一种新的脑胶质瘤分子分型方法
文献发布时间:2023-06-19 10:48:02
技术领域
本发明涉及脑胶质瘤技术领域,具体为一种新的脑胶质瘤分子分型方法。
背景技术
脑胶质瘤是最常见的颅内恶性肿瘤,具有高度的异质性,手术治疗辅以放疗和化疗的标准化方案是当前脑胶质瘤的主要治疗手段,但整体情况仍不理想,预后极差,特别是胶质母细胞瘤,在体细胞中,各种机制引起DNA损伤,进而发生体细胞突变,使得细胞基因组不断变化,各种类型的突变不断积累,形成一个个独特的突变积累组合,每种组合即为一种“突变特征”,突变特征系统地表征了导致癌症的突变积累,并将突变过程与DNA损伤机制、临床特点等联系起来,为深入分析和掌握肿瘤的分子特征提供了新的机会,鉴于此,我们以突变特征着手进行研究,从基因组变异的角度提出一种新的脑胶质瘤分子分型方法并进行初步转化,建立预测个体预后及免疫治疗反应的评估体系。
随着生物信息学的迅速发展和分子诊断技术的兴起,精密治疗和免疫治疗使胶质瘤的治疗摆脱目前的困境成为可能,将胶质瘤分为不同的分子表型,如IDH突变型和IDH野生型胶质瘤,1p/19q共缺失和1p/19q完整性胶质瘤,积累的证据表明,IDH突变和1p/19q 共缺失的胶质瘤患者对放疗和化疗相对敏感,预后良好,然而,这种分类只关注一种或几种基因组改变特征,它缺乏全局性的视角,也不能完全顾及胶质瘤的高分子异质性,因此,有必要对胶质瘤的基因组改变进行系统的研究,进行更加精细的分型传统的脑胶质瘤分子分型方法的局限性较大,无法精准的对患者进行分类,提升了患者治疗的难度。
因此亟需设计一种新的脑胶质瘤分子分型方法来解决上述问题。
发明内容
本发明的目的在于提供一种新的脑胶质瘤分子分型方法,以解决上述背景技术中提出的传统脑胶质瘤分子分型方法精准度低的问题。
为实现上述目的,本发明提供如下技术方案:一种新的脑胶质瘤分子分型方法,包括如下步骤:
步骤一:收集数据
从数据库中获取基因表达谱和临床信息数据,五个胶质瘤队列纳入本研究:TCGA-GBMLGG队列(n=892),三个CGGA队列(mRNA-array (n=301),mRNAseq_325(n=325)和mRNAseq_693(n=693))和 Rembrandt队列(n=475),从公开数据库收集了四个独立的免疫治疗队列,包括:①Roh队列:抗CTLA-4、抗PD-1治疗队列;② GSE100797:过继性T细胞治疗队列;③GSE78220:抗PD-1治疗队列;④GSE35640:抗MAGE-A3治疗队列,根据recistv1.1标准,完全缓解(CR)或部分缓解(PR)患者被视为免疫治疗有反应者,疾病稳定(SD)或疾病进展(PD)患者被视为免疫治疗无反应者,不可评估(NE)患者则被剔除;
步骤二:突变特征谱
突变特征库(第二版)聚焦于碱基置换突变,突变点的碱基置换包含六种类型:C>A,C>G,C>T,T>A,T>C和T>G,突变点两侧(5' 和3'端)各可搭配四种碱基(A、T、C、G),最终可产生96种可能的突变类型(6种突变位点的碱基替换类型×4种,5'碱基×4种, 3'碱基),在体细胞中,各种机制引起DNA损伤,进而发生体细胞突变,使得细胞基因组不断变化,各种类型的突变不断积累,最终,在96种突变类型上具有不同的积累,形成一个个独特的突变积累组合,检测到的每种组合即为一种“突变特征”;
步骤三:亚型识别
(一)、数据获取:从COSMIC网站获取获取每种突变特征的特征数据信息,从TCGA-LGG和TCGA-GBM获得的体细胞突变数据在去除沉默突变后用于构建每个个体的突变特征谱,参考基因组为h38
(二)、R包:DeconstructSigs和NMFpackage
(三)、方法
①移除突变数据中的沉默突变
②利用计算机技术将移除沉默突变的突变数据转化为突变环境矩阵
