一种单氟甲基依托昔布及其制备方法
文献发布时间:2023-06-19 19:00:17
技术领域
本发明属于化学合成技术领域,具体涉及一种新的单氟甲基依托昔布及其制备方法。
背景技术
在现代医学的发展过程中,人们在动物和人身体中发现一类不饱和脂肪酸组成的活性物质,并将其命名为前列腺素。前列腺素可以与特异的受体结合后在介导细胞增殖,分化等一系列细胞活动以及调节生殖功能。此外,前列腺素也参与炎症、癌症、多种心血管疾病的病理过程。但是,前列腺素也会引起疼痛,发烧等和炎症反应相关的症状。后来,科学家们发现人体内存在的环氧化酶是一种催化花生四烯酸转化为前列腺素的关键酶。这种酶存在两种形态,COX-1为结构型,主要存在于胃肠道等组织内,保护胃肠黏膜。而COX-2为诱导型,在炎症条件下进行表达。而传统的非甾体类药物往往是同时抑制两种酶,从而在抑制炎症时产生胃肠功能紊乱的不良反应。因此,选择性抑制COX-2的抗炎药物是市场主流。默沙东公司研制生产的依托昔布正是一种可以选择性抑制COX-2酶的抗炎药物,主要的药理作用就是体内制造前列腺素的COX-2酶,却不会抑制COX-1酶的表达,从而对胃肠功能的副作用大大降低。依托昔布的苯环上的甲磺酰基结构对COX-2受体有高选择性,对COX-1没有抑制作用。因此,针对甲磺酰基结构的修饰较少,由于氟具有与氢的空间相似性和极强的电负性,氟被广泛用于调节药物分子的生物学特性,如酸度、碱度、蛋白质结合亲和力和亲脂性。氟的引入可以提高有机分子的代谢稳定性,因为破坏碳-氟键形成碳-氧键的能量成本是不利的。由于C-F键的偶极矩较大,氟取代还可以通过各种立体电子相互作用引起构象的显著变化,从而改变有机分子的生物活性。因此,在保留依托昔布结构的基础上引入氟原子会使依托昔布的药物特性有较大的变化。
目前,单氟甲基依托昔布未见文献报道。
发明内容
本发明的目的是提供一种新结构的单氟甲基依托昔布,结构中具有两个吡啶环和一个苯环构成三环结构,在中心吡啶环的间位连有氯原子,另一个吡啶环的邻位连有甲基基团,且中心吡啶环上连有单氟甲基苯磺酰基作为主要活性中心。
本发明第二个目的是提供一种所述单氟甲基依托昔布的制备方法,该方法成本低廉、步骤简洁、操作简单。
为了实现上述目的,本发明采用的技术方案如下:
本发明的第一方面提供了一种单氟甲基依托昔布,结构如下所示:
所述单氟甲基依托昔布的分子式为C
本发明的第二方面提供了一种所述单氟甲基依托昔布的制备方法,包括以下步骤:
第一步,三氟乙酰基化:
在氮气保护下,将氢化钠溶解于四氢呋喃中,并加入三氟乙酸乙酯,加入依托昔布的四氢呋喃溶液,依托昔布、氢化钠与三氟乙酸乙酯的摩尔比为1:(5~6):(2~3)(优选为1:5:2),回流反应,TLC监控原料点消失,得到粗产品中间体A;
第二步,氟化反应
在氮气保护下,将粗产品中间体A、三水合硝酸铜溶解于乙腈中,TLC监控原料点消失,将选择性氟试剂溶解于乙腈和水的混合溶剂中加入上述反应液中,乙腈与水的体积比为3:2,中间体A、三水合硝酸铜与选择性氟试剂的摩尔比为1:(0.1~0.3):(1.1~1.3),TLC监控中间体A消失后结束反应,得到中间体B;
第三步,脱三氟甲基乙二醇基团:
在氮气保护下,将摩尔比为(0.4~0.8):1的中间体B、氢氧化钾溶解于四氢呋喃中,保持室温反应,TLC监控原料点消失,得到单氟甲基依托昔布。
所述第二步中,中间体A、三水合硝酸铜与选择性氟试剂的摩尔比为1:0.2:1.2。
所述第三步中,中间体B、氢氧化钾的摩尔比为1:2。
本发明的第三方面提供了一种所述单氟甲基依托昔布在制备抗炎镇痛药物中的应用。
由于采用上述技术方案,本发明具有以下优点和有益效果:
