一种基于测序和机器学习的构建疾病风险预测模型的方法和系统
文献发布时间:2023-06-19 11:27:38
技术领域
本发明属于生物医学领域,涉及一种基于测序和机器学习的构建疾病风险预测模型的方法和系统。
背景技术
随着测序技术的发展,成本的降低,在人类健康领域,人基因组测序成为今后的主流趋势,精准医疗将是测序的最终目的。因此,如何准确的发掘测序结果成为了实现精准医疗的必要手段。
结直肠癌(Colorectal cancer,CRC)是世界范围内第三大常见的癌症,也是造成癌症相关死亡的第四大常见原因,其发病迅速,预后较差,且发病率逐年增加。据统计,结直肠癌家族史阳性和五十岁以上的人群发生CRC的风险显著增加,炎症性肠病史和溃疡性结肠炎患者患大肠癌的风险相比健康人群增加3.7%。目前临床上区分溃疡与癌变通过肠镜抓取组织块结合活检病理学诊断基本可以明确,然而对于早期和晚期病变的甄别目前还没有很好的分子标志物。CRC患者的预后主要取决于诊断时疾病的分期。CRC的早期发现会降低相关的死亡率,其前体病变的检测甚至可以降低发病率。
正常组织向癌细胞的转变是逐步进展的,特别是癌变早期、疾病发生期和疾病进展期细胞内的基因表达模式会发生显著改变。RNAseq有助于全面捕捉发病关键基因的表达信号、替代基因剪接转录本、转录后修饰、基因融合、突变/SNP,以及随着时间的推移基因表达变化。转录组是疾病发生早期和发展过程中的直接信号。通过患者与正常人的RNAseq测序对比分析可以快速全面掌握细胞内与疾病发生有关的基因表达模式的改变,对疾病的诊断和治疗提供重要解决策略。
本研究基于机器学习方法,从数学建模、数据挖掘和系统设计实现三个方面,构建疾病发病风险评估的临床辅助诊断系统,并以结直肠癌为例,通过对RNAseq测序数据的标准化预处理、分析定量,构建机器学习分类模型,获得了一个基于人类全部转录基因表达为特征的疾病分类模型,可以区分正常组织、结直肠息肉组织、早期结直肠癌组织和晚期结直肠癌组织。
发明内容
本发明目的之一在于提供一种构建疾病诊断模型的方法,本发明的方法以机器学习和多种统计方法为基础,能在多种类型的数据上展现出较高的灵敏性和特异性。
本发明的目的之二在于提供一种AI分子诊断产品,该产品可用于疾病的早期筛查,预测疾病的发展,为临床医学的进一步诊疗提供辅助。
为了实现上述目的,本发明采用如下技术方案:
本发明第一方面提供了一种构建疾病风险预测模型的方法,所述方法包括:
获取样本测序数据;
对基因进行表达量定量,构建表达量矩阵;
基于MLseq工具进行模型训练,选择分类效果最佳的算法进行风险预测模型的训练和构建。
进一步,所述方法还包括对测序数据进行处理和质控。
进一步,所述方法还包括将处理后的数据比对至人类参考基因组上。
进一步,所述方法还包括将临床信息按照样本分组信息进行特征标记。
进一步,所述方法还包括在模型训练前将样本随机分为训练集和测试集。
进一步,所述方法还包括对风险预测模型进行验证,得到预测分类结果。
进一步,所述模型训练前还包括建模数据处理。
进一步,建模数据处理包括特征剔除、批次效应校正。
进一步,特征剔除的步骤包括:将所有基因单独进行过滤,计算表达量为0的样本数量,并统计其在总样本数中所占的比例,剔除高于特定比例的特征。
进一步,批次效应校正的步骤包括:将样本的分组信息和批次标记信息保存为向量,对表达量矩阵、分组信息向量、批次标记信息向量进行矩阵建模,估计代表批次效应的参数,将原始数据映射到预期的分布,进而生成新的表达量矩阵。
进一步,建模数据处理还包括离群样本剔除。
进一步,离群样本剔除的步骤包括:进行主成分分析,将剔除批次效应的表达量矩阵作为参数传入,进行数据的基因特征的维度映射,生成降低维度的主成分的数值矩阵,绘制PCA图,删除远离群体的样本。
进一步,所述表达量矩阵为M*N的基因表达量矩阵,表达量矩阵中的第i行第j列的数值表示第j个样本对应第i个基因的表达量count值,其中1≤i≤M,1≤j≤N;M表示检测基因的数量,N表示分析样本的数量。
进一步,表达量矩阵的定量标准类型为基因ID。
进一步,构建表达量矩阵的步骤还包括合并数据集。
进一步,利用基因ID合并数据集。
本发明第二方面提供了一种构建结直肠疾病风险预测模型的方法,包括:
获取样本测序数据;
对基因进行表达量定量,构建表达量矩阵;
基于MLseq工具进行模型训练,选择分类效果最佳的算法进行风险预测模型的训练和构建。
进一步,所述方法还包括对测序数据进行处理和质控。
进一步,所述方法还包括将处理后的数据比对至人类参考基因组上。
进一步,所述方法还包括将临床信息按照样本分组信息进行特征标记。
进一步,所述方法还包括在模型训练前将样本随机分为训练集和测试集。
进一步,所述方法还包括对风险预测模型进行验证,得到预测分类结果。
进一步,所述模型训练前还包括建模数据处理。
进一步,所述测序数据来源于TCGA数据库和SRA数据库。
进一步,所述模型训练前还包括建模数据处理。
进一步,建模数据处理包括特征剔除、批次效应校正。
进一步,特征剔除的步骤包括:使用featurefilter函数将所有基因单独进行过滤,计算表达量为0的样本数量,并统计其在总样本数中所占的比例,剔除高于特定比例的特征。
进一步,所述特定比例为0.05。
进一步,批次效应校正的步骤包括:将样本的分组信息和批次标记信息保存为向量,对表达量矩阵、分组信息向量、批次标记信息向量进行矩阵建模,估计代表批次效应的参数,将原始数据映射到预期的分布,进而生成新的表达量矩阵。
进一步,批次校正的函数为ComBat-Seq函数。
进一步,建模数据处理还包括离群样本剔除。
