一种基于自适应寻优模型的血糖变化趋势预测系统
文献发布时间:2023-06-19 09:30:39
技术领域
本发明涉及血糖预测领域,具体是一种血糖变化趋势预测系统。
背景技术
近年来,随着社会经济的发展和居民生活水平的提高,国民人均寿命不断延长,在老龄化趋势的影响下,在老年人群体中糖尿病的发病率及患病率逐年升高,成为威胁人民健康的重大社会问题,引起各国政府、卫生部门以及广大医务工作者的关注和重视。糖尿病是由遗传因素、免疫功能紊乱、微生物感染等各种致病因子导致的代谢紊乱综合征,临床上以高血糖为主要特征。糖尿病危害巨大,不仅影响患者的生活质量,而且给患者带来沉重的心理负担,糖尿病并发症对患者的健康和生命构成威胁,可导致残废和早亡。若是能够一定的方法对人们的血糖变化趋势进行提前的预测,从而提出相应的干预对策,可以极大的降低糖尿病病发的几率,也能减少巨大的资金和资源上的浪费。
人工智能(AI)逐渐在各行各业展露头角,计算机能够像人类一样思考和学习,又称为机器学习,在医疗领域上有莫大发展潜力。甚至在消费化时代扮演要角,透过提升病患和医生的医疗体验,来改善整个医疗体制。机器学习也是精准医疗(precision medicine)的幕后功臣,将每位病患独特的基因组成、环境因素、生活模式和家族史纳入考量,进而提高诊断的准确度,提供个人化的疾病治疗和预防计划。机器学习亦有助于分析病患资料,把资料化为可视和可行的信息,人们也在进行相关方面的研究。
发明内容
本发明实施例的目的在于提供一种血糖变化趋势预测系统,以解决上述背景技术中提出的问题。
为实现上述目的,本发明实施例提供如下技术方案:
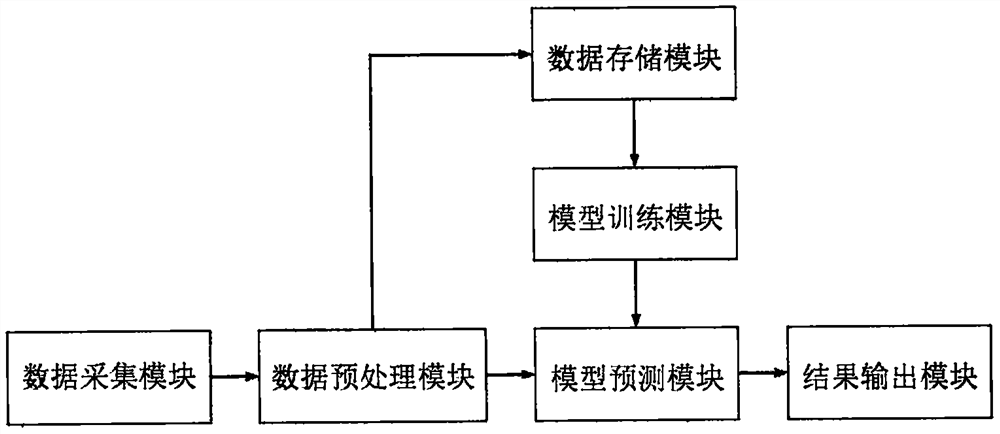
一种血糖变化趋势预测系统,包括数据采集模块、数据预处理模块、数据存储模块、模型训练模块、模型预测模块和结果输出模块,所述数据采集模块与数据预处理模块相连,模型预测模块分别与数据预处理模块、模型训练模块和结果输出模块相连,数据存储模块分别与数据预处理模块和模型训练模块相连,数据采集模块用于对患者血液的采集以及对血液相关成分分析,数据存储模块用于存贮和更新患者的历史数据,丢弃陈旧数据,模型训练模块用于生成训练模型,模型预测模块用于对数据进行预测,最终集成得到最后的预测输出,结果输出模块用于输出最终的血糖趋势预测结果以及相应的医疗建议,数据预处理模块用于分析数据的分布特征,过滤原始数据中的异常值,对有必要的缺失值进行填充,构建新特征。
作为本发明实施例进一步的方案:模型训练模块包括分类模型模块和回归模型模块。
作为本发明实施例进一步的方案:分类模型模块采用Bagging算法建立多个子模型,每个子模型由GBDT和LR组合而成,先利用GBDT构建新特征,并进行独热编码,然后作为新特征输入LR,最后得到分类模型。
作为本发明实施例进一步的方案:回归模型模块使用Xgboost算法建模,然后进行集成。
所述血糖变化趋势预测系统的工作流程,具体步骤如下:
步骤一,数据采集模块采集用户的数据并且作为原始训练集,用户输入的数据作为原始数据;
步骤二,数据预处理模块对原始训练集进行分析和预处理,得到预处理后的数据;
步骤三,模型训练模块依据预处理后的数据建立血糖值的预测模型;
步骤四,模型预测模块利用得到的血糖值的预测模型对原始数据进行预测,获得血糖预测值,结果输出模块把血糖预测值的结果输出。
作为本发明实施例进一步的方案:步骤二中分析包括统计缺失值和分析异常值。
作为本发明实施例进一步的方案:步骤二中预处理包括缺失值处理、异常值处理和构造新特征。
作为本发明实施例进一步的方案:缺失值处理为对原始训练集的特征数据进行遍历,对特征数据中特征的缺失程度大于或等于70%以上的特征数据进行丢弃,对特征数据中特征的缺失程度低于70%的特征数据视为可接受缺失范围,对可接受缺失范围的特征数据,计算未缺失的特征数据的平均值,并用该平均值来填补该特征数据里的缺失数据。
作为本发明实施例进一步的方案:异常值处理为过滤血糖值大于30的原始训练集。
作为本发明实施例进一步的方案:构造新特征为根据已有特征手动构造相关特征,可以深一步地挖掘出原始训练集中的信息。