本发明的单氟甲基依托昔布的活性基团与依托昔布类似,且有氟原子改变了分子的空间性、电负性、生物学特性等,在一定程度上保持了其对环氧化酶COX-2的抑制性和选择性,可作为潜在的抗炎镇痛药物,具有广泛的应用价值和市场价值。
本发明首次合成了具有一定药用价值的单氟甲基依托昔布,且在一定程度上保持了依托昔布的药用活性,使该新化合物成为潜在的镇痛和抗炎的药物,本发明的合成方法操作简单,成本低廉,易放大生产。
本发明合成的单氟甲基依托昔布在药物活性测试中表现出了和依托昔布相近的活性,对环氧化酶COX-2的抑制活性相近,可作为潜在的依托昔布替代药物。
目前尚无单氟甲基依托昔布的合成方法,本发明作为首次合成的方法步骤简单,仅需三步且中间体无需纯化即可进行下一步反应,所用药品及溶剂可商业购买且价格便宜,另外此合成方法合成条件简单,对反应环境要求小,反应温度仅需回流温度或室温,整体过程较为安全。相较于其他类似化合物的氟化方法而言,步骤简单,过程安全,成本低廉。
附图说明
图1为本发明的单氟甲基依托昔布的反应路线示意图。
图2为本发明的单氟甲基依托昔布的核磁氢谱图。
图3为本发明的单氟甲基依托昔布的核磁氟谱图。
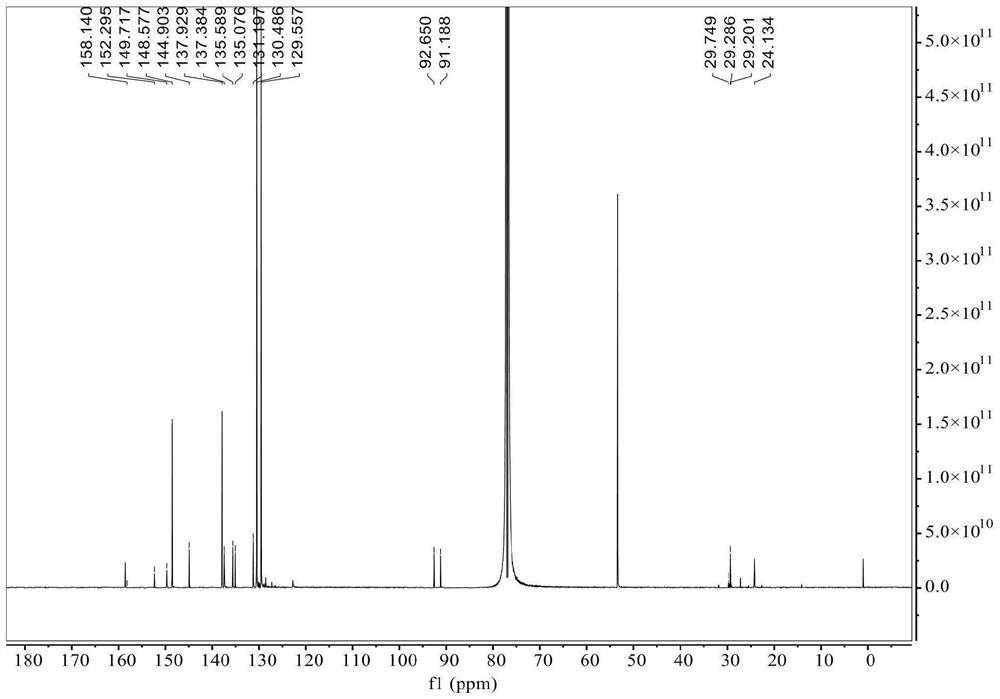
图4为本发明的单氟甲基依托昔布的核磁碳谱图。
图5为本发明的单氟甲基依托昔布的质谱图。
图6为本发明的单氟甲基依托昔布的高效液相色谱图。
具体实施方式
为了更清楚地说明本发明,下面结合优选实施例对本发明做进一步的说明。本领域技术人员应当理解,下面所具体描述的内容是说明性的而非限制性的,不应以此限制本发明的保护范围。
实施例1
一种单氟甲基依托昔布的制备方法,包括以下步骤:如图1所示,图1为本发明的单氟甲基依托昔布的反应路线示意图。
第一步,三氟乙酰基化:
在氮气保护下,将氢化钠(5mmol,120mg)溶解于0.75mL四氢呋喃中,并加入三氟乙酸乙酯(2mmol,0.24ml),加入依托昔布(1mmol,358mg)的四氢呋喃溶液0.75mL,控温75℃保持溶剂的回流。反应12个小时,TLC监控原料点消失,加入1M的盐酸淬灭反应,乙酸乙酯萃取三次,收集有机相,无水硫酸钠干燥,减压旋蒸除去溶剂后得到中间体A的粗产品486mg。
第二步,氟化反应
在氮气保护下,将粗产品中间体A(1mmol,454mg)、三水合硝酸铜(0.2mmol,48mg)溶解于2mL乙腈中,反应两个小时,TLC监控原料点消失。将选择性氟试剂(1.2mmol,425mg)溶解于0.6mL乙腈和0.4mL水的混合溶剂中加入反应液中,乙腈与水的体积比为3:2,反应24小时,TLC监控中间体A消失后结束反应。乙酸乙酯萃取三次,收集有机相,无水硫酸钠干燥,旋干溶液后收集粗品中间体B523mg,无需纯化,直接投入下一步。
第三步,脱三氟甲基乙二醇基团:
在氮气保护下,将中间体B(1mmol,491mg)、氢氧化钾(2mmol,112mg)溶解于1mL四氢呋喃中,保持室温反应12个小时,TLC监控原料点消失,减压旋蒸除去溶剂,用PE:EA=1:1的洗脱液进行柱层析纯化,得到单氟甲基依托昔布130mg,总收率为34.5%,纯度为99.6%。
制备的单氟甲基依托昔布,结构如下所示:
单氟甲基依托昔布的分子量为376.83,熔点156.8℃,固体状态为淡黄色粉末。
核磁数据如图2~图4所示,图2为本发明的单氟甲基依托昔布的核磁氢谱图。
质谱图如5所示,图5为本发明的单氟甲基依托昔布的质谱图。HRMS(ESI)Calcdfor C
实施例2