进一步,离群样本剔除的步骤包括:进行主成分分析,将剔除批次效应的表达量矩阵作为参数传入,进行数据的基因特征的维度映射,生成降低维度的主成分的数值矩阵,绘制PCA图,删除远离群体的样本。
进一步,所述表达量矩阵为M*N的基因表达量矩阵,表达量矩阵中的第i行第j列的数值表示第j个样本对应第i个基因的表达量count值,其中1≤i≤M,1≤j≤N;M表示检测基因的数量,N表示分析样本的数量。
进一步,表达量矩阵的定量标准类型为基因ID。
进一步,表达量矩阵的定量模式为intersection-nonempty。
进一步,构建表达量矩阵还包括合并数据集。
进一步,利用基因ID合并数据集。
进一步,临床信息处理的步骤包括:
stage特征中I、IA、IB标记为TNM1;II、IIA、IIB、IIC标记为TNM2;III、IIIA、IIIB、IIIC标记为TNM3;IV、IVA、IVB标记为TNM4;
stage字段重命名为subclass;
肠道息肉样本的subtype字段重命名为subclass,并标记为polyps;
无患病正常样本的subtype字段重命名为subclass,并标记为normal;
添加字段source,标记样本来源TCGA或者是SRA。
进一步,所述模型包括10个子模型,模型训练的算法为voomNSC算法和svm算法。
进一步,使用voomNSC算法和svm算法进行模型训练包括:构建用于子模型训练的表达量矩阵和设计矩阵。
进一步,设计矩阵的样本ID和临床表型一一对应。
进一步,模型训练还包括将表达量矩阵和设计矩阵封装为DESeqDataSetFromMatrix对象。
进一步,模型训练使用十折交叉进行验证。
进一步,十折交叉的参数设置如下:
-Method:repeatedcv;
-Number:10;
-Repeats:10;
进一步,voomNSC算法进行模型训练还包括构建voomControl控制器。
进一步,voomNSC算法使用classify方法进行模型训练。
进一步,传入参数为DESeqDataSetFromMatrix对象、voomControl控制器;
进一步,参数设置如下:
-Method:voomNSC,
-preProcessing:TMM;
进一步,svm算法进行模型训练还包括构建trainControl控制器。
进一步,svm算法使用classify方法进行模型训练。
进一步,传入参数为DESeqDataSetFromMatrix对象、trainControl控制器。
进一步,参数设置如下:
-Method:svmLinear,
-preProcessing:TMM;
进一步,使用模型对测试集进行验证的步骤包括:构建表达量矩阵和设计矩阵。
进一步,对测试集进行验证的步骤还包括将表达量矩阵和设计矩阵封装为DESeqDataSetFromMatrix对象。
进一步,使用predict法预测分类结果。
进一步,传入参数为DESeqDataSetFromMatrix对象。
进一步,预测分类结果为m*n矩阵,m为子模型的数量,n为待测样本的数量。
进一步,预测分类结果的判定标准为:待测样本的子模型1预测结果为normal且子模型2-5预测结果为normal的数量≥3,该样本判定分类为normal;待测样本的子模型6-9预测结果为polyps的数量≥3,该样本判定分类为polyps;待测样本的子模型10预测结果为stage1,则该样本的判定分类为结直肠癌早期;反之,该样本的判定分类为结直肠癌晚期。
本发明第三方面提供了如下如下任一种风险预测模型:
1)由本发明第一方面所述的方法构建而成的疾病风险预测模型;
2)由本发明第二方面所述的方法构建而成的结直肠疾病风险预测模型。
本发明第四方面提供了如下任一项所述的应用:
1)本发明第一方面所述的方法在制备疾病风险预测系统/产品中的应用;
2)本发明第二方面所述的方法在制备结直肠疾病风险预测系统/产品中的应用;
3)本发明第三方面所述的风险预测模型在制备疾病/结直肠疾病风险预测系统/产品中的应用。
本发明第五方面提供了如下任一诊断系统/产品:
1)一种疾病风险预测系统/产品,其特征在于,所述系统/产品包括风险预测模块,所述风险预测模块嵌入了本发明第一方面所述的方法构建的模型;
2)一种结直肠疾病风险预测系统/产品,其特征在于,所述系统/产品包括风险预测模块,所述风险预测模块嵌入了本发明第二方面所述的方法构建的模型。
进一步,所述的系统/产品还包括数据采集模块,所述数据采集模块用于采集样本的测序数据。
进一步,a)或b)所述的系统/产品还包括数据采集模块,所述数据采集模块用于采集样本的测序数据。
进一步,a)或b)所述的系统/产品还包括数据处理模块,所述数据处理模块用于处理测序数据,获得基因表达量矩阵。
进一步,结直肠疾病风险预测系统/产品风险判定标准为:待测样本的子模型1预测结果为normal且子模型2-5预测结果为normal的数量≥3,该样本判定分类为normal;待测样本的子模型6-9预测结果为polyps的数量≥3,该样本判定分类为polyps;待测样本的子模型10预测结果为stage1,则该样本的判定分类为结直肠癌早期;反之,该样本的判定分类为结直肠癌晚期。
本发明第六方面提供了如下任一种计算机可读存储介质:
1)其存储有程序,该程序用于执行本发明第一方面所述的方法或本发明第二方面所述的方法;
2)其存储有程序,该程序用于执行本发明第三方面所述的风险预测模型。
进一步,2)中所述程序还用于处理数据构建基因表达量矩阵。
进一步,处理数据的步骤包括:
对测序数据进行处理和质控,得到cleandata;
将cleandata比对至人类参考基因组上;
结合注释文件,对比对后数据进行基因的表达量的定量,构建表达量矩阵。
