5、12.5、25ng/μL)、缓冲液2μL(10X),BstUI 2μL(20U)和14μL无DNA/RNA酶水,60℃反应30分钟,得到靶DNA的酶切产物。用RPA试剂盒对上述步骤的消化产物进行扩增,得到双链DNA(dsDNA)扩增产物,其中,扩增步骤和体系参考实施例一中RPA扩增。最后,参照实施例一中电泳分析,将RPA反应产物进行PAGE分析,得到目标gDNA酶切效果。如图4A所示,PAGE实验结果表明,当底物量为100-500ng(5/12.5/25ng/μL时,靶DNA出现了特异性的扩增,说明此条件下Jurkat gDNA消化不完全。而在50ng(2.5ng/μL)组没有出现目的条带,这表明在此条件下,说明此条件下Jurkat gDNA被完全消化,因此选择2.5ng/μL Jurkat gDNA(50ng)作为MSRE实验的最优酶切底物浓度。
2.BstUI酶浓度优化:我们首先将酶切反应时间设定为30min、底物浓度设为2.5ng/μL,以优化最佳BstUI酶浓度。MSRE(甲基化敏感限制性内切酶)实验BstUI酶浓度条件优化:将2μL不同浓度的BstUI酶(即0、0.1、0.25、0.5、0.75、1U/μL)、缓冲液2μL(10×),BstUI 2μL(20U)和14μL无DNA/RNA酶水,60℃反应30分钟,得到靶DNA的酶切产物。用RPA试剂盒对上述步骤的消化产物进行扩增,得到双链DNA(dsDNA)扩增产物,其中,扩增步骤和体系参考实施例一中RPA扩增。最后,将RPA反应产物经步骤五处理,进行PAGE分析,得到目标gDNA酶切效果。如图4B所示,PAGE实验结果表明,当BstUI酶浓度为0、0.1、0.25、0.5U/μL)时,靶DNA出现了特异性的扩增,说明此条件下Jurkat gDNA消化不完全。而在0.75、1U/μL时没有出现目的条带,这表明在此条件下Jurkat gDNA被完全消化,鉴于BstUI切割能力与背景信号有关,消化不完全会导致高背景信号,因此选择1U/μL BstUI酶作为MSRE实验的最优酶浓度。
(二)RPA优化实验
1.Mg
2.引物浓度优化:我们首先配制不同浓度的引物(0、100、200、400、500和600nM),以优化引物的浓度。具体步骤如下:按照说明书,取29.5μL的RPA反应缓冲液重悬RPA冻干粉,得到均一且透明的RPA反应液;然后向RPA反应液中加入取2μL经上述步骤得到的酶切产物(无需纯化)、2μL的反向引物(不同浓度)、2μL的正向引物(不同浓度)和12.85μL无DNA/RNA酶水,充分混匀。最后加入2μL醋酸镁(终浓度10mM),总反应体积为50μL。在42℃条件下反应30分钟,得到反应产物(dsDNA)。其中,底物消化步骤和体系参考步骤三。最后,将RPA反应产物经步骤五处理,进行PAGE分析,得到目标gDNA扩增效果。如图4D所示,PAGE实验结果表明,当引物浓度为100、200、400、500和600nM时,靶DNA出现了特异性的扩增。而在0nM时,没有出现目的条带,说明扩增的特异性。此外,发现在400nM条件下,靶DNA条带的亮度最大,说明此条件下靶DNA扩增效率最高,因此选择400nM作为RPA实验的最优引物浓度。