③使用DeconstructSigs包,分析每个样本中30个signature 的组成比例,参考的signature是COSMIC,cutoff值设置为0.06,标准化方式使用“exome2genome”,最终我们得到了一个矩阵(行为30个signature,列为每个样本,cell的值为signature在每个样本的比例,所有signature加起来刚好为1)
④使用NMF包进行提取和聚类分析,设置潜在的ranks=2:5,运算执行次数设置为50,method设置为‘lee’,最终通过从cophenetic 系数和轮廓系数决定最佳rank=4,如图1,即根据TCGA-GBMLGG队列中每个患者的突变特征谱,将胶质瘤分为四种分子亚型最优
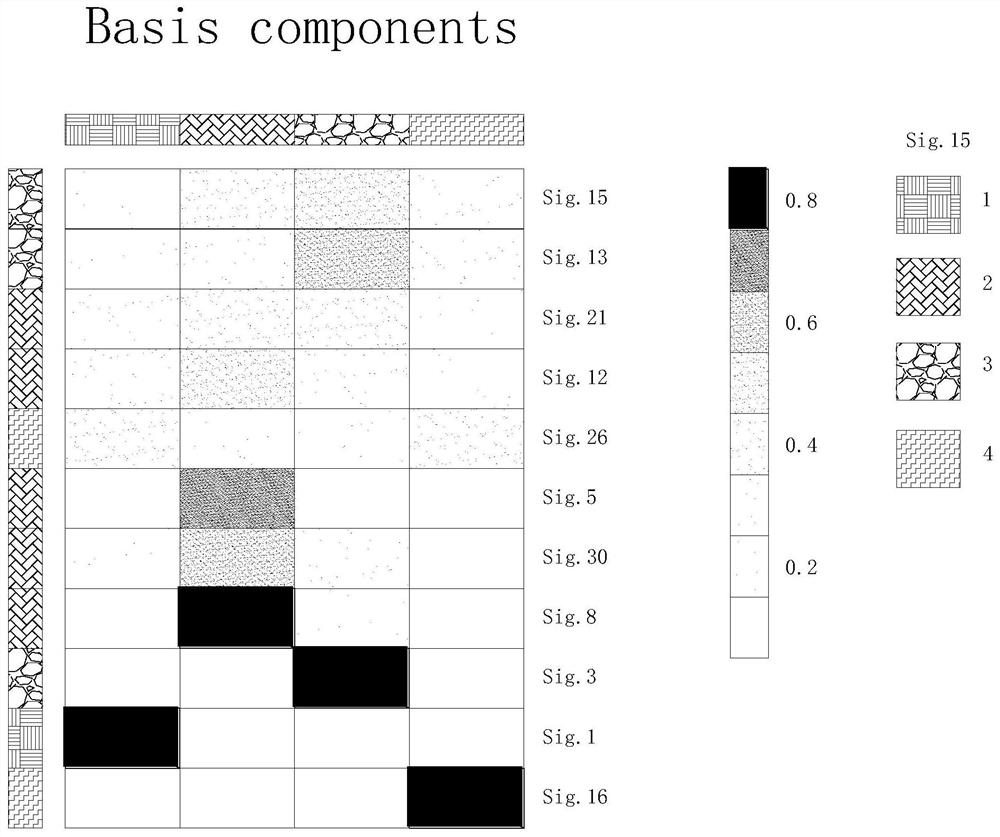
⑤非负矩阵分解的一个特性是倾向于产生观察数据的稀疏表示,导致双聚类的自然应用,通过少量特征表征样本组,在NMF模型中,根据对每个样本贡献最大的基础组分(即在系数矩阵的每列中具有最大系数的基础组分)对样本进行分组,然后,通过根据基础矩阵计算的基础特异性评分选择的一组特征对每组样本进行表征,上述过程由NMF包实现,根据所有患者的突变特征谱,构建NMF 模型,并通过extractFeatures函数(方法设置为“max”)对最基本的特定特征进行提取,最终,患者分为4个基础组,并提取出11 种可表征每组样本的关键突变特征(mutational signature 1,3, 5,8,12,13,15,16,21,26和30),结果如图2所示,每种亚型都有特异的突变特征变量,而后,根据提取出的这11种最基础的突变特征,进行NMF聚类分析,将TCGA-GBMLGG队列的所有患者分为四种亚型,命名为C1,C2,C3和C4,如图3所示;
步骤四:胶质瘤风险指数(glioma risk index,GRI)的构建
①数据分析,TCGA-GBMLGG胶质瘤队列作为训练集进行建模;三个CGGA队列(mRNA-array(n=301),mRNAseq_325(n=325)和mRNAseq_693(n=693))
②筛选4种亚型之间的共同差异表达基因(differentially expressed genes,DEGs):分别将每一种亚型组与其他三个亚型组配对比较,使用edgeR软件包进行基因表达差异分析,标准为校正p值<0.05和|log2 FC|>1,结果:鉴定出四组DEGs后取交集,共识别出708个DEGs
③对708个DEGs做单因素cox回归分析,|1-HR|>0.5和 P-adjust<0.05的基因作为预后相关基因纳入下一步分析,(HR:危险比;P-adjust:校正P值),结果:一共提取到226个基因纳入下一步分析
④将这226个基因两两组合,形成基因对(gene pair),每对基因包含两个基因,A和B,表示为A|B,在一个样本中,若基因A 的表达值高于基因B,那么该A|B基因对的值标记为1,反之为0,这样的赋值设计的优势在于,只需要关注两个基因mRNA表达之间的数学关系,完全忽略了不同平台之间的批次效应,不需要定义截断值(cut-off值),增加了临床适用性,在TCGA队列中,通过上述赋值方法对每个样本中的所有基因对进行赋值,并剔除在80%以上样本中评分为全为0或全为1的基因对,最终得到一个由样本和基因对构成的二进制0/1矩阵,用于下一步骤的分析
⑤根据上述0/1矩阵,对其中包含的基因对进行Lasso回归,以降维并建模,最优模型由惩罚系数λ决定,当惩罚系数 lambda=0.07094148时,模型最优,最优模型包含了由36个基因组成的44个基因对基于这44个基因对
⑥GRI计算公式设计如下:
GRI=∑β