本发明的第七方面提供了如下任一种电子设备:
1)一种电子设备,所述电子设备包括处理器,所述处理器用于执行可执行程序,所述可执行程序包括完成本发明一方面或本发明第二方面所述的方法;
2)一种电子设备,其特征在于,所述电子设备包括处理器,所述处理器用于执行可执行程序,所述可执行程序用于运行本发明第三方面所述的风险预测模型。
进一步,所述电子设备还包括输入装置,与处理器相连,用于接受用户的输入信息;
进一步,所述电子设备还包括存储器,存储器用于存储文件;
进一步,所述电子设备还包括显示装置,根据输入装置和处理器的指令将风险预测结果进行显示。
进一步,2)中所述的处理器还用于构建基因表达量矩阵,使得将基因的特征指标输入模型中,进而得到风险预测结果。
进一步,构建基因表达量矩阵的步骤包括:
对测序数据进行处理和质控,得到cleandata;
将cleandata比对至人类参考基因组上;
结合注释文件,对比对后数据进行基因的表达量的定量,构建表达量矩阵。
本发明的优点和有益效果:
本发明中提供了一种构建疾病风险预测模型的方法,采用该方法构建的疾病风险模型具有较高的诊断敏感性和特异性。
本发明提供了的风险预测模型,嵌入该风险模型的风险预测系统/产品以及计算机可读存储介质和电子设备的预测准确性高,可以有效的实现疾病与正常,以及不同阶段的疾病的区分。
附图说明
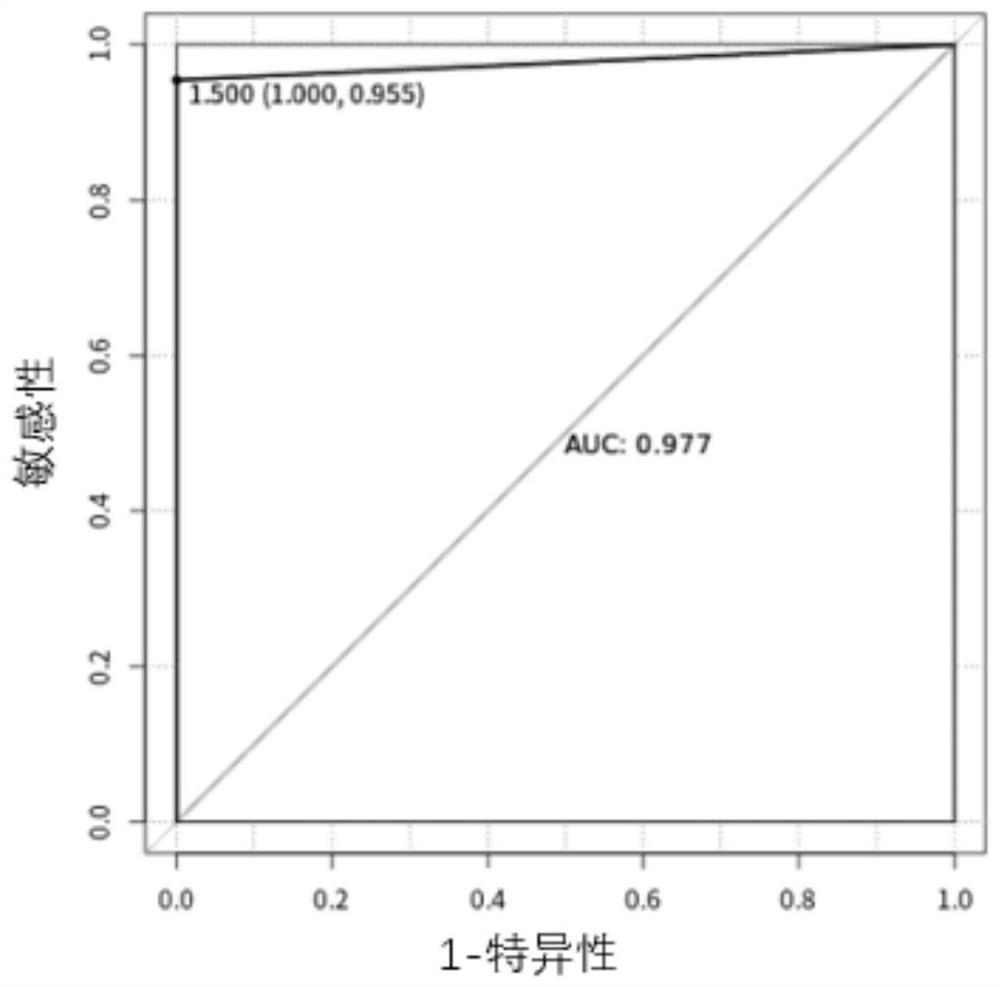
图1是子模型1的预测结果图;
图2是子模型2的预测结果图;
图3是子模型3的预测结果图;
图4是子模型4的预测结果图;
图5是子模型5的预测结果图;
图6是子模型6的预测结果图;
图7是子模型7的预测结果图;
图8是子模型8的预测结果图;
图9是子模型9的预测结果图;
图10是子模型10的预测结果图。
具体的实施方式
本公开在对多种实施方案的详细描述中,出于说明的目的,阐述了许多具体细节以提供对所公开的实施方案的透彻理解。然而,本领域技术人员将理解,可以在具有或没有这些具体细节的情况下实践这些不同的实施方案。此外,本领域技术人员可以容易地理解,方法被提供及进行的具体顺序是说明性的,并且设想顺序可以不同并且仍然保持在本文公开的多种实施方案的范围内。
如本文使用的,术语“测序”是指用于确定生物分子,例如核酸,诸如DNA或RNA的序列的若干种技术中的任一种。示例性测序方法包括但不限于靶向测序、单分子实时测序、外显子测序、基于电子显微术的测序、panel测序、晶体管介导的测序、直接测序、随机鸟枪法测序、Sanger双脱氧终止测序、全基因组测序、杂交测序、焦磷酸测序、毛细管电泳、凝胶电泳、双链体测序、循环测序、单碱基延伸测序、固相测序、高通量测序、大规模并行信号测序(massively parallel signature sequencing)、乳液PCR、低变性温度共扩增PCR(COLD-PCR)、多重PCR、可逆染料终止子测序、配对末端测序、近末端测序(near-termsequencing)、外切核酸酶测序、连接测序、短读段测序、单分子测序、合成测序、实时测序、反向终止子测序、纳米孔测序、454测序、Solexa基因组分析仪测序、SOLiD测序、MS-PET测序及其组合。在一些实施方案中,测序可以通过基因分析仪进行,例如可从Illumina或Applied Biosystems商业上获得的基因分析仪。
术语“下一代测序”NGS是指与传统的基于Sanger和毛细管电泳的方法相比具有增加的通量的测序技术,例如,具有一次产生数十万个相对较小的序列读段的能力。下一代测序技术的一些实例包括但不限于合成测序、连接测序和杂交测序。
本公开内容提供了被编程为实现本公开内容的方法的系统。所述系统被编程或以其他方式配置为分析序列数据、构建基因的表达量矩阵。所述系统可以调控本公开内容的序列分析的各个方面,诸如,例如将数据针对已知序列进行匹配。所述系统可以是用户的电子装置或相对于该电子装置远程定位的计算机系统。电子装置可以是移动电子装置。
所述系统包括处理器,其可以是单核或多核处理器或用于并行处理的多于一个处理器。所述系统还包括存储器(例如,随机存取存储器、只读存储器、闪速存储器)、电子存储单元(例如,硬盘)、用于与一个或更多个其他系统进行通信的通信界面(例如,网络适配器)和外围装置,诸如高速缓冲存储器、其他存储器、数据存储和/或电子显示适配器。存储器、电子储存单元、通信界面和外围装置与处理器通过通信总线(实线),诸如主板通信。存储单元可以是用于存储数据的数据存储单元(或数据储存库)。所述系统可以借助于通信界面被可操作地耦合至计算机网络。网络可以是互联网、内联网和/或外联网、或与互联网通信的内联网和/或外联网。在一些情况下,网络为通信和/或数据网络。网络可以包括一个或更多个计算机服务器,这可以支持分布式计算,诸如云计算。在一些情况下,借助于系统,网络可以实现对等网络,其可以使耦合至系统的装置能够作为客户端或服务器运行。
所述处理器可以执行一系列的机器可读指令,该机器可读指令可以以程序或软件来体现。指令可以被存储于存储器位置,诸如存储器中。指令可以被导向处理器,该指令可以随后编程或以其他方式配置处理器以实现本公开内容的方法。由处理器进行的操作的实例可以包括读取、解码、执行和写回。
处理器可以是电路诸如集成电路的一部分,系统的一个或更多个其他组件可以被包含在电路中,在一些情况下,电路为专用集成电路。
电子存储单元可以存储文件,诸如驱动程序、库和保存的程序。电子存储单元可以存储用户数据,例如,用户偏好和用户程序。在一些情况下,系统可以包括一个或更多个另外的数据存储单元,该数据存储单元在计算机系统的外部,诸如位于通过内联网或互联网而与系统通信的远程服务器上。