与现有技术相比,本发明实施例的有益效果是:
本发明采集用户之前一段时间的各种生理指标数据和基础信息,分析相关生理指标本与血糖值隐形联系,挖掘出用户过去一段时间血糖变化的趋势,,预测用户一年内的血糖变化趋势,从而可以对可能出现高血糖的用户提出预防建议,有利于降低糖尿病的发病率;
本发明将不同的算法合理的组合,扬长避短,可以更好更充分地挖掘出数据中的信息,同时保证模型的精确度、普适性和鲁棒性,使用前景广阔。
附图说明
图1为血糖变化趋势预测系统的结构示意图。
图2为血糖变化趋势预测系统的工作流程图。
具体实施方式
下面结合具体实施方式对本专利的技术方案作进一步详细地说明。
实施例1
一种血糖变化趋势预测系统,包括数据采集模块、数据预处理模块、数据存储模块、模型训练模块、模型预测模块和结果输出模块,所述数据采集模块与数据预处理模块相连,模型预测模块分别与数据预处理模块、模型训练模块和结果输出模块相连,数据存储模块分别与数据预处理模块和模型训练模块相连,数据采集模块用于对患者血液的采集以及对血液相关成分分析,数据存储模块用于存贮和更新患者的历史数据,丢弃陈旧数据,模型训练模块用于生成训练模型,模型预测模块用于对数据进行预测,最终集成得到最后的预测输出,结果输出模块用于输出最终的血糖趋势预测结果以及相应的医疗建议,数据预处理模块用于分析数据的分布特征,过滤原始数据中的异常值,对有必要的缺失值进行填充,构建新特征。模型训练模块包括分类模型模块和回归模型模块。分类模型模块采用Bagging算法建立多个子模型,每个子模型由GBDT和LR组合而成,先利用GBDT构建新特征,并进行独热编码,然后作为新特征输入LR,最后得到分类模型。回归模型模块使用Xgboost算法建模,然后进行集成。
所述血糖变化趋势预测系统的工作流程,具体步骤如下:
步骤一,从某医院获取了m个体检者的体检数据,作为本方法的原始训练集,其中每个体检者的体检信息包括性别、年龄,以及不同时期(之前半个年内每个月,之前一个月每个星期)的血常规、肝功能、肾功能、血脂等个人以及体检相关数据总共325维特征,体检信息具体包括性别、年龄、舒张压、天门东氨酸转移酶、丙氨酸氨基转移酶、碱性磷酸酶、r-谷氨酰基转移酶、淋巴细胞总数、总蛋白、白蛋白、球蛋白、白球比例、甘油三酯、总胆固醇、低密度脂蛋白胆固醇、高密度脂蛋白胆固醇、尿素、肌酐、尿酸、乙肝表面抗体、乙肝表面抗原、乙肝e抗原、乙肝抗体、乙肝核心抗体、白细胞计数、红细胞计数、血红蛋白、红细胞压积、红细胞平均体积、红细胞平均血红蛋白量、红细胞平均血红蛋白浓度、红细胞体积分布宽度、血小板计数、血小板平均体积、血小板体积分布宽度、血小板比积、中性粒细胞的数量百分比、淋巴细胞的数量百分比、单核细胞的数量百分比、嗜酸细胞的数量百分比和嗜碱细胞的数量百分比。
步骤二,对原始训练集进行处理,包括初步分析、缺失值处理、异常值处理和构造新特征。初步分析:通过分析原始训练集的数据,以判断原始训练集是否适用当前设计的算法模型,原始训练集的分析包括:统计缺失值,分析异常值,通过对特征数据分布分析,计算偏度、峰度,发现有部分字段偏离标准整体分布较为严重,使用log转换,对这些字段做log转换之后计算和标签变量的相关系数。缺失值处理:(1)对特征数据进行遍历,对特征数据中特征的缺失程度大于或等于70%以上的特征数据进行丢弃;(2)对特征数据进行遍历,对特征数据中特征的缺失程度低于70%的特征数据视为可接受缺失范围,对可接受缺失范围的特征数据计算未缺失的特征数据的平均值,并用该平均值来填补该特征数据里的缺失数据。异常值处理:过滤血糖值大于30的原始训练集。构造新特征:查询相关病理知识,根据已有特征手动构造相关特征,可以深一步地挖掘出原始训练集中的信息,本发明构造的新特征如下:data[’总酶’]=data[’*天门冬氨酸氨基转换酶’]+data[’*丙氨酸氨基转换酶’]+data[’*碱性磷酸酶’]+data[’*r-谷氨酰基转换酶’]
data[’*天门冬氨酸氨基转换酶ratio’]=data[’*天门冬氨酸氨基转换酶’]/np.maximum(data[″总酶″].astype(″float″),1)