其中i为Lasso回归得到的关键基因对,GPV是i的赋值(0/1),β是i对应的Lasso回归系数,最终,GRI计算公式为:GRI=0.022 ×GPV(AGXT|BPIFB4)+0.002×GPV(AGXT|STMND1)+0.040×GPV (C5orf46|CSAG3)+0.031×GPV(CD70|FMO1)+0.070×GPV (DCSTAMP|FMO1)+-0.087×GPV(EDARADD|MAGED4)+0.051×GPV (EMP3|SOCS2)+0.234×GPV(EN1|FAT2)+0.076×GPV(EN1|PXDNL) +0.016×GPV(EN1|TDO2)+-0.020×GPV(ESR2|MAGED4)+0.138× GPV(FAM92B|FCAMR)+-0.285×GPV(FAT2|ITGBL1)+-0.160×GPV (FAT2|TMEM71)+-0.244×GPV(FAT2|WISP1)+-0.058×GPV (FMO1|HIST1H2BH)+-0.021×GPV(FMO1|HOXD11)+0.401×GPV (GPR1|KLRC1)+0.156×GPV(HCRT|PRSS48)+0.115×GPV (HCRT|SLC6A18)+0.055×GPV(HIST1H2AJ|SLC6A18)+0.019×GPV (HIST1H3C|RIPPLY3)+0.161×GPV(HIST1H3F|USP29)+0.270×GPV (HIST1H3G|PRSS48)+0.030×GPV(HIST1H3G|SLC6A18)+0.040× GPV(HIST1H3G|SLCO1B1)+0.098×GPV(HIST1H4B|SLCO1B1)+0.038 ×GPV(HIST1H4D|SLC6A18)+0.260×GPV(HIST1H4D|SPRR2A)+0.177 ×GPV(HOXA6|SLC6A18)+0.022×GPV(HOXA6|SPRR2A)+0.026×GPV (HOXD11|POTEF)+0.130×GPV(HOXD11|PRSS48)+0.038×GPV (HOXD11|TCF23)+0.007×GPV(HOXD11|UCN2)+0.207×GPV (IGFBP2|SLC29A1)+0.005×GPV(IL36B|SLCO1B1)+0.057×GPV (MAGED4|MOCOS)+-0.041×GPV(METTL1|PLA2G5)+0.093×GPV (NPIPA7|SLC6A18)+0.243×GPV(PAEP|SLCO1B1)+0.115×GPV(PLEKHN1|TNFSF11)+0.016×GPV(POTEI|POTEJ)+0.004×GPV (RBP1|SOCS2)
⑦用survminer包确定GRI最佳截断点0.8321341,根据此截断值,可将患者分为高、低GRI两组,Kaplan-Meier生存分析表明高 GRI患者的预后生存情况比低GRI患者差。
优选的,所述步骤一中数据来源从癌症基因组图谱(TCGA)和胶质瘤基因组图谱(CGGA)数据库获取,所述TCGA-GBM和TCGA-LGG 对应的体细胞突变数据、拷贝数变异数据和甲基化450K数据亦从 TCGA数据库中获取。
优选的,所述步骤二中“突变特征”收集的数量为30种,且将 30种“突变特征”绘制成表格进行对比。
优选的,所述临床特征包括胶质瘤级别分类(胶质母细胞瘤GBM/ 低级别胶质瘤LGG)、年龄(<60岁/≥60岁)、性别(男/女)、 IDH状态(突变型/野生型)、1p/19q(共缺失/无共缺失)、7+/10- (共发生/无共发生)和MGMT启动子甲基化(甲基化/无甲基化)在四种亚型中分布不同,从C1至C4,老年患者和高级别胶质瘤(GBM) 患者的百分比呈下降趋势,4个集群的性别分布无显著差异,C4的 IDH突变、1p/19q共缺失和MGMT启动子甲基化百分比最高。