系统可以与一个或更多个远程计算机系统通过网络进行通信。例如,系统可以与用户(例如,医师)的远程计算机系统进行通信。远程计算机系统的实例包括个人计算、板型或平板PC、电话、智能电话或个人数字助理。用户可经由网络访问系统。
如本文描述的方法可以通过机器(例如,计算机处理器)可执行代码的方式实现,该机器可执行代码被存储在系统的电子存储位置,例如存储器或电子存储单元上。机器可执行代码或机器可读代码可以以软件的形式提供。在使用期间,代码可以由处理器执行。在一些情况下,代码可以从电子存储单元检索并存储在存储器上,以用于由处理器即时访问。在一些情况下,可以排除电子存储单元,而将机器可执行指令存储于存储器中。
代码可以被预编译并配置为用于与具有适于执行该代码的处理器的机器一起使用,或者可以在运行时间期间被编译。代码可以以编程语言的形式提供,该编程语言可以被选择使得代码能够以预编译的或按编译原样的方式被执行。
本文提供的系统和方法的各方面,诸如系统,可以以编程来体现。技术的多个方面可以被认为是通常呈一种机器可读介质执行或体现的机器(或处理器)可执行代码和/或相关数据的形式的产品。机器可执行代码可以被存储于电子存储单元诸如存储器(例如,只读存储器、随机存取存储器、闪速存储器)或硬盘上。“存储”型介质可以包括计算机、处理器等的任何或所有有形存储器,或其相关模块,诸如多种半导体存储器、磁带驱动器、磁盘驱动器等,其可以在任何时间为软件编程提供非暂时性存储。软件的所有或部分有时可以通过互联网或多种其他通信网络进行通信。例如,此类通信可以使得将软件从一个计算机或处理器加载到另一个计算机或处理器中,例如,从管理服务器或主机加载到应用服务器的计算机平台中。因此,能够携带软件元件的另一类型的介质包括诸如在本地装置之间的物理界面、通过有线和光纤陆线网络以及在多种空中链路上使用的光波、电波和电磁波。携带此类波的物理元件,诸如有线或无线链路、光链路等,也可被认为是携带软件的介质。如本文使用的,除非被限制为非暂时性的、有形的“存储”介质,否则术语诸如计算机或机器“可读介质”是指参与将指令提供至处理器用于执行的任何介质。
因此,机器可读介质,诸如计算机可执行代码,可以采取多种形式,包括但不限于有形存储介质、载波介质或物理传输介质。非易失性存储介质包括,例如光盘或磁盘,诸如在任何计算机等中的任何存储设备,易失性存储介质包括动态存储器,诸如此类计算机平台的主存储器。有形的传输介质包括同轴电缆;铜线和光纤,包括构成计算机系统内的总线的导线。载波传输介质可以采取电信号或电磁信号或者声波或光波的形式,诸如在射频和红外数据通信期间生成的那些。因此,计算机可读介质的常见形式包括例如:软盘、软性磁盘、硬盘、磁带、任何其他磁介质、CD-ROM、DVD或DVD-ROM、任何其他光学介质、穿孔卡片纸带、具有孔模式的任何其他物理存储介质、RAM、ROM、PROM和EPROM、FLASH-EPROM、任何其他存储器芯片或盒、传输数据或指令的载波、传输此类载波的缆线或链路,或者计算机可以从其读取编程代码和/或数据的任何其他介质。这些计算机可读介质的形式中的许多形式可以参与向处理器传送一个或更多个指令的一个或更多个序列以用于执行。
所述系统可以包括电子显示器或与之通信,该电子显示器包括用户界面(UI),用于提供例如关于分析结果的信息。UI的实例包括但不限于图形用户界面(GUI)和基于网络的用户界面。
在一个方面,本文提供了包括计算机的系统,计算机包括处理器和计算机存储器,其中计算机与通信网络通信,并且其中计算机存储器包括代码,当代码由处理器执行时,通过通信网络将序列数据接收到计算机存储器中;使用本文所述的方法,判定序列数据是否与疾病相关以及与疾病的何种阶段相关,以及通过通信网络报告出该判定结果。
通信网络可以是连接到互联网的任何可用网络。通信网络可以利用例如高速传输网络,包括但不限于电力线宽带、电缆调制解调器、数字用户线路、光纤、卫星和无线电。
在一个方面,本文提供了一种系统,该系统包括:局域网;一个或更多个DNA测序仪,包括被配置为存储DNA序列数据的连接到局域网的计算机存储器;生物信息学计算机,包括计算机存储器和处理器,该计算机连接到局域网;其中所述计算机还包括代码,当所述代码被执行时,复制存储在DNA测序仪上的DNA序列数据、将复制的数据写入生物信息学计算机中的存储器、并进行如本文描述的步骤。
本公开内容的方法和系统可以通过一个或更多个算法来实现。算法可以在由处理器执行后通过软件来实现。
在本发明中,程序和代码可以互换使用。
术语“样本”可以是从受试者分离的任何生物样本。例如,样本可以包括但不限于体液、全血、血小板、血清、血浆、粪便、红细胞、白细胞或白血球、内皮细胞、组织活组织检查、滑液、淋巴液、腹水、间质或细胞外液、细胞间空间的液体,包括龈沟液、骨髓、脑脊液、唾液、粘液、痰、精液、汗液、尿液、鼻刷液、巴氏涂片液或任何其他体液。体液可以包括唾液、血液或血清。例如,多核苷酸可以是从体液例如血液或血清分离的无细胞DNA。样本也可以是肿瘤样本,肿瘤样本可以通过各种方法从受试者获得,所述方法包括但不限于静脉穿刺、排泄、射精、按摩、活组织检查、针抽吸、灌洗、刮擦、手术切口或介入或其他方法。样本可以是无细胞样本(例如,不包含任何细胞)。