使用试剂:DMSO(N,N-二甲基亚砜),COX-2检测缓冲液(100mM Tris-HCl,pH 8.0,含有1.25μg/rxn COX-2酶),参考抑制剂DUP-697(5-溴-2-(4-氟苯基)-3-(4-甲基磺酰基苯基)噻吩),COX-2酶的ELISA(Caymen INC.)检测试剂盒。
测试方法:以10nM的单氟甲基依托昔布和依托昔布的测试过程为例
将10nM单氟甲基依托昔布与依托昔布分别溶解在10%DMSO溶液中,并将其稀释10×10倍,取10μl稀释液加入到反应中,确保在所有反应中DMSO的最终浓度为0.5%。向反应中加入COX-2酶检测缓冲液(100mM Tris-HCl,pH 8.0,含有1.25μg/rxn COX-2),并温育10min,之后再向反应中添加底物花生四烯酸(200μM终浓度,1%EtOH终浓度),对于阴性对照试验,使用不含COX-2酶的检测缓冲液。通过COX-2酶ELISA(Caymen INC.)试剂盒检测形成的前列腺素PGE2和荧光强度。使用Tecan Infinite M1000读板器测量发光信号。在每个浓度下进行一式两份的抑制测定。使用计算机软件Graphpad Prism分析发光数据。
结果:将不同摩尔量的单氟甲基依托昔布和依托昔布分别作为测试抑制剂进行COX-2抑制活性测试,以COX-2酶活性平均值作为其活性判断依据。
COX-2酶的活性值计算公式为:%activity=[(F-Fb)/(Ft-Fb)]×100
其中,F:加入各个浓度的抑制剂的测量值;Fb:本底值;Ft:未加抑制剂的测量值。
将单氟甲基依托昔布和依托昔布分别作为测试抑制剂进行COX-2抑制活性测试,以不同浓度下的COX-2酶活性平均值作为其活性判断依据。
表1
表2
比较表1和表2两种测试抑制剂的活性数据,可以看出在五种不同的测试浓度下,COX-2的活性百分值是相似的,由此推断出单氟甲基依托昔布和依托昔布对COX-2酶的抑制活性相似,由此也确认单氟甲基依托昔布的抗炎镇痛效果也和依托昔布类似,所以单氟甲基依托昔布可作为潜在的抗炎镇痛药物或者依托昔布的替代药物。
对比例1
将实施例1中的依托昔布、氢化钠与三氟乙酸乙酯的摩尔比为1:5:2替换为1:7:2进行对比实验。
在氮气保护下,将氢化钠(7mmol,168mg)溶解于0.75mL四氢呋喃中,并加入三氟乙酸乙酯(2mmol,0.24ml),加入依托昔布(1mmol,358mg)的四氢呋喃溶液0.75mL,控温75℃保持溶剂的回流。反应12个小时,TLC监控原料点消失,加入1M的盐酸淬灭反应,乙酸乙酯萃取三次,收集有机相,无水硫酸钠干燥,减压旋蒸除去溶剂,TLC点板确认存在副反应,副产物增多,且PE:EA=1:1的洗脱液洗脱后收集确定各副产物结构,核磁确认反应并未得到中间体A。因此改变其比例无法进行后续反应。
对比例2
将实施例1中的依托昔布、氢化钠与三氟乙酸乙酯的摩尔比为1:5:2替换为1:5:1进行对比实验。
在氮气保护下,将氢化钠(5mmol,120mg)溶解于0.75mL四氢呋喃中,并加入三氟乙酸乙酯(1mmol,0.24ml),加入依托昔布(1mmol,358mg)的四氢呋喃溶液0.75mL,控温75℃保持溶剂的回流。反应12个小时,TLC监控原料点并未消失,继续反应12个小时后TLC监控反应原料点消失,加入1M的盐酸淬灭反应,乙酸乙酯萃取三次,收集有机相,无水硫酸钠干燥,减压旋蒸除去溶剂后,TLC点板确认副产物增多,且PE:EA=1:1的洗脱液洗脱后收集确定各副产物结构,仅得到中间体A 12mg,仅有0.026mmol的反应物,摩尔量太小无法进行后续反应。