优选的,所述步骤四中随机抽取一组数据,且将Rembrandt队列(n=475)作为测试集进行验证。
优选的,所述步骤四中利用计算机将数据输入,生成GRI初始模型,在利用计算机技术得到GRI的计算公式。
优选的,所述步骤四⑥中的GPV(A|B)即基因对的赋值,且这 44个A|B基因对即Lasso回归得到的44个关键基因对。
优选的,所述步骤四⑦中利用GRI预测1年、3年、5年、7年和9年生存状态的AUC分别为0.921、0.958、0.941、0.925和0.908,并与数据库中后期患者的健康程度进行对比,验证GRI预测的精准度。
与现有技术相比,本发明的有益效果是:
1、该脑胶质瘤分子分型方法通过大量数据得到GRI值,不仅可以对不同的患者脑胶质瘤分子进行划分,而且提高了脑胶质瘤分子划分的精准度,利用GRI对患者进行划分,方便辅助医生对患者进行不同类型的划分,利用不同的治疗方式对患者进行科学的治疗,提高患者的治疗康复率,解决了传统脑胶质瘤分子分型精准度低的问题。
2、该脑胶质瘤分子分型方法通过步骤一中数据来源从癌症基因组图谱(TCGA)和胶质瘤基因组图谱(CGGA)数据库获取,且 TCGA-GBM和TCGA-LGG对应的体细胞突变数据、拷贝数变异数据和甲基化450K数据亦从TCGA数据库中获取,提供研究数据的来源,避免数据造假,提高研究成果的可靠性,通过步骤二中“突变特征”收集的数量为30种,且将30种“突变特征”绘制成表格进行对比,提高数据分析的精准度,利用多组数据进行分析,避免研究出现偏差,造成个例的出现,提高科研的成果的精准度,通过使用 pRRophetic包,预测4个集群对吉西他滨和硼替佐米的整体敏感性,吉西他滨或硼替佐米与标准化疗药物替莫唑胺联合应用可使胶质瘤患者生存获益,本研究中通过半数抑制浓度(IC50)定量药物敏感性,IC50越低,敏感性越高,结果发现,C1亚型对硼替佐米更敏感, C3亚型对吉西他滨更敏感,方便不同类型的患者利用不同类型的治疗方式进行治疗,提高患者的康复率。
3、该脑胶质瘤分子分型方法通过Rembrandt队列(n=475)作为测试集进行验证,提高科研的严谨性,避免以点概面的显现出现,验证科研成果的精准度,通过利用计算机技术方便简化科研流程,同时提高数据分析的精准度,有利于快速的得到GRI的公式,通过公式中的变量进行解释,方便其他非科研人员对数据的理解,通过 GRI预测患者之后1年、3年、5年、7年和9年生存状态,并与数据库中后期患者的健康程度进行对比,验证GRI预测的精准度。
附图说明
图1为本发明cophenetic系数和轮廓系数的关系图;
图2为本发明多组突变特征变化特征图;
图3为本发明结构分析图。
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
请参阅图1-3,本发明提供的一种实施例:
一种新的脑胶质瘤分子分型方法,包括如下步骤:
步骤一:收集数据
从数据库中获取基因表达谱和临床信息数据,五个胶质瘤队列纳入本研究:TCGA-GBMLGG队列(n=892),三个CGGA队列(mRNA-array (n=301),mRNAseq_325(n=325)和mRNAseq_693(n=693))和 Rembrandt队列(n=475),从公开数据库收集了四个独立的免疫治疗队列,包括:①Roh队列:抗CTLA-4、抗PD-1治疗队列;② GSE100797:过继性T细胞治疗队列;③GSE78220:抗PD-1治疗队列;④GSE35640:抗MAGE-A3治疗队列,根据recistv1.1标准,完全缓解(CR)或部分缓解(PR)患者被视为免疫治疗有反应者,疾病稳定(SD)或疾病进展(PD)患者被视为免疫治疗无反应者,不可评估(NE)患者则被剔除;