在本发明中,所述疾病包括但不限于自身免疫疾病或免疫相关疾病、肿瘤、障碍疾病。自身免疫疾病或免疫相关疾病类型包括但不限于炎症、抗磷脂综合征、系统性红斑狼疮、类风湿性关节炎、自身免疫性血管炎、乳糜泻、自身免疫性甲状腺炎、输血后免疫、母体胎儿不相容(maternal-fetal incompatibility)、输血反应、免疫缺陷诸如IgA缺陷、常见变异型免疫缺陷、药物诱导性狼疮、糖尿病、I型糖尿病、II型糖尿病、幼年型糖尿病、幼年型类风湿性关节炎、银屑病性关节炎、多发性硬化、免疫缺陷、过敏、哮喘、银屑病、特应性皮炎、过敏性接触性皮炎、慢性皮肤病、肌萎缩侧索硬化、化疗所致损伤、移植物抗宿主病(graft-vs-host diseases)、骨髓移植排斥、强直性脊柱炎、特应性湿疹、天疱疮、白塞病、慢性疲劳综合症纤维肌痛、化疗所致损伤、重症肌无力、肾小球肾炎、过敏性视网膜炎、系统性硬化、亚急性皮肤型红斑狼疮、包括冻疮样红斑狼疮的皮肤型红斑狼疮、干燥综合征、自身免疫性肾炎、自身免疫性血管炎、自身免疫性肝炎、自身免疫性心脏炎、自身免疫性脑炎、自身免疫介导的血液病、lc-SSc(局限性皮肤型硬皮病)、dc-SSc(弥漫性皮肤型硬皮病)、自身免疫性甲状腺炎(AT)、格雷夫斯病(GD)、重症肌无力、多发性硬化(MS)、强直性脊柱炎、移植排斥、免疫衰老、风湿性/自身免疫性疾病、混合型结缔组织病、脊柱关节病、银屑病、银屑病性关节炎、肌炎、硬皮病、皮肌炎、自身免疫性血管炎、混合型结缔组织病、特发性血小板减少性紫癜、克罗恩病、人类佐剂病、骨性关节炎、幼年型慢性关节炎、脊柱关节病、特发性炎性肌病、系统性血管炎、结节病、自身免疫性溶血性贫血、自身免疫性血小板减少症、甲状腺炎、免疫介导的肾病、中枢或外周神经系统脱髓鞘病、特发性脱髓鞘多神经病、格林-巴利综合征、慢性炎性脱髓鞘多神经病、肝胆疾病、传染性或自身免疫性慢性活动性肝炎、原发性胆汁性肝硬化、肉芽肿性肝炎、硬化性胆管炎、炎性肠病、谷蛋白敏感性肠病、惠普尔病、自身免疫性或免疫介导的皮肤病、大疱性皮肤病、多形性红斑、过敏性鼻炎、特应性皮炎、食物过敏、荨麻疹、肺部免疫性疾病、嗜酸性肺炎、特发性肺纤维化、过敏性肺炎、移植相关疾病、移植物排斥或移植物抗宿主病、银屑病性关节炎、银屑病、皮炎、多肌炎/皮肌炎、中毒性表皮坏死松解症、系统性硬皮病和硬化、与炎性肠病相关的应答、克罗恩病、溃疡性结肠炎、呼吸窘迫综合征、成人型呼吸窘迫综合征(ARDS)、脑膜炎、脑炎、葡萄膜炎、结肠炎、肾小球肾炎、过敏性状况、湿疹、哮喘、涉及T细胞浸润和慢性炎性应答的状况、动脉粥样硬化、自身免疫性心肌炎、白细胞黏附缺陷症、过敏性脑脊髓炎、与由细胞因子和T淋巴细胞介导的急性和迟发性过敏相关的免疫应答、结核病、结节病、包括韦格纳肉芽肿病的肉芽肿病、粒细胞缺乏症、血管炎(包括ANCA)、再生障碍性贫血、Diamond Blackfan贫血、包括自身免疫性溶血性贫血(AIHA)的免疫性溶血性贫血、恶性贫血、纯红细胞再生障碍(PRCA)、因子VIII缺乏症、血友病A、自身免疫性中性粒细胞减少症、全血细胞减少症、白细胞减少症、涉及白细胞渗出的疾病、中枢神经系统(CNS)炎性紊乱、多器官损伤综合征、重症肌无力、抗原-抗体复合物介导的疾病、抗肾小球基膜病、抗磷脂抗体综合征、过敏性神经炎、白塞病、Castleman综合征、Goodpasture综合征、兰伯特-伊顿肌无力综合征、雷诺综合征、干燥综合征、斯-约综合征、大疱性类天疱疮、天疱疮、自身免疫性多内分泌腺疾病、Reiter病、僵人综合征、巨细胞性动脉炎、免疫复合物性肾炎、IgA肾病、IgM多神经病或IgM介导的神经病、特发性血小板减少性紫癜(ITP)、血栓性血小板减少性紫癜(TTP)、自身免疫性血小板减少症、包括自身免疫性睾丸炎和卵巢炎的睾丸和卵巢自身免疫性疾病、原发性甲状腺功能减退症、包括自身免疫性甲状腺炎的自身免疫性内分泌疾病、慢性甲状腺炎(桥本甲状腺炎)、亚急性甲状腺炎、特发性甲状腺功能减退症、爱迪生氏病、格雷夫斯病、自身免疫性多内分泌腺综合征(或多腺性内分泌病综合征)、席汉氏综合征、自身免疫性肝炎、淋巴细胞性间质性肺炎(HIV)、闭塞性细支气管炎(非移植)对NSIP、格林-巴利综合征、大血管血管炎(包括风湿性多肌痛和巨细胞性(高安)动脉炎)、中血管血管炎(包括川崎病和结节性多动脉炎)、强直性脊柱炎、Berger病(IgA肾病)、快速进行肾小球肾炎、原发性胆汁性肝硬化、口炎性腹泻(Celiac sprue)(谷蛋白肠病)、冷球蛋白血症、和肌萎缩侧索硬化(ALS)。
肿瘤类型包括但不限于急性成淋巴细胞性白血病(ALL)、急性髓性白血病、肾上腺皮质癌、成人急性髓性白血病、成人原发部位不明癌、成人恶性间皮瘤、艾滋病相关癌症、艾滋病相关淋巴瘤、肛门癌、阑尾癌、星形细胞瘤、儿童小脑或大脑癌、基底细胞癌、胆管癌、膀胱癌、骨肿瘤、骨肉瘤/恶性纤维组织细胞瘤、脑癌、脑干胶质瘤、乳腺癌、支气管腺瘤/类癌、伯基特淋巴瘤、类癌瘤、原发性不明的癌、中枢神经系统淋巴瘤、小脑星形细胞瘤、大脑星形细胞瘤/恶性神经胶质瘤、宫颈癌、儿童急性髓性白血病、儿童原发部位不明的癌症、儿童癌症、儿童大脑星形细胞瘤、儿童间皮瘤、软骨肉瘤、慢性淋巴细胞白血病、慢性髓细胞性白血病、慢性骨髓增生性紊乱、结肠癌、皮肤T细胞淋巴瘤、促结缔组织增生性小圆细胞肿瘤、子宫内膜癌、子宫内膜子宫癌、室管膜瘤、上皮样血管内皮瘤(EHE)、食管癌、尤因肿瘤肉瘤家族、尤因肿瘤家族中的尤因肉瘤、颅外生殖细胞肿瘤、性腺外生殖细胞肿瘤、肝外胆管癌、眼癌、眼内黑素瘤、胆囊癌、胃(gastric)(胃(stomach))癌、胃类癌、胃肠道类癌肿瘤、胃肠道间质瘤(GIST)、妊娠性滋养层细胞瘤、脑干胶质瘤、胶质瘤、毛细胞白血病、头颈癌、心脏癌、肝细胞(肝)癌、霍奇金淋巴瘤、下咽癌、下丘脑和视觉途径胶质瘤、胰岛细胞癌(内分泌胰腺)、卡波西肉瘤、肾癌(肾细胞癌)、喉癌、急性成淋巴细胞性白血病(也称为急性淋巴细胞白血病)、急性髓性白血病(也称为急性髓细胞性白血病)、慢性淋巴细胞性白血病(也称为慢性