因此技术方案中的依托昔布,氢化钠与三氟乙酸乙酯的摩尔比为1:5:2为最佳反应比例,降低或增加其比例都无法得到最佳收率下的目标产物。
对比例3
将实施例1中的第二步中间体A、三水合硝酸铜与选择性氟试剂的摩尔比为1:0.2:1.2替换为1:1:0.8进行对比试验。
第一步,三氟乙酰基化:
在氮气保护下,将氢化钠(5mmol,120mg)溶解于0.75mL四氢呋喃中,并加入三氟乙酸乙酯(2mmol,0.24ml),加入依托昔布(1mmol,358mg)的四氢呋喃溶液0.75mL,控温75℃保持溶剂的回流。反应12个小时,TLC监控原料点消失,加入1M的盐酸淬灭反应,乙酸乙酯萃取三次,收集有机相,无水硫酸钠干燥,减压旋蒸除去溶剂后得到中间体A的粗产品486mg。
第二步,氟化反应
在氮气保护下,将粗产品中间体A(1mmol,454mg)、三水合硝酸铜(1mmol,241mg)溶解于2mL乙腈中,反应两个小时,TLC监控原料点消失。将选择性氟试剂(0.8mmol,283mg)溶解于0.6mL乙腈和0.4mL水的混合溶剂中,乙腈与水的体积比为3:2,反应24小时,TLC监控中间体A消失后结束反应。乙酸乙酯萃取三次,收集有机相,无水硫酸钠干燥,减压旋蒸除去溶剂后TLC点板确认存在多种副产物,核磁确认结构后证明反应未生成中间体B。因此,改变其比例无法得到目标产物中间体B。
对比例4
将实施例1中的第三步中的中间体B与氢氧化钾的摩尔比为1:2替换为2:1进行对比试验。
第一步,三氟乙酰基化:
在氮气保护下,将氢化钠(5mmol,120mg)溶解于0.75mL四氢呋喃中,并加入三氟乙酸乙酯(2mmol,0.24ml),加入依托昔布(1mmol,358mg)的四氢呋喃溶液0.75mL,控温75℃保持溶剂的回流。反应12个小时,TLC监控原料点消失,加入1M的盐酸淬灭反应,乙酸乙酯萃取三次,收集有机相,无水硫酸钠干燥,减压旋蒸除去溶剂后得到中间体A的粗产品486mg。
第二步,氟化反应
在氮气保护下,将粗产品中间体A(1mmol,454mg)、三水合硝酸铜(0.2mmol,48mg)溶解于2mL乙腈中,反应两个小时,TLC监控原料点消失。将选择性氟试剂(1.2mmol,425mg)溶解于0.6mL乙腈和0.4mL水的混合溶剂中,乙腈与水的体积比为3:2,反应24小时,TLC监控中间体A消失后结束反应。乙酸乙酯萃取三次,收集有机相,无水硫酸钠干燥,旋干溶液后收集粗品中间体B523mg,无需纯化,直接投入下一步。
第三步,脱三氟甲基乙二醇基团:
在氮气保护下,将中间体B(1mmol,491mg)、氢氧化钾(0.5mmol,28mg)溶解于1mL四氢呋喃中,保持室温反应12个小时,TLC监控原料点未消失,继续反应24小时后原料点仍未消失,结束反应,减压旋蒸除去溶剂,TLC确认原料未反应完,用PE:EA=1:1的洗脱液进行柱层析纯化,得到单氟甲基依托昔布11mg。总收率为2.9%,纯度为99.6%。收率过低。改变其比例后得不到产物的最高收率,证明技术方案中的比例为最佳比例。
以上所述仅是本发明的较佳实施例而已,并非对本发明作任何形式上的限制,虽然本发明已以较佳实施例揭露如上,然而并非用以限定本发明,任何熟悉本专利的技术人员在不脱离本发明技术方案范围内,当可利用上述提示的技术内容作出些许更动或修饰为等同变化的等效实施例,但凡是未脱离本发明技术方案的内容,依据本发明的技术实质对以上实施例所作的任何简单修改、等同变化与修饰,均仍属于本发明方案的范围内。
- 一种帕瑞昔布钠新晶型及其制备方法
- 一种制备依托昔布中间体-6-甲基烟酸及其酯的方法
- 一种制备依托昔布中间体-6-甲基烟酸及其酯的新方法