步骤二:突变特征谱
突变特征库(第二版)聚焦于碱基置换突变,突变点的碱基置换包含六种类型:C>A,C>G,C>T,T>A,T>C和T>G,突变点两侧(5' 和3'端)各可搭配四种碱基(A、T、C、G),最终可产生96种可能的突变类型(6种突变位点的碱基替换类型×4种,5'碱基×4种, 3'碱基),在体细胞中,各种机制引起DNA损伤,进而发生体细胞突变,使得细胞基因组不断变化,各种类型的突变不断积累,最终,在96种突变类型上具有不同的积累,形成一个个独特的突变积累组合,检测到的每种组合即为一种“突变特征”;
步骤三:亚型识别
(一)、数据获取:从COSMIC网站获取获取每种突变特征的特征数据信息,从TCGA-LGG和TCGA-GBM获得的体细胞突变数据在去除沉默突变后用于构建每个个体的突变特征谱,参考基因组为h38
(二)、R包:DeconstructSigs和NMFpackage
(三)、方法
①移除突变数据中的沉默突变
②利用计算机技术将移除沉默突变的突变数据转化为突变环境矩阵
③使用DeconstructSigs包,分析每个样本中30个signature 的组成比例,参考的signature是COSMIC,cutoff值设置为0.06,标准化方式使用“exome2genome”,最终我们得到了一个矩阵(行为30个signature,列为每个样本,cell的值为signature在每个样本的比例,所有signature加起来刚好为1)
④使用NMF包进行提取和聚类分析,设置潜在的ranks=2:5,运算执行次数设置为50,method设置为‘lee’,最终通过从cophenetic 系数和轮廓系数决定最佳rank=4,如图1,即根据TCGA-GBMLGG队列中每个患者的突变特征谱,将胶质瘤分为四种分子亚型最优
⑤非负矩阵分解的一个特性是倾向于产生观察数据的稀疏表示,导致双聚类的自然应用,通过少量特征表征样本组,在NMF模型中,根据对每个样本贡献最大的基础组分(即在系数矩阵的每列中具有最大系数的基础组分)对样本进行分组,然后,通过根据基础矩阵计算的基础特异性评分选择的一组特征对每组样本进行表征,上述过程由NMF包实现,根据所有患者的突变特征谱,构建NMF 模型,并通过extractFeatures函数(方法设置为“max”)对最基本的特定特征进行提取,最终,患者分为4个基础组,并提取出11 种可表征每组样本的关键突变特征(mutational signature 1,3, 5,8,12,13,15,16,21,26和30),结果如图2所示,每种亚型都有特异的突变特征变量,而后,根据提取出的这11种最基础的突变特征,进行NMF聚类分析,将TCGA-GBMLGG队列的所有患者分为四种亚型,命名为C1,C2,C3和C4,如图3所示;
步骤四:胶质瘤风险指数(glioma risk index,GRI)的构建
①数据分析,TCGA-GBMLGG胶质瘤队列作为训练集进行建模;三个CGGA队列(mRNA-array(n=301),mRNAseq_325(n=325)和 mRNAseq_693(n=693))
②筛选4种亚型之间的共同差异表达基因(differentially expressed genes,DEGs):分别将每一种亚型组与其他三个亚型组配对比较,使用edgeR软件包进行基因表达差异分析,标准为校正p值<0.05和|log2 FC|>1,结果:鉴定出四组DEGs后取交集,共识别出708个DEGs
③对708个DEGs做单因素cox回归分析,|1-HR|>0.5和P-adjust<0.05的基因作为预后相关基因纳入下一步分析,(HR:危险比;P-adjust:校正P值),结果:一共提取到226个基因纳入下一步分析