data[’*天门冬氨酸氨基转换酶ratio’].loc[data[’*天门冬氨酸氨基转换酶ratio’]<0]=0
data[’*天门冬氨酸氨基转换酶ratio’].loc[data[’*天门冬氨酸氨基转换酶ratio’]>1]=1
data[’*丙氨酸氨基转换酶ratio’]=data[’*丙氨酸氨基转换酶’]/np.maximum(data[″总酶″].astype(″float″),1)
data[’*丙氨酸氨基转换酶ratio’].loc[data[’*丙氨酸氨基转换酶ratio’]<0]=0
data[’*丙氨酸氨基转换酶ratio’].loc[data[’*丙氨酸氨基转换酶ratio’]>1]=1
data[’*碱性磷酸酶ratio’]=data[’*碱性磷酸酶’]/np.maximum(data[″总酶″].astype(″float″),1)
data[’*碱性磷酸酶ratio’].loc[data[’*碱性磷酸酶ratio’]<0]=0
data[’*碱性磷酸酶ratio’].loc[data[’*碱性磷酸酶ratio’]>1]=1
data[’*r-谷氨酰基转换酶ratio’]=data[’*r-谷氨酰基转换酶’]/np.maximum(data[″总酶″].astype(″float″),1)
data[’*r-谷氨酰基转换酶ratio’].loc[data[’*r-谷氨酰基转换酶ratio’]<0]=0
data[’*r-谷氨酰基转换酶ratio’].loc[data[’*r-谷氨酰基转换酶ratio’]>1]=1
data[’白蛋白ratio’]=data[’白蛋白’]/np.maximum(data[″*总蛋白″].astype(″float″),1)
data[’白蛋白ratio’].loc[data[’白蛋白ratio’]<0]=0
data[’白蛋白ratio’].loc[data[’白蛋白ratio’]>1]=1
data[’*球蛋白ratio’]=data[’*球蛋白’]/np.maximum(data[″*总蛋白″].astype(″float″),1)
data[’*球蛋白ratio’].loc[data[’*球蛋白ratio’]<0]=0
data[’*球蛋白ratio’].loc[data[’*球蛋白ratio’]>1]=1
data[’高密度脂蛋白胆固醇ratio’]=data[’高密度脂蛋白胆固醇’]/np.maximum(data[″总胆固醇″].astype(″float″),1)
data[’高密度脂蛋白胆固醇ratio’].loc[data[’高密度脂蛋白胆固醇ratio’]<0]=0
data[’高密度脂蛋白胆固醇ratio’].loc[data[’高密度脂蛋白胆固醇ratio’]>1]=1
data[’低密度脂蛋白胆固醇ratio’]=data[’低密度脂蛋白胆固醇’]/np.maximum(data[″总胆固醇″].astype(″float″),1)
data[’低密度脂蛋白胆固醇ratio’].loc[data[’低密度脂蛋白胆固醇ratio’]<0]=0
data[’低密度脂蛋白胆固醇ratio’].loc[data[’低密度脂蛋白胆固醇ratio’]>1]=1
data[’null_count’]=data.isnull().sum(axis=1)
data[’*r-谷氨酰基转换酶-尿酸’]=data[’*r-谷氨酰基转换酶’]-data[’尿酸’]
data[’*r-谷氨酰基转换酶*年龄’]=data[’*r-谷氨酰基转换酶’]*data[’年龄’]
data[’*r-谷氨酰基转换酶*总胆固醇’]=data[’*r-谷氨酰基转换酶’]*data[’总胆固醇’]
data[’*丙氨酸氨基转换酶**天门冬氨酸氨基转换酶’]=data[’*丙氨酸氨基转换酶’]*data[’*天门冬氨酸氨基转换酶’]
data[’*丙氨酸氨基转换酶+*天门冬氨酸氨基转换酶’]=data[’*丙氨酸氨基转换酶’]+data[’*天门冬氨酸氨基转换酶’]