淋巴细胞白血病)、白血病(leukaemia)、慢性髓细胞性白血病(也称为慢性髓性白血病)、毛细胞白血病(leukemia)、唇及口腔癌、脂肪肉瘤、肝癌(原发性)、非小细胞肺癌、小细胞肺癌、淋巴瘤(艾滋病相关)、淋巴瘤、巨球蛋白血症、男性乳腺癌、骨恶性纤维组织细胞瘤/骨肉瘤、髓母细胞瘤、黑素瘤、梅克尔细胞癌、原发灶隐匿转移性颈部鳞状癌、口癌、多发性内分泌肿瘤综合征、儿童多发性骨髓瘤(骨髓癌)、多发性骨髓瘤/浆细胞赘生物、蕈样肉芽肿、骨髓增生异常综合征、骨髓增生异常/骨髓增生性疾病、慢性髓细胞性白血病、粘液瘤、鼻腔和副鼻窦癌、鼻咽癌、神经母细胞瘤、非霍奇金淋巴瘤、非小细胞肺癌、少突神经胶质瘤、口腔癌、口咽癌、骨肉瘤/骨恶性纤维组织细胞瘤、卵巢癌、卵巢上皮癌(表面上皮间质肿瘤)、卵巢生殖细胞瘤、卵巢低恶性潜能肿瘤、胰腺癌、胰岛细胞癌、副鼻窦和鼻腔癌、甲状旁腺癌、阴茎癌、咽癌、嗜铬细胞瘤、松果体星形细胞瘤、松果体生殖细胞瘤、松果体母细胞瘤和幕上原始神经外胚层肿瘤、垂体腺瘤、浆细胞赘生物/多发性骨髓瘤、胸膜肺母细胞瘤、原发性中枢神经系统淋巴瘤、前列腺癌、直肠癌、肾细胞癌(肾癌)、肾盂和输尿管移行细胞癌、视网膜母细胞瘤、横纹肌肉瘤、唾液腺癌、Sézary综合征、皮肤癌(黑素瘤)、皮肤癌(非黑素瘤)、梅克尔细胞皮肤癌、小细胞肺癌、小肠癌、软组织肉瘤、鳞状细胞癌、原发灶隐匿转移性颈部鳞状癌、胃癌、幕上原始神经外胚层肿瘤、皮肤T细胞淋巴瘤、睾丸癌、喉癌、胸腺瘤和胸腺癌、胸腺瘤、甲状腺癌、肾盂和输尿管移行细胞癌、输尿管和肾盂移行细胞癌、尿道癌、子宫肉瘤、阴道癌、视觉途径和下丘脑神经胶质瘤、儿童视觉途径和下丘脑神经胶质瘤、外阴癌、巨球蛋白血症和肾母细胞瘤(肾癌)。
障碍疾病的类型包括但不限于神经性障碍(例如偏头痛;癫痫症;阿尔茨海默病;帕金森病;脑损伤;中风;脑血管疾病(包括脑动脉硬化症、脑淀粉样蛋白血管病、遗传性脑出血和脑缺氧缺血);脊髓性肌萎缩;侧索硬化;多发性硬化;认知障碍(包括健忘症、老年性痴呆、HIV相关性痴呆、阿尔茨海默病相关性痴呆、亨廷顿病相关性痴呆、Lewy体痴呆、血管性痴呆、药物相关性痴呆、谵妄和轻度认知缺损);与帕金森病和抑郁症相关的认知功能障碍;心理缺陷(包括唐氏综合征和脆性X染色体综合征);精神病(包括精神分裂症(例如连续性或发作性、偏执型、青春型、紧张型、分化不良型和残留型精神分裂性障碍)、情感性分裂症、精神分裂症样精神病和妄想症。
下面结合具体的实施例和附图进一步说明本发明,本发明的实施例仅用于解释本发明,并不意味着限制本发明的保护范围。
下述实施例中所使用的实验方法如无特殊说明,均为常规方法。
实施例1疾病预测模型的构建
1、获取数据
采集样本的测序数据。
2、测序数据的处理
使用fastp软件进行接头处理和质控,得到cleandata。
3、序列比对
使用ICGC软件将cleandata比对至人类参考基因组(版本GRCh38.d1.vd1)得到bam文件。
4、构建表达量矩阵
使用htseq软件,结合注释文件,对比对后bam文件进行基因的表达量的定量,根据基因的ID,多样本的表达量合并构建M*N的基因表达量矩阵,基因表达量矩阵中的第i行第j列的数值表示第j个样本对应第i个基因的表达量count值,其中1≤i≤M,1≤j≤N;M表示检测基因的数量,N表示分析样本的数量。将表达量矩阵保存为.Rdata对象文件。
5、临床信息处理
将临床信息按照样本分组信息进行特征标记,如将无患病正常样本命名为subclass并标记为normal,将疾病样本标记为disease等。
添加字段source,标记样本来源。将count表达量矩阵和临床信息保存为.Rdata对象文件。
6、数据分组
将.Rdata对象文件对样本集合进行拆分,分为训练集和测试集。针对临床信息
subclass字段的每一分类,分别随机取部分样本作为测试集,剩余样本作为训练集,将拆分好的count训练集、count测试集、训练集临床信息、测试集临床信息保存为.Rdata对象文件。
7、建模数据处理
1)特征剔除
使用featurefilter函数将所有基因单独进行过滤,计算表达量为0的样本数量,并统计其在总样本数中所占的比例,剔除特定比例(如>0.05)的特征,将剩余的特征保存为.Rdata对象文件。
2)批次效应校正
将待处理数据的分组信息和批次标记信息保存为向量,将待处理数据的表达量矩阵、分组信息向量、批次标记信息向量输入ComBat-Seq函数中进行运算,使用负二项式回归模型对count值进行建模,估计代表批次效应的参数,将原始count值映射到预期的分布,生成新的表达量矩阵,保存为.Rdata文件待用。
3)离群样本剔除
使用prcomp函数进行主成分分析,将剔除批次效应的表达量矩阵作为参数传入,进行数据的基因特征的维度映射,生成降低维度的主成分的数值矩阵;
选择主成分PC1和PC2的数值,并结合样本的分组信息,构建绘制pca plot所需要的长格式的数据集合;
计算主成分PC1和PC2的方差/所有主成分的方差,作为PC1和PC2的对于变异的解释度;
选择主成分PC1和PC2使用ggplot2绘制样本分布图,根据样本的分组信息标记颜色和形状,样本点标记样本ID;
选择远离群体的点作为待删除样本;
在表达量矩阵中删除以上被挑选的样本,重新保存为.Rdata文件待用。
8、模型训练
基于MLseq工具进行模型的训练,模型保存为fit.Rdata对象文件。模型训练算法如表1所示。模型的设计思路是将不同分组的样本进行细分,预测最终的目标是预测受试者是否患有疾病以及预测疾病的进展,选择分类效果最好的算法进行模型的训练和构建。
表1模型训练算法
9、模型验证