3.温度优化:我们首先设置不同温度(38℃、39℃、40℃、41℃、42℃和43℃),以优化反应的温度。具体步骤如下:按照说明书,取29.5μL的RPA反应缓冲液重悬RPA冻干粉,得到均一且透明的RPA反应液;然后向RPA反应液中加入取2μL经上述步骤得到的酶切产物(无需纯化)、2μL的反向引物(终浓度为400nM)、2μL的正向引物(终浓度为400nM)和12.85μL无DNA/RNA酶水,充分混匀。最后加入2μL醋酸镁(终浓度10mM),总反应体积为50μL。在不同温度下反应30分钟,得到反应产物(dsDNA)。其中,底物消化步骤和体系参考步骤三。最后,将RPA反应产物经步骤五处理,进行PAGE分析,得到目标gDNA扩增效果。如图4E所示,PAGE实验结果表明,当温度为38℃、39℃、40℃、41℃、42℃和43℃时,靶DNA出现了特异性的扩增。而在42℃条件下,靶DNA条带的亮度最大,说明此条件下靶DNA扩增效率最高,因此选择42℃作为RPA实验的最优反应温度。
4.时间优化:此外,RPA扩增时间也是一个重要反应参数,与甲基化基因检测的灵敏度密切相关。我们首先设置不同反应时间(0、5、10、15、20和30min),以优化反应的时间。具体步骤如下:按照说明书,取29.5μL的RPA反应缓冲液重悬RPA冻干粉,得到均一且透明的RPA反应液;然后向RPA反应液中加入取2μL经上述步骤得到的酶切产物(无需纯化)、2μL的反向引物(终浓度为400nM)、2μL的正向引物(终浓度为400nM)和12.85μL无DNA/RNA酶水,充分混匀。最后加入2μL醋酸镁(终浓度10mM),总反应体积为50μL。在42℃条件下反应不同时间,得到反应产物(dsDNA)。其中,底物消化步骤和体系参考步骤三。最后,将RPA反应产物经步骤五处理,进行PAGE分析,得到目标gDNA扩增效果。如图4F所示,PAGE实验结果表明,而在0min和5min时,没有出现目的条带,说明扩增的特异性。当时间为5min、10min、15min和30min时,靶DNA出现了特异性的扩增。而在15min条件下,靶DNA条带的亮度已达到峰值,说明此条件下靶DNA扩增效率最高,因此选择15min作为RPA实验的最优反应时间。
(三)CRISPR/Cas12a优化实验
1.温度优化:我们首先设置不同温度(35℃、37℃、39℃、41℃、43℃和45℃),以优化反应的浓度。具体步骤如下:反应总体积为20μL:8μL无DNA/RNA酶水,2μL Cas12a酶反应缓冲液,4μL的crRNA(1μM),2μL的Cas12a(1μM),2μL的荧光探针(5μM FAM-DNA-BHQ1)和2μLRPA反应产物,不同温度下孵育15分钟。其中,RPA实验步骤和体系参考实施例一。对于高通量检测来说,加入80μL的无DNA/RNA酶水,在480nm激发下用多功能酶标仪(SpectraMax M5)测定其在525nm下荧光信号强度。对于可视化检测来说,将CRISPR/Cas12a切割产物(无需稀释)置于紫外灯下,用智能手机或数码相机拍照。如图4G-4H所示,从35℃到45℃,随着温度的增加,荧光信号逐渐增强。且当反应温度为45℃时,荧光信号强度最高(图4G)。可视化结果(图4H)同样发现反应温度为45℃时,绿色荧光信号最强。因此接下来的实验Cas12a反应温度为45分钟。
实施例三MeCRISPR系统检测DNA甲基化的性能研究
与现有技术的DNA甲基化检测方法相比,当前其他技术用的都是线性的DNA检测难度较低,没有用具有超螺旋结构的基因组DNA,因此本发明所述方法用的是基因组DNA更贴近临床应用,且检出限较低,检测范围较宽。此外,整个反应可以在1h内完成,因此非常适合即时检测(POCT)。这些优异的性能可能得益于以下原因:(1)BstUI酶的特异性识别,且在本发明所选片段中有3个酶切位点,使甲基化和非甲基化DNA能够有效区分。(2)RPA的高效的扩增能力可以在短时间内产生大量的dsDNA。(3)获得的dsDNA,因PAM序列位点的存在,使其可以被Cas12a特异性识别,增加了其检测的特异性。而CRISPR/Cas12a系统的高特异性和自放大能力进一步提高了本检测系统的检测性能。