④将这226个基因两两组合,形成基因对(gene pair),每对基因包含两个基因,A和B,表示为A|B,在一个样本中,若基因A 的表达值高于基因B,那么该A|B基因对的值标记为1,反之为0,这样的赋值设计的优势在于,只需要关注两个基因mRNA表达之间的数学关系,完全忽略了不同平台之间的批次效应,不需要定义截断值(cut-off值),增加了临床适用性,在TCGA队列中,通过上述赋值方法对每个样本中的所有基因对进行赋值,并剔除在80%以上样本中评分为全为0或全为1的基因对,最终得到一个由样本和基因对构成的二进制0/1矩阵,用于下一步骤的分析
⑤根据上述0/1矩阵,对其中包含的基因对进行Lasso回归,以降维并建模,最优模型由惩罚系数λ决定,当惩罚系数 lambda=0.07094148时,模型最优,最优模型包含了由36个基因组成的44个基因对基于这44个基因对
⑥GRI计算公式设计如下:
GRI=∑β
其中i为Lasso回归得到的关键基因对,GPV是i的赋值(0/1),β是i对应的Lasso回归系数,最终,GRI计算公式为:GRI=0.022 ×GPV(AGXT|BPIFB4)+0.002×GPV(AGXT|STMND1)+0.040×GPV (C5orf46|CSAG3)+0.031×GPV(CD70|FMO1)+0.070×GPV (DCSTAMP|FMO1)+-0.087×GPV(EDARADD|MAGED4)+0.051×GPV (EMP3|SOCS2)+0.234×GPV(EN1|FAT2)+0.076×GPV(EN1|PXDNL) +0.016×GPV(EN1|TDO2)+-0.020×GPV(ESR2|MAGED4)+0.138× GPV(FAM92B|FCAMR)+-0.285×GPV(FAT2|ITGBL1)+-0.160×GPV (FAT2|TMEM71)+-0.244×GPV(FAT2|WISP1)+-0.058×GPV (FMO1|HIST1H2BH)+-0.021×GPV(FMO1|HOXD11)+0.401×GPV (GPR1|KLRC1)+0.156×GPV(HCRT|PRSS48)+0.115×GPV (HCRT|SLC6A18)+0.055×GPV(HIST1H2AJ|SLC6A18)+0.019×GPV (HIST1H3C|RIPPLY3)+0.161×GPV(HIST1H3F|USP29)+0.270×GPV (HIST1H3G|PRSS48)+0.030×GPV(HIST1H3G|SLC6A18)+0.040× GPV(HIST1H3G|SLCO1B1)+0.098×GPV(HIST1H4B|SLCO1B1)+0.038 ×GPV(HIST1H4D|SLC6A18)+0.260×GPV(HIST1H4D|SPRR2A)+0.177 ×GPV(HOXA6|SLC6A18)+0.022×GPV(HOXA6|SPRR2A)+0.026×GPV (HOXD11|POTEF)+0.130×GPV(HOXD11|PRSS48)+0.038×GPV (HOXD11|TCF23)+0.007×GPV(HOXD11|UCN2)+0.207×GPV (IGFBP2|SLC29A1)+0.005×GPV(IL36B|SLCO1B1)+0.057×GPV (MAGED4|MOCOS)+-0.041×GPV(METTL1|PLA2G5)+0.093×GPV (NPIPA7|SLC6A18)+0.243×GPV(PAEP|SLCO1B1)+0.115×GPV(PLEKHN1|TNFSF11)+0.016×GPV(POTEI|POTEJ)+0.004×GPV (RBP1|SOCS2)