读取数据分组步骤生成的.Rdata数据文件,使用测试集count表达量矩阵和测试集临床信息的数据,封装为DESeqDataSetFromMatrix对象。使用predict方法,需要传参DESeqDataSetFromMatrix对象。测试集数据表达量进行转化,转换到和训练集相同的scale,进行后验概率的计算,得到预测分类结果。
实施例2结直肠疾病诊断模型的构建
1、数据来源与获取
构建结直肠癌发病风险模型的所有数据下载自TCGA和NCBI-SRA数据库,其中结直肠癌的癌症和癌旁的表达量文件下载自TCGA数据库,肠道息肉raw数据下载自NCBI-SRA数据库。检索获得共选取443个结直肠癌病例样本,31个肠道息肉的样本,72个正常样本,共计546个样本数据用于进一步筛选和质控。
2、Raw data的处理
使用fastp软件进行接头处理和质控,得到cleandata,步骤包括:
a.接头处理
利用fastp软件双端序列自动检测模式进行接头处理;
b.数据修剪和质控
最低N碱基数量阈值为5,reads最低长度阈值为15,碱基质量阈值Q15,低质量碱基百分比阈值为40%,以4个碱基为单位滑动窗口过滤,窗口平均质量阈值Q20。
3、序列比对
分析得到的clean data使用ICGC软件(https://github.com/akahles/icgc_rnaseq_align)比对到人类参考基因组,参考基因组版本为GRCh38.d1.vd1,基因组注释文件版本为gencode.v22.annotation.gtf,比对后得到bam格式数据文件。运行参数设置:
outFilterMultimapScoreRange:1
outFilterMultimapNmax:20
outFilterMismatchNmax:10
alignIntronMax:500000
alignMatesGapMax:1000000
sjdbScore:2
limitBAMsortRAM:0
alignSJDBoverhangMin:1
genomeLoad:NoSharedMemory
outFilterMatchNminOverLread:0.33
outFilterScoreMinOverLread:0.33
twopass1readsN:-1
sjdbOverhang:100
outSAMstrandField:intronMotif
outSAMunmapped:Within
比对得到的bam文件使用samtools的sort指令进行排序:
排序标准:name。
4、构建表达量矩阵
使用htseq软件,结合注释文件,对bam文件进行基因的表达量的定量。
定量模式:intersection-nonempty;
定量标准类型:gene_id;
链特异性:非特异性。
SRA数据库来源的所有样本的Count值表达量文件按照基因ID进行合并,构建M*N的基因表达量矩阵,基因表达量矩阵中的第i行第j列的数值表示第j个样本对应第i个基因的表达量count值,其中1≤i≤M,1≤j≤N;M表示检测基因的数量,N表示分析样本的数量。将表达量矩阵保存为.Rdata对象文件。
不同数据来源的表达量矩阵的构建:
TCGA数据库来源count值表达量矩阵和SRA数据库来源样本的count值表达量矩阵(Rdata)按照基因ID进行合并。
5、临床信息处理
将临床信息进行特征标记。
stage特征中I、IA、IB标记为TNM1;II、IIA、IIB、IIC标记为TNM2;III、IIIA、IIIB、IIIC标记为TNM3;IV、IVA、IVB标记为TNM4;
stage字段重命名为subclass;
肠道息肉样本的subtype字段重命名为subclass,并标记为polyps;
无患病正常样本的subtype字段重命名为subclass,并标记为normal;
添加字段source,标记样本来源TCGA或者是SRA。将count表达量矩阵和临床信息保存为.Rdata对象文件。
6、数据分组
将.Rdata对象文件对样本集合进行拆分,分为训练集和测试集。针对临床信息subclass字段的每一分类,分别随机取样30%的样本作为测试集,剩余70%作为训练集,将拆分好的count训练集、count测试集、训练集临床信息、测试集临床信息保存为.Rdata对象文件。
7、建模数据处理
1)特征剔除
使用featurefilter函数将所有基因单独进行过滤,计算表达量为0的样本数量,并统计其在总样本数中所占的比例,剔除比例>0.05的特征,将剩余的特征保存为.Rdata对象文件。
2)批次效应校正
将样本分组信息和批次标记信息保存为向量,样本分组参照临床信息的source字段,批次标记信息参照临床信息的subclass字段;将待处理数据的表达量矩阵、分组信息向量、批次标记信息向量输入ComBat-Seq函数中进行运算,使用负二项式回归模型对count值进行建模,估计代表批次效应的参数,将原始count值映射到预期的分布,生成新的表达量矩阵,保存为.Rdata文件待用。
3)离群样本剔除
使用prcomp函数进行主成分分析,将剔除批次效应的表达量矩阵作为参数传入,进行数据的基因特征的维度映射,生成降低维度的主成分的数值矩阵;
选择主成分PC1和PC2的数值,并结合样本的分组信息,构建绘制pca plot所需要的长格式的数据集合;
计算主成分PC1和PC2的方差/所有主成分的方差,作为PC1和PC2的对于变异的解释度;
选择主成分PC1和PC2使用ggplot2绘制样本分布图,根据样本的分组信息标记颜色和形状,样本点标记样本ID;
选择远离群体的点作为待删除样本;