data[’*丙氨酸氨基转换酶/*天门冬氨酸氨基转换酶’]=data[’*丙氨酸氨基转换酶’]/data[’*天门冬氨酸氨基转换酶’]
data[’*天门冬氨酸氨基转换酶/*总蛋白’]=data[’*天门冬氨酸氨基转换酶’]/data[’*总蛋白’]
data[’*天门冬氨酸氨基转换酶-*球蛋白’]=data[’*天门冬氨酸氨基转换酶’]-data[’*球蛋白’]
data[’*球蛋白/甘油三酯’]=data[’*球蛋白’]/data[’甘油三酯’]
data[’年龄*红细胞计数/红细胞体积分布宽度-红细胞计数’]=data[’年龄’]*data[’红细胞计数’]/(data[’红细胞体积分布宽度’]-data[’红细胞计数’])
data[’尿酸/肌酐’]=data[’尿酸’]/data[’肌酐’]
data[’肾’]=data[’尿素’]+data[’肌酐’]+data[’尿酸’]
data[’红细胞计数*红细胞平均血红蛋白量’]=data[’红细胞计数’]*data[’红细胞平均血红蛋白量’]
data[’红细胞计数*红细胞平均血红蛋白浓度’]=data[’红细胞计数’]*data[’红细胞平均血红蛋白浓度’]
data[’红细胞计数*红细胞平均体积’]=data[’红细胞计数’]*data[’红细胞平均体积’]data[’嗜酸细胞’]=data[’嗜酸细胞%’]*100
data[’年龄*中性粒细胞%/尿酸*血小板比积’]=data[’年龄’]*data[’中性粒细胞%’]/(data[’尿酸’]*data[’血小板比积’])
data[’丙氨-高密1’]=data[’*丙氨酸氨基转换酶’]-data[’高密度脂蛋白胆固醇’]
data[’丙氨-高密2’]=data[’*丙氨酸氨基转换酶’]/data[’高密度脂蛋白胆固醇’]
data[’丙氨-高密3’]=(data[’*丙氨酸氨基转换酶’]-data[’高密度脂蛋白胆固醇’])/data[’高密度脂蛋白胆固醇’]
data[’碱性-高密1’]=data[’*碱性磷酸酶’]-data[’高密度脂蛋白胆固醇’]
data[’碱性-高密2’]=data[’*碱性磷酸酶’]/data[’高密度脂蛋白胆固醇’]
data[’碱性-高密3’]=(data[’*碱性磷酸酶’]-data[’高密度脂蛋白胆固醇’])/data[’高密度脂蛋白胆固醇’]
data[’r-谷-高密1’]=data[’*r-谷氨酰基转换酶’]-data[’高密度脂蛋白胆固醇’]
data[’r-谷-高密2’]=data[’*r-谷氨酰基转换酶’]/data[’高密度脂蛋白胆固醇’]
data[’r-谷-高密3’]=(data[’*r-谷氨酰基转换酶’]-data[’高密度脂蛋白胆固醇’])/data[’高密度脂蛋白胆固醇’]。
步骤三,通过对原始训练集的分布的初步分析可知,将血糖值以7为界,小于7视为血糖正常,大于等于7视为血糖异常,两类数据存在明显的差异,若视其为一个整体单独建立一个模型,预测效果不佳,所以本发明先将血糖值进行分类,然后对两类数据分别建立血糖值的预测模型。
训练预测模型时先将原始训练集划分为训练集和测试集,由训练集训练模型,测试集评价模型,最终以MSE作为评价指标。
分类模型以Bagging作为整体框架,使用GBDT和LR构建子模型。每个子模型的训练数据是从原始训练数据中随机抽取得到的,将血糖值大于等于7的标记为1,反之标记为0。GBDT用以构建新特征,如此构建得到的特征可视为原始特征的线性、非线性组合,由于GBDT是有标签的学习算法,所以这样得到的新特征还更具有导向性。得到的新特征经过独热编码后传入LR得到分类结果,包括当前子模型判断的结果和属于异常类的概率。如此循环可得到N个分类子模型。
GBDT和LR算法都会涉及一些参数设置,本发明使用自定义的网格搜索进行寻优,结合5折交叉验证,以F1作为参数寻优的评价指标,其中F1值就是精确值和召回率的调和均值,公式如下:
其中P代表精确率,R表示召回率,公式如下:
其中TP表示被正确预测为异常类的样本(实际为异常类),FP表示被错误预测为异常类的样本(实际为正常类),FN表示实际为异常类但被判断为正常类的样本。