⑦用survminer包确定GRI最佳截断点0.8321341,根据此截断值,可将患者分为高、低GRI两组,Kaplan-Meier生存分析表明高GRI患者的预后生存情况比低GRI患者差,通过大量数据得到GRI 值,不仅可以对不同的患者脑胶质瘤分子进行划分,而且提高了脑胶质瘤分子划分的精准度,利用GRI对患者进行划分,方便辅助医生对患者进行不同类型的划分,利用不同的治疗方式对患者进行科学的治疗,提高患者的治疗康复率,解决了传统脑胶质瘤分子分型精准度低的问题。
步骤一中数据来源从癌症基因组图谱(TCGA)和胶质瘤基因组图谱(CGGA)数据库获取,TCGA-GBM和TCGA-LGG对应的体细胞突变数据、拷贝数变异数据和甲基化450K数据亦从TCGA数据库中获取,通过步骤一中数据来源从癌症基因组图谱(TCGA)和胶质瘤基因组图谱(CGGA)数据库获取,且TCGA-GBM和TCGA-LGG对应的体细胞突变数据、拷贝数变异数据和甲基化450K数据亦从TCGA数据库中获取,提供研究数据的来源,避免数据造假,提高研究成果的可靠性。
步骤二中“突变特征”收集的数量为30种,且将30种“突变特征”绘制成表格进行对比,通过步骤二中“突变特征”收集的数量为30种,且将30种“突变特征”绘制成表格进行对比,提高数据分析的精准度,利用多组数据进行分析,避免研究出现偏差,造成个例的出现,提高科研的成果的精准度。
临床特征包括胶质瘤级别分类(胶质母细胞瘤GBM/低级别胶质瘤LGG)、年龄(<60岁/≥60岁)、性别(男/女)、IDH状态(突变型/野生型)、1p/19q(共缺失/无共缺失)、7+/10-(共发生/无共发生)和MGMT启动子甲基化(甲基化/无甲基化)在四种亚型中分布不同,从C1至C4,老年患者和高级别胶质瘤(GBM)患者的百分比呈下降趋势,4个集群的性别分布无显著差异,C4的IDH突变、 1p/19q共缺失和MGMT启动子甲基化百分比最高,通过使用pRRophetic包,预测4个集群对吉西他滨和硼替佐米的整体敏感性,吉西他滨或硼替佐米与标准化疗药物替莫唑胺联合应用可使胶质瘤患者生存获益,本研究中通过半数抑制浓度(IC50)定量药物敏感性,IC50越低,敏感性越高,结果发现,C1亚型对硼替佐米更敏感, C3亚型对吉西他滨更敏感,方便不同类型的患者利用不同类型的治疗方式进行治疗,提高患者的康复率。
步骤四中随机抽取一组数据,且将Rembrandt队列(n=475)作为测试集进行验证,通过Rembrandt队列(n=475)作为测试集进行验证,提高科研的严谨性,避免以点概面的显现出现,验证科研成果的精准度。
步骤四中利用计算机将数据输入,生成GRI初始模型,在利用计算机技术得到GRI的计算公式,通过利用计算机技术方便简化科研流程,同时提高数据分析的精准度,有利于快速的得到GRI的公式。
步骤四⑥中的GPV(A|B)即基因对的赋值,且这44个A|B基因对即Lasso回归得到的44个关键基因对,通过公式中的变量进行解释,方便其他非科研人员对数据的理解。
步骤四⑦中利用GRI预测1年、3年、5年、7年和9年生存状态的AUC分别为0.921、0.958、0.941、0.925和0.908,并与数据库中后期患者的健康程度进行对比,验证GRI预测的精准度,通过 GRI预测患者之后1年、3年、5年、7年和9年生存状态,并与数据库中后期患者的健康程度进行对比,验证GRI预测的精准度。
对于本领域技术人员而言,显然本发明不限于上述示范性实施例的细节,而且在不背离本发明的精神或基本特征的情况下,能够以其他的具体形式实现本发明。因此,无论从哪一点来看,均应将实施例看作是示范性的,而且是非限制性的,本发明的范围由所附权利要求而不是上述说明限定,因此旨在将落在权利要求的等同要件的含义和范围内的所有变化囊括在本发明内。不应将权利要求中的任何附图标记视为限制所涉及的权利要求。
- 一种新的脑胶质瘤分子分型方法
- 一种针对新冠肺炎临床分型的CT影像对比特征学习方法