为了研究该甲基化检测检测系统的性能,我们配制了不同浓度的mSEPT9(0、1、2.5、5、10、10
现有研究表明,哺乳动物基因组中甲基化修饰的比例通常小于0.1%,绝大多数均处于非甲基化修饰状态。因此,在大量非甲基化样本中,如何检测出含量极低的甲基化基因尤为重要。为了解决以上问题,我们首先将非甲基化靶DNA(umSEPT9)检测量设置为50ng,然后将不同量的甲基化靶DNA(mSEPT9)加入到非甲基化SEPT9的反应体系中,配制一系列浓度梯度的mSEPT9溶液(0%,0.01%,0.05%,0.1%,1%,10%,and 100%),在最优实验条件下,进一步研究MeCRISPR系统对mSEPT9检测的性能。如图5B所示,在480nm激射光刺激下,0.01%,0.05%,0.1%,1%,10%,and 100%mSEPT9在525nm波长下的荧光信号数值逐渐增加,且明显高于对照组(0%mSEPT9)。以上结果表明,本发明采用的MeCRISPR系统在甲基化基因检测中具有较高的准确性和敏感性。
实施例四MeCRISPR系统对DNA甲基化检测的可视化研究
肿瘤的可视化检测具有直观、便捷等的优点,因此对于建立肉眼可视化的甲基化基因检测平台在肿瘤检测中具有极大的应用价值。因此,本发明将MeCRISPR系统与便携式手提紫外灯结合,以实现检测的可视化。根据MeCRISPR可视化的检测原理(图6A),当存在甲基化靶DNA出现时(mSEPT9)时,试管在紫外灯下会出现绿色。而在非甲基化的DNA(mSEPT9)中不会有明显的绿色荧光出现。接下来,本发明研究了基于MeCRISPR系统在甲基化基因可视化检测中的灵敏度。如图6B所示,在紫外灯下,随着mSEPT9浓度从0到10
实施例五真实样本检测
为了评估MeCRISPR系统检测mSEPT9在宫颈癌临床诊断中的效果,以宫颈癌无创诊断中常用的临床样本-宫颈脱落细胞为材料,进行MeCRISPR实验检测mSEPT9,并与宫颈癌诊断的金标准-病理学检测作对比,探究其检测效果。首先以提取的宫颈脱落细胞的基因组DNA样本为材料,将其浓度调整为25ng/μL,靶DNA(mSEPT9)采用MeCRISPR法检测。如图7A所示,在相同量的基因组DNA(25ng/μL)条件下,宫颈癌患者的脱落细胞样本的荧光信号值明显高于非宫颈癌患者样本的信号值。此外,在相同数量的基因组DNA (25ng/μL)下,用肉眼观察紫外灯下的试管荧光强度,发现宫颈癌患者的基因组DNA中荧光亮度明显高于非癌患者的样本(图7B所示)。最后,测试了53例真实样本,本发明在宫颈癌检测中的灵敏度为100%、特异性为92.3%,准确度为96.2%,极佳的检测性能可适用于复杂的真实样品的检测。以上结果表明本发明所述方法对细胞DNA甲基化的检测在宫颈癌诊断中具有极高的灵敏度和特异性。
应该理解到披露的本发明不仅仅限于描述的特定的方法、方案和物质,因为这些均可变化。还应理解这里所用的术语仅仅是为了描述特定的实施方式方案的目的,而不是意欲限制本发明的范围,本发明的范围仅受限于所附的权利要求。
本领域的技术人员还将认识到,或者能够确认使用不超过常规实验,在本文中所述的本发明的具体的实施方案的许多等价物。这些等价物也包含在所附的权利要求中。
- 基板处理装置、基板保持件以及半导体装置的制造方法
- 基板反转装置、基板处理装置及基板支撑装置以及其方法
- 基板处理装置及基板处理方法
- 基板处理方法和基板处理装置
- 基板处理装置、喷嘴安装件、基板处理方法以及半导体装置的制造方法
- 喷嘴的清洁方法、基板处理方法、半导体装置的制造方法、基板处理装置和记录介质