5折交叉验证:将数据集平均划分为5分,先用第1份作为验证数据,其余4份作为训练数据,第2次用第2份作为验证数据,其余4份作为训练数据,如此循环5次,保证模型精度的同时,可以防止模型过拟合。
可利用网格搜索选优的参数包括GBDT模型的相关主要参数如下:
n_estimators:控制弱学习器的数量
max_depth:设置树深度,深度越大可能过拟合
max_leaf_nodes:最大叶子节点数
learning_rate:更新过程中用到的收缩步长,(0,1]
max_features:划分时考虑的最大特征数,如果特征数非常多,我们可以灵活使用其他取值来控制划分时考虑的最大特征数,以控制决策树的生成时间。
min_samples_split:内部节点再划分所需最小样本数,这个值限制了子树继续划分的条件,如果某节点的样本数少于min_samples_split,则不会继续再尝试选择最优特征来进行划分。
min_samples_leaf:叶子节点最少样本数,这个值限制了叶子节点最少的样本数,如果某叶子节点数目小于样本数,则会和兄弟节点一起被剪枝。
min_weight_fraction_leaf:叶子节点最小的样本权重和,这个值限制了叶子节点所有样本权重和的最小值,如果小于这个值,则会和兄弟节点一起被剪枝。
min_impurity_split:节点划分最小不纯度,使用min_impurity_decrease替代。
min_impurity_decrease:如果节点的纯度下降大于了这个阈值,则进行分裂。
subsample:采样比例,取值为(0,1]。
LR的主要相关参数:
C:正则化系数λ的倒数
solver:优化算法选择参数,只有五个可选参数,即newton-cg,lbfgs,liblinear,sag,saga。默认为liblinear。
class_weight:用于标示分类模型中各种类型的权重
random_state:随机数种子
子模型构建完成后选择阈值,有两种方式。一种是根据N个模型判定为异常类的票数进行投票,设阈值为Y1,若大于Y1则视为异常类。另一种是根据N个模型预测的属于异常类的概率的均值与阈值进行比较,设阈值为Y2,若概率均值大于等于Y2则视为异常类。最终从两种方式再择优。
分类模型得到的结果会进行分析,从而对分类模型模型再作相应的调整,例如子模型的数量和建子模型的训练数据抽样比。
回归模型使用Xgboost算法分别对正常和异常两类建模,并保存模型。过程中也需要设置一些参数,该参数为连续型浮点数。此处,本发明使用自定义的粒子群函数进行寻优,以MSE作为评价指标。主要参数有:
min_child_weight:决定最小叶子节点样本权重和。
max_depth:树的最大深度。
max_leaf_nodes:树上最大的节点或叶子的数量,可以代替max_depth的作用。
Gamma:在节点分裂时,只有在分裂后损失函数的值下降了,才会分裂这个节点。
max_delta_step:这参数限制每颗树权重改变的最大步长。
回归模型得到的结果会进行分析,包括对预测误差较大的样本的统计,从而对回归模型再作相应的调整。
步骤四,模型预测模块利用得到的预测模型对输入的体检数据进行预测,获得血糖预测值,并把结果反馈给用户。
本发明实施例提出的一种基于树模型(GBDT和Xgboost)和回归模型的血糖值预测方法,结合个体的血常规、肝功能、血脂、等体检数据,挖掘出用户过去一段时间血糖变化的趋势,预测出按照当前趋势发展,1年后用户的血糖值,可以对糖尿病高风险患者进行有效预警。
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。不应将权利要求中的任何附图标记视为限制所涉及的权利要求。
此外,应当理解,虽然本说明书按照实施方式加以描述,但并非每个实施方式仅包含一个独立的技术方案,说明书的这种叙述方式仅仅是为清楚起见,本领域技术人员应当将说明书作为一个整体,各实施例中的技术方案也可以经适当组合,形成本领域技术人员可以理解的其他实施方式。