在表达量矩阵中删除以上被挑选的样本,重新保存为.Rdata文件待用。
8、模型训练
基于MLseq工具的进行模型的训练,模型保存为fit.Rdata对象文件。模型训练算法如表1所示。模型的设计思路是将结直肠癌、肠道息肉、正常样本进一步细分,模型拆分为10个子模型,预测的最终目标是区分结直肠癌早期、晚期、肠道息肉和正常样本。
子模型列举如下:息肉组织vs正常组织(子模型1),TNM1vs正常组织(子模型2),TNM2vs正常组织(子模型3),TNM3vs正常组织(子模型4),TNM4vs正常组织(子模型5),TNM1vs息肉组织(子模型6),TNM2vs息肉组织(子模型7),TNM3vs息肉组织(子模型8),TNM4vs息肉组织(子模型9),TNM1+2vsTNM4(子模型10),选择分类效果最好的算法进行模型的训练和构建。
经过严格评估和筛选,选择voomNSC算法和SVM算法分类模型。
voomNSC算法子模型的训练:
读取建模数据处理步骤生成的.Rdata数据文件,使用训练集count表达量矩阵和训练集临床信息的数据。从count矩阵中选取对应分类样本,构建用于子模型训练的count表达量矩阵。从训练集临床信息的数据中选取对应分类样本,构建用于子模型训练的设计矩阵。设计矩阵需要样本ID和临床表型一一对应,临床表型字段设置为condition,因子型。
将上述处理好的表达量矩阵和设计矩阵,封装为DESeqDataSetFromMatrix对象。
构建模型训练的控制器,使用voomControl方法进行封装。模型训练使用十折交叉验证,参数设置如下:
Method:repeatedcv;
Number:10;
Repeats:10。
模型训练使用classify方法,需要传入参数DESeqDataSetFromMatrix对象、voomControl控制器。参数设置如下:
Method:voomNSC,
preProcessing:TMM。
对count表达矩阵进行TMM标准化处理,计算表达量的log-cpm值,估计基因的均值方差关系,为每个基因生成精确性权重,继而使用log-cpm值和精确性权重计算加权差异分值。使用soft-thresholding方法收缩加权差异分值,收缩阈值为0,选择未收缩至阈值的基因作为模型训练的特征。预处理后的训练集表达量数据随机拆分为10份,选择其一作为验证集,进行NSC模型训练和验证,反复十次择优。最优模型保存为fit.Rdata对象文件。
svm算法子模型的训练:
读取建模数据处理步骤生成的Rdata数据文件,使用训练集count表达量矩阵和训练集临床信息的数据。从count表达量矩阵中选取对应分类样本,构建用于子模型训练的count表达量矩阵。从训练集临床信息的数据中选取对应分类样本,构建用于子模型训练的设计矩阵。设计矩阵需要样本ID和临床表型一一对应,临床表型字段设置为condition,因子型。
将上述处理好的表达量矩阵和设计矩阵,封装为DESeqDataSetFromMatrix对象。
构建模型训练的控制器,使用trainControl方法进行封装。模型训练使用十折交叉验证,参数设置如下:
-Method:repeatedcv;
-Number:10;
-Repeats:10。
模型训练使用classify方法,需要传入参数DESeqDataSetFromMatrix对象、trainControl控制器,参数设置如下:
-Method:svmLinear;
-preProcessing:TMM。
对count表达矩阵进行TMM标准化处理。预处理后的训练集表达量数据随机拆分为10份,选择其一作为验证集,进行SVM模型训练和验证,反复十次择优。最优模型保存为fit.Rdata对象文件。
9、模型验证
测试集数据需要经10个子模型分别处理。
读取数据分组步骤生成的.Rdata数据文件,使用测试集count表达量矩阵和测试集临床信息的数据构建的设计矩阵,封装为DESeqDataSetFromMatrix对象。使用predict方法,需要传入参数DESeqDataSetFromMatrix对象。测试集数据表达量进行转化,转换到和训练集相同的scale,进行后验概率的计算,得到预测分类结果。对模型的预测结果进行ROC分析,计算得到AUC值和ROC曲线。
将10个子模型的预测分类结果汇总成m*n矩阵,m为子模型的数量,n为待测样本的数量。对每个样本进行单独的预测分类结果判定,判定标准如下,需按照此顺序:待测样本的子模型1预测结果为normal且子模型2-5预测结果为normal的数量大于等于3,该样本判定分类为normal;待测样本的子模型6-9预测结果为polyps的数量大于等于3,该样本判定分类为polyps;待测样本的子模型10预测结果为stage1,则该样本的判定分类为结直肠癌早期;反之,该样本的判定分类为结直肠癌晚期。
将测试样本的最终预测判定结果与实际临床表型进行对比,预测正确的样本数量与测试集样本总数的比例,作为模型整体的准确度。
10、结果
子模型使用voomNSC和svmLiner两种模型,voomNSC用于检测正常组织、息肉组织和癌组织,svmLinear用于区分结直肠癌早晚期,各子模型纳入的基因部分情况和预测结果分别如表2和图1-10所示。
表2子模型纳入基因
上述实施例的说明只是用来理解本发明的技术方案。应当指出,对于本领域的普通技术人员来说,在不脱离本发明原理的前提下,可以对本发明进行若干改进和修饰,这些改进和修饰也将落入本发明权利要求的保护范围内。
- 一种基于测序和机器学习的构建疾病风险预测模型的方法和系统
- 一种基于机器学习方法的肝脏纤维化预测模型的构建方法、预测系统、设备